

Abstract
Background
Red blood cell (RBC) physiology is directly linked to many human disorders associated with low tissue oxygen levels or anemia including chronic obstructive pulmonary disease, congenital heart disease, sleep apnea and sickle cell anemia. Parasites such as Plasmodium spp. and phylum Apicomplexa directly target RBCs, and surface molecules within the RBC membrane are critical for pathogen interactions. Proteomics of RBC membrane ‘ghost’ fractions has therefore been of considerable interest, but protocols described to date are either suboptimal or too extensive to be applicable to a larger set of clinical cohorts.
Methods
Here, we describe an optimised erythrocyte isolation protocol from blood, tested for various storage conditions and explored using different fractionation conditions for isolating ghost RBC membranes. Liquid chromatography mass spectrometry (LC–MS) analysis on a Q-Exactive Orbitrap instrument was used to profile proteins isolated from the comparative conditions. Data analysis was run on the MASCOT and MaxQuant platforms to assess their scope and diversity.
Results
The results obtained demonstrate a robust method for membrane enrichment enabling consistent MS based characterisation of > 900 RBC membrane proteins in single LC–MS/MS analyses. Non-detergent based membrane solubilisation methods using the tissue and supernatant fractions of isolated ghost membranes are shown to offer effective haemoglobin removal as well as diverse recovery including erythrocyte membrane proteins of high and low abundance.
Conclusions
The methods described in this manuscript propose a medium to high throughput framework for membrane proteome profiling by LC–MS of potential applicability to larger clinical cohorts in a variety of disease contexts.
Electronic supplementary material
The online version of this article (10.1186/s12014-018-9190-4) contains supplementary material, which is available to authorized users.
Keywords: Erythrocyte ghosts, Shotgun proteomics, Tandem mass spectrometry, Membrane proteome, Haemoglobinopathies
Background
Whereas erythrocyte proteins, in particular globins, have been studied extensively, the application of proteomics methods to erythrocyte membrane ‘ghost’ preparations is a relatively recent phenomenon with the first experiments combining gel separation with Matrix Associated Laser Desorption Ionization Time-of-Flight (MALDI-TOF) analysis published in 2002 [1]. Since then, a growing number of original research papers have highlighted the benefits of RBC proteomics, in particular the membrane fraction, as well as drawing light to some of the challenges faced in its implementation and practice. In particular, the difficulties faced in obtaining pure fractions of RBC’s from whole blood, reducing cytosolic haemoglobin contamination as well as overcoming the wide dynamic range in protein expression. To facilitate the collection of pure sample fractions; a large number of methodologies utilizing centrifugation [2], filtration [3, 4], columns [5], flow cytometry [6], multiple enzyme digests combined with fractionation [7] as well as in vitro whole blood incubation to enable reticulocytes to mature over 72–96 h have been presented [3].
One of the more comprehensive published protocols proposes a combination of some of these approaches along with the use of a haemoglobin depletion kit to allow the collection of an enriched RBC fraction with minimal cytosolic contamination [8]. In addition to the diversity of erythrocyte isolation and purification methods, there exist no established protocols for the extraction, digestion and quantification of RBC membrane proteins by liquid chromatography based mass spectrometry (LC–MS). The major areas of differentiation in the sequence of steps required for comprehensive RBC ghost preparation for LC MS are in choices related to the use or not of detergents [9–11] membrane isolation procedures including the use of leucocyte filters [12] and sample collection and storage conditions most notably the impact of using RBC preservative fluids [13].
The anuclear erythrocyte is unique amongst all human cell types in that its diversity is primarily determined by its membrane and associated components. Red cell membranes (RCM) are composed of mainly protein and lipid based structures impacting their transport, antigen profiles and mechanical characteristics, all of which can be altered in disease. These mechanical functions involve oxygen transport, adhesion interactions with other cells as well as receptor signalling processes which have been implicated in infection and inflammatory diseases [14]. Red cells constitute the core oxygen transport molecules in humans and are generated via a process of erythropoiesis. This is divided into four stages in a process triggered by low oxygen tension in cells which in turn activate the hormone erythropoietin in a negative feedback loop [15]. Myeloid progenitor cells in the bone marrow become locked into an erythropoietic fate and are released into the blood as reticulocytes which mature into fully fledged erythrocytes staying in circulation for 90–120 days in healthy individuals [16]. Erythrocyte survival and morphology can be severely altered in a number of diseases making them an important target for study [17, 18].
