

 This work is licensed under a
This work is licensed under a Abstract
Heart failure is a major cause of