Ghost RCM’s constitute a collection of erythrocytes devoid of their cytosolic content with the retainment of their membrane and cytoskeletal structures. LC–MS based proteomics offers sensitive methods with the capacity to identify a significant proportion of integral and associated membrane proteins. An overview of current literature shows maximal identifications of several hundred proteins at best using described methods [4, 8, 19–21]. A recent publication has proposed combined identifications of over 2500 membrane proteins using an amalgamation of fractionation approaches combined with three sets of multi-enzyme digests [7]. Though insightful, this latest method proposed is labour intensive and unlikely to be applicable for mid-to large-scale clinical discovery cohorts. In this paper, we assess the impact of varying storage conditions on erythrocyte proteomic expression as well as the stability of major membrane proteins and propose a low intervention, high recovery methodology for the comprehensive isolation and characterization of the protein content of erythrocyte membranes. Few investigations have been made into the impact of prolonged storage on red blood cells with the only significant paper using LCMS published choosing to focus on morphological changes and their resultant impact on the quantity and composition of phospholipids [22]. The fundamental approach of the proposed method is in its preference for the limited usage of chemical modulators such as detergents in the purification and recovery of RBC ghosts from whole blood. Despite this, a demonstrated capacity to reproducibly cover a broad spectrum of known membrane and membrane associated proteins in human whole blood is achieved.
Methods
Sample collection: stability studies
Stability studies were conducted on whole blood collected from three volunteer anonymized healthy donors who gave informed consent. Four vials of 10 millilitres of whole blood were collected into K2EDTA vacutainers via venepuncture. The following storage conditions were assessed on each sample: (1) whole blood kept at 4 °C with packed erythrocyte membranes isolated within 24 h of venepuncture; (2) whole blood kept at room temperature with packed erythrocyte membranes isolated within 24 h of venepuncture (3) whole blood kept at 4 °C for 7 days with packed erythrocyte membranes isolated on day 8 and; (4) whole blood kept at room temperature for 7 days with membrane isolation on day 8 (Fig. 1).
Fig. 1.

Summary of experimental process
Sample collection: RBC proteome coverage optimization
Twelve samples were selected from the wider Proteomics Pathways of Sickle Cell Anaemia (P2SCA) study cohort. The study involved the recruitment of one-hundred and twenty one individuals identified from the Muhimbili Sickle Cell Programme database which has since 2004 surveilled Sickle Cell Anaemia (SCA) cases and controls from outpatient clinics and admissions at the Muhimbili National Hospital in Dar es Salaam, Tanzania. Potential participants were approached and informed of the study objectives and those willing to participate gave informed consent. Whole blood was collected into 10 ml K2EDTA vacutainers via venepuncture and the samples shipped within 72 h to the University of Oxford at ambient temperature. Samples were received, logged and processed to isolate plasma, erythrocyte and buffy coat fractions within 7 days of collection. The P2SCA study was granted full ethical clearance by the review boards at the Muhimbili University of Allied Sciences (MUHAS) and the Tanzania Nation Institute for Medical Research (NIMR).
Erythrocyte ‘ghost’ membrane isolation
Ten millilitres of whole blood was spun for 10 min at 3000 RPM (1731G) to separate packed red cells, plasma and buffy coat. The latter two fractions were removed via aspiration and frozen at − 80 °C, leaving behind the packed erythrocytes which were taken forth for extensive washing and lysis steps to isolate ghost erythrocyte membrane cells.
Approximately 2–4.5 ml of packed erythrocytes were transferred to a 50 ml falcon tube and washed in tenfold volume of 1X PBS. The suspension was mixed by hand inversion and spun at 3000 RPM at 4 °C thrice. Erythrocytes were pelleted after each centrifugation step and the resultant supernatant aspirated. Washed cells from this point were kept on ice for the remaining steps. A lysis buffer of 5 mM Na2HPO4 at pH 7.4 was prepared and kept at 4 °C before use. The washed RBC pellets were transferred to 50 ml ultracentrifuge tubes; 30 ml of cold lysis buffer were added to each erythrocyte pellet, mixed by strong inversion and spun on a Beckman Coulter ultracentrifuge at 14,000 RPM at 4 °C for 10 min with slow deceleration to avoid ghost pellet disruption. The resulting supernatant was removed and the steps repeated for a total of five washes. 0.5 mM of the protease inhibitor phenylmethylsulfonyl fluoride (PMSF) was added to the lysis buffer at the last wash cycle. The resultant ghost membranes were promptly collected and frozen at − 80 °C.
Ghost membrane preparations for stability studies
Ghost membranes were resuspended 3:1 in 4× Laemmli Buffer (200 mM Tris–HCl, pH 6.8, 400 mM DTT, 8% SDS, 40% Glycerol and 0.4% Bromophenol Blue) and a Pierce BCA assay kit used to determine protein concentration. Using 80 μg of starting protein amount, the resuspended ghosts underwent reduction-alkylation, double methanol-chloroform precipitation to remove excess detergent and overnight digest with trypsin [23]. Peptide desalting was performed on a Waters C18 solid phase extraction column. Briefly, the column was equilibrated with organic buffer (65% ACN; 35% H2O, 0.1% TFA) followed by addition into aqueous buffer (65% H2O; 35% ACN, 0.1% TFA) of the peptide digest sample. The bound peptides were then washed with the aqueous buffer and eluted in 1200 μl of organic solution. The eluate was dried down in a vacuum centrifuge and resuspended in aqueous buffer to a final concentration of 2 μg/μl.
Ghost membrane preparations for RBC proteome optimization and analysis
Ghost membrane suspensions from sample P2S1 were spun down and 1:1 volumes of the semi solid pellet (tissue) and its’ supernatant combined as starting material for the reduction-alkylation, methanol chloroform precipitation and overnight in-solution digestion with trypsin steps. The resultant peptides were quantified using a Thermo kit (Pierce™ Quantitative Colorimetric Peptide Assay) and 50ug from each patient sample loaded on a C18 based SOLA Hydrophobic Reversed Phase (HRP) column. Briefly, the column was equilibrated with organic buffer (65% ACN; 35% H2O, 0.1% TFA) followed by addition into aqueous buffer (65% H2O; 35% ACN, 0.1% TFA) of the peptide digest sample. The bound peptides were then washed with the aqueous buffer and eluted in 600 μl of organic solution. The eluate was dried down in a vacuum centrifuge and resuspended in aqueous buffer to a final concentration of 2 μg/μl.
Protein and peptide quantitation assays
Pierce BCA Protein and Quantitative Colorimetric Peptide Assay Kits (Thermo Scientific) were respectively utilized to quantify the protein and peptide concentrations of sample aliquots. Standard kit protocols were followed for each assay with each sample run in duplicate and read on a TECAN microplate reader. The protein BCA assays were read at a wavelength of 570 nm after a 30 min plate incubation at 37 °C. The colorimetric peptide assays were read at 480 nm after a 15 min incubation at 37 °C. Raw data readouts were blank subtracted and the unknown sample absorbances extrapolated from a four parameter non-linear regression quadratic curve fit.
Mass spectrometry analysis and protein identification
Ultra-performance liquid chromatography (UPLC) separation was performed on a Dionex Ultimate 3000 coupled to a Thermo Scientific Q-Exactive High Field Mass Spectrometer for tandem MS–MS analysis. The UPLC was fitted with an EASY-Spray PepMap C18 column (500 mm × 75 μm, 2 μm particle size) over a 60 min gradient of 2–35% acetonitrile in 5% DMSO, 0.1% formic acid at a flow rate of 250 nl/min. MS1 scans were acquired at a resolution of 60,000 at 200 m/z with the top 12 most abundant precursors targeted for HCD fragmentation. Raw files from each injection were converted to a MASCOT Generic Format (MGF) and searched on the MASCOT server (version 2.5.1) against the UPR Homosapiens database (dated 2017/05/10). The search parameters allowed for 1 missed cleavage, a peptide mass tolerance of ± 10 ppm with carbamidomethyl as a fixed modification on cysteine and the following variable modifications; deamidation (N), deamidation (Q) and oxidation (M). Fragment mass tolerance was set to ± 0.05 Da.
Further analysis of MS raw data was performed using the MaxQuant (v1.6.0.1) [24] and Perseus (v1.6.0.2) [25] programs. Proteins were identified by searching raw data against the UniProt Reference human proteome database (May 2017 version, 93591 sequences) with carbamidomethylation of cysteine as a fixed modification. The variable modifications allowed were oxidation of methionine, and deamidation of asparagine and glutamine. Cleavage specificity was tryptic, allowing for a maximum of two missed cleavages. Enabling the “match between runs” option allowed for identification transfer between samples. Label-free protein quantitation was performed with a minimum ratio count of 2. All other settings used default parameters, which include a peptide and protein false discovery (FDR) rate cut-off of 1%.
The protein groups text files from MaxQuant were used for onward quantitative protein expression analysis across experimental groups. Those not meeting the FDR threshold or identified from the reverse database or contaminant hits were removed prior to further analysis. The resultant list was analysed as per published instructions by the software developers [25]. Statistical analysis on log transformed data were also performed on Perseus.
Results
Effects of whole blood storage temperature on erythrocyte membrane protein expression
Comparative analysis of the number of protein hits obtained from erythrocyte ghost membrane samples isolated from three volunteers show no significant loss in total proteins identified in whole blood samples stored for up to 7 days (Fig. 2). On a global scale, total protein expression remains relatively stable in whole blood collected and stored for up to 7 days. The results obtained from the stability experiments suggest ambient storage to be more robust for RBC proteomics in comparison to refrigeration. Targeted analysis of the major membrane proteins of different classes are highlighted in Fig. 3a showing that they demonstrate relative stability across conditions. Correlation curves show strong linearity between conditions, as displayed in Fig. 3b i–iii.
Fig. 2.
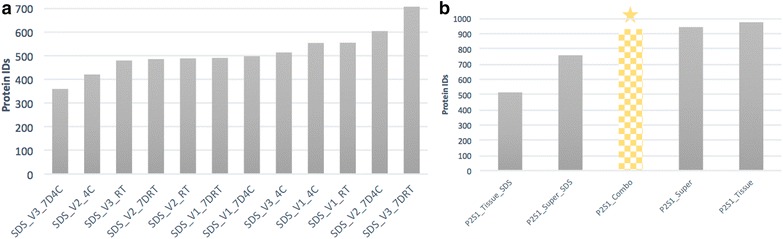
Bar plot showing the total protein numbers identified from MASCOT searches of raw data obtained from: a Protein extractions from RBC membranes kept for up to 7 days in different storage conditions; and b comparative detergent and non-detergent based RBC membrane extraction methods applied to membrane supernatant “Super”, semi-solid tissue pellets referred to as “Tissue” and a combination of both in samples from the P2SCA cohort. P2S1 acronym stands for Proteomics Pathways Sample #1
Fig. 3.

a Profile plot showing trends in the quantitative expression levels of major membrane proteins across distinct pre-processing conditions. Lines labelled as follows V (Volunteer) # (participant number) 4C (storage at 4 °C for 24 h); V# 7D4C (storage at 4 °C for 7 days); V# 7DRT (storage at room temperature for 7 days); V# RT (storage at room temperature for 24 h). b i–iii Pearson correlation plots of comparative conditions within individual donors
Assessing the effectiveness of utilizing detergent and non-detergent based buffers for membrane solubilization
The second assessment made in these experiments were the optimal methods to be adopted for extensive coverage of the membrane proteome using detergent and non-detergent assisted solubilization (Fig. 4). A sizeable increase in the number of identifiable proteins was seen by adapting protocols from resuspending ghost pellets in SDS laced buffer to utilizing direct combinations of the semi solid membrane pellets combined with their soluble supernatants (Fig. 2b). The latter approach was tested by assessing the adaptation of tissue versus supernatant preparations. The results obtained show some diversity in the scope of proteins captured (Fig. 5) resulting in the adaptation of a protocol combining both fractions.
Fig. 4.
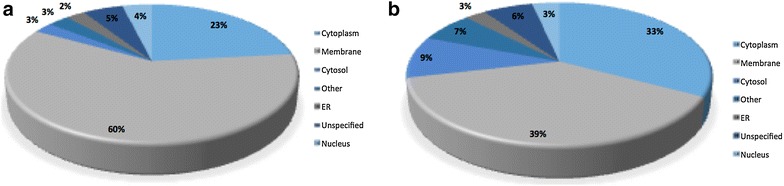
a Pie chart summarizing cellular localization of proteins identified from SDS assisted solubilized method. b Pie chart summarizing cellular localization of proteins identified from non-SDS assisted solubilized method
Fig. 5.

Diagram showing the degree of overlapping proteins expressed in the non-detergent solubilized supernatant and membrane tissue fragments collected following hypotonic lysis assisted ghost membrane preparation of sample P2S1. ‘Super’ refers to the supernatant isolated from isolated ghost membrane cells following centrifugation. ‘Tissue’ refers to the semi-solid pellet collected after this step
Assessment of membrane proteome coverage and the impact of possible haemoglobin contamination
Manual analysis of the localization of the total range of proteins identified from the optimization experiments show a majority of them categorized as “membrane” or “membrane associated”. The other major proportion were the cytoplasmic fractions, constituting 23% in the SDS-assisted and 33% in the non-SDS assisted method. One of the major concerns in the MS analysis of erythrocyte ghosts is potential contamination with haemoglobin which constitutes > 98% of RBCs. The results obtained from the proposed protocol suggest that the comprehensive washing and lysis steps described above sufficiently lower haemoglobin concentrations thus allowing the quantifiable expression of the diverse erythrocyte proteome. Known membrane proteins such as glycophorins, spectrin (alpha & beta), ankyrin, calmodulin, band 3 anion, glyceraldehyde-3-phosphate dehydrogenase, actin and beta-adducin were the predominant hits obtained in all MASCOT searches with haemoglobin featuring lower down with lower scores. Our method also consistently identified low-abundant membrane proteins such as tropomodulin, several isoforms of the solute carrier family proteins SLC40, SLC43, SLC2A1, ADP/ATP translocase, transmembrane emp24 domain containing protein and calpastatin.
Robustness of proposed method across serial measurements of proteins from clinical blood samples
Ten individual clinical samples from the P2SCA cohort were searched on the MASCOT server and the sequence coverage of six key membrane proteins tracked as a measure of the robustness of the utilized preparatory and LC–MS analysis methods. Table 1 shows comparable sequence coverage rates across ten independently prepared and analysed patient samples. Analysis of variation (ANOVA) tests on iBAQ quantitative values obtained on Perseus showed no significant difference across protein expression levels of ten independent samples of the same Hb genotype (data not shown). From a starting set of a total 2081 proteins identified across ten samples; several key filtration steps as described above were applied. A rule was set which only allowed proteins identified in 80% of the samples to be included in the final statistical analysis; this yielded a final list of 1296 proteins for comparison. These were normally distributed across the board (Additional file 1: Figure 1).
Table 1.
Summary of attained sequence coverage across ten independent samples prepared with the finalized detergent free method demonstrating robust identification of major membrane proteins
| Accession | Name | Function | Sequence coverage (%) samples 1–10 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |||
| P02549 | Spectrin alpha chain | RBC membrane skeleton | 95 | 88 | 88 | 92 | 89 | 90 | 90 | 91 | 93 | 93 |
| P11277 | Spectrin beta chain | RBC membrane skeleton | 90 | 89 | 90 | 89 | 88 | 91 | 90 | 94 | 90 | 95 |
| P16157 | Human Ankyrin | Integral membrane protein | 95 | 90 | 93 | 94 | 90 | 91 | 90 | 94 | 95 | 93 |
| P11171 | Protein 4.1 | RBC membrane skeleton | 77 | 89 | 73 | 71 | 72 | 71 | 75 | 78 | 69 | 76 |
| P02730 | Band 3 anion transport | Ion exchange | 53 | 59 | 67 | 58 | 77 | 69 | 67 | 52 | 67 | 53 |
| P04406 | Glyceraldehyde-3-phosphate dehydrogenase | Glycolytic enzyme | 98 | 84 | 94 | 95 | 94 | 94 | 94 | 93 | 95 | 97 |
Discussion
Human erythrocytes represent an important cell type relevant to several disease models. Disease and infection associated morbidity has been known to impact the lifespan and morphology of these cells. In their role as oxygen carriers, healthy RBC’s undergo routine cycles of deformation and reformation allowing them to pass through capillaries much smaller in diameter. RBC’s are anuclear cells with their cytosols primarily composed of haemoglobin, this renders their membranes as their major site of differentiation reflecting changes effected by genotype and environmental differences. That said, there exist few methodologies which allow medium to high throughput capacity study of the membrane structures of these cells in a clinical context. The methods proposed here aim to make a contribution towards this shortfall by allowing researchers to undertake these experiments in a reproducible manner.
The high throughput method proposed for comprehensive proteomic analysis of the RBC membrane proteome is novel standing out as a research publication which has assessed the implications of storage duration and condition on the recovery of proteins from erythrocyte ghosts. In addition, with the demonstrable impact of this an optimal protocol with minimal sample interference is also presented allowing for the robust selection of a broad spectrum of membrane-associated proteins without fractionation. The potential impact of the presentation of this simplified and streamlined approach for ghost membrane analysis cannot be overstated and should act as an important step in allowing the analysis of this key fraction of whole blood in numerous disease contexts,
Conclusions
The results presented demonstrate the stability of the erythrocyte proteome over a defined period; a point which should facilitate sample collection and transport across a range of clinical studies. In the area of sample preparation, the protocol proposed offers low intervention methods compatible with established protein isolation techniques applicable to large sample numbers. Without the use of fractionation and multi-enzyme digestion methods, a large number of robustly identifiable proteins are still reliably covered and quantifiable using standard proteomics analysis packages and tools.
Additional file
Additional file 1: Figure 1 Histogram showing the distribution of the Intensity Based Absolute Quantification (iBAQ) values for identified peptides across samples 1 to 10 (represented by iBAQ 1-10 respectively) listed in Table 1.
Authors’ Contributions
HF prepared the full manuscript including figures and tables. HF also performed all the experiments detailed in this manuscript. PM was responsible for obtaining informed consent from potential participants as well as bleeding them. LB provided training to HF on a basic ghost isolation protocol from RBC’s. MLT assisted with LC–MS analysis. SD assisted with LC–MS analysis. RF assisted with method development. GLR assisted with participant recruitment and the shipment of samples from Tanzania. JM assisted with study design, participant overview and identification. BMK provided the laboratory space and equipment for the completion of these works. BMK also provided detailed editing of earlier drafts of this manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors would like to acknowledge the contributions of Dr’s Furahini Tluway and Raphael Sangeda from MUHAS for their assistance in participant recruitment and data management.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This study received full ethical approval with all participants giving informed consent prior to enrolment.
Funding
This study was supported by a fellowship award from the Africa Research Excellence Fund (Number: AREF-157-0004-F-FYE). These funds were used to complete all aspects of work related to study design, data collection and analysis.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- ANOVA
analysis of variation
- FDR
false discovery rate
- HRP
hydrophobic reversed phase
- LC–MS
liquid chromatography mass spectrometry
- MALDI-TOF
Matrix Associated Laser Desorption Ionization Time-of-Flight
- MUHAS
Muhimbili University of Allied Sciences
- NIMR
National Institute for Medical Research
- PMSF
phenylmethylsulfonyl fluoride
- P2SCA
Proteomics Pathways of Sickle Cell Anaemia
- RBC
red blood cell
- RCM
red cell membrane
- SCA
sickle cell anaemia
- UPLC
ultra-performance liquid chromatography
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12014-018-9190-4) contains supplementary material, which is available to authorized users.
References
- 1.Low TY, Seow TK, Chung MC. Separation of human erythrocyte membrane associated proteins with one-dimensional and two-dimensional gel electrophoresis followed by identification with matrix-assisted laser desorption/ionization-time of flight mass spectrometry. Proteomics. 2002;2(9):1229–1239. doi: 10.1002/1615-9861(200209)2:9<1229::AID-PROT1229>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 2.Brosseron F, et al. Stepwise isolation of human peripheral erythrocytes, T lymphocytes, and monocytes for blood cell proteomics. Proteom Clin Appl. 2012;6(9–10):497–501. doi: 10.1002/prca.201200032. [DOI] [PubMed] [Google Scholar]
- 3.Pasini EM, et al. In-depth analysis of the membrane and cytosolic proteome of red blood cells. Blood. 2006;108(3):791–801. doi: 10.1182/blood-2005-11-007799. [DOI] [PubMed] [Google Scholar]
- 4.Pesciotta EN, et al. A label-free proteome analysis strategy for identifying quantitative changes in erythrocyte membranes induced by red cell disorders. J Proteom. 2012;76:194–202. doi: 10.1016/j.jprot.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ringrose JH, et al. Highly efficient depletion strategy for the two most abundant erythrocyte soluble proteins improves proteome coverage dramatically. J Proteome Res. 2008;7(7):3060–3063. doi: 10.1021/pr8001029. [DOI] [PubMed] [Google Scholar]
- 6.Goodman SR, et al. The isolation of reticulocyte-free human red blood cells. Exp Biol Med (Maywood) 2007;232(11):1470–1476. doi: 10.3181/0706-RM-163. [DOI] [PubMed] [Google Scholar]
- 7.Bryk AH, Wisniewski JR. Quantitative analysis of human red blood cell proteome. J Proteome Res. 2017;16:2752–2761. doi: 10.1021/acs.jproteome.7b00025. [DOI] [PubMed] [Google Scholar]
- 8.Alvarez-Llamas G, et al. A novel methodology for the analysis of membrane and cytosolic sub-proteomes of erythrocytes by 2-DE. Electrophoresis. 2009;30(23):4095–4108. doi: 10.1002/elps.200900046. [DOI] [PubMed] [Google Scholar]
- 9.Churchward MA, et al. Enhanced detergent extraction for analysis of membrane proteomes by two-dimensional gel electrophoresis. Proteome Sci. 2005;3(1):5. doi: 10.1186/1477-5956-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prete PS, et al. Multiple stages of detergent-erythrocyte membrane interaction—a spin label study. Biochim Biophys Acta. 2011;1808(1):164–170. doi: 10.1016/j.bbamem.2010.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Yu J, Fischman DA, Steck TL. Selective solubilization of proteins and phospholipids from red blood cell membranes by nonionic detergents. J Supramol Struct. 1973;1(3):233–248. doi: 10.1002/jss.400010308. [DOI] [PubMed] [Google Scholar]
- 12.Pietersz RN, van der Meer PF, Seghatchian MJ. Update on leucocyte depletion of blood components by filtration. Transfus Sci. 1998;19(4):321–328. doi: 10.1016/S0955-3886(98)00056-3. [DOI] [PubMed] [Google Scholar]
- 13.D’Amici GM, et al. Red blood cell storage in SAGM and AS3: a comparison through the membrane two-dimensional electrophoresis proteome. Blood Transfus. 2012;10(Suppl 2):s46–s54. doi: 10.2450/2012.008S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrison T, et al. Erythrocyte G protein-coupled receptor signaling in malarial infection. Science. 2003;301(5640):1734–1736. doi: 10.1126/science.1089324. [DOI] [PubMed] [Google Scholar]
- 15.Fried W. Erythropoietin and erythropoiesis. Exp Hematol. 2009;37(9):1007–1015. doi: 10.1016/j.exphem.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 16.Dzierzak E, Philipsen S. Erythropoiesis: development and differentiation. Cold Spring Harb Perspect Med. 2013;3(4):a011601. doi: 10.1101/cshperspect.a011601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barcellini W, et al. Hereditary red cell membrane defects: diagnostic and clinical aspects. Blood Transfus. 2011;9(3):274–277. doi: 10.2450/2011.0086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mohandas N, Gallagher PG. Red cell membrane: past, present, and future. Blood. 2008;112(10):3939–3948. doi: 10.1182/blood-2008-07-161166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alvarez-Llamas G, et al. A role for the membrane proteome in human chronic kidney disease erythrocytes. Transl Res. 2012;160(5):374–383. doi: 10.1016/j.trsl.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 20.Kakhniashvili DG, Bulla LA, Jr, Goodman SR. The human erythrocyte proteome: analysis by ion trap mass spectrometry. Mol Cell Proteom. 2004;3(5):501–509. doi: 10.1074/mcp.M300132-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Kakhniashvili DG, et al. The proteomics of sickle cell disease: profiling of erythrocyte membrane proteins by 2D-DIGE and tandem mass spectrometry. Exp Biol Med (Maywood) 2005;230(11):787–792. doi: 10.1177/153537020523001102. [DOI] [PubMed] [Google Scholar]
- 22.Almizraq R, et al. Storage of red blood cells affects membrane composition, microvesiculation, and in vitro quality. Transfusion. 2013;53(10):2258–2267. doi: 10.1111/trf.12080. [DOI] [PubMed] [Google Scholar]
- 23.Kaisar M, et al. Plasma degradome affected by variable storage of human blood. Clin Proteom. 2016;13:26. doi: 10.1186/s12014-016-9126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 25.Tyanova S, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13(9):731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure 1 Histogram showing the distribution of the Intensity Based Absolute Quantification (iBAQ) values for identified peptides across samples 1 to 10 (represented by iBAQ 1-10 respectively) listed in Table 1.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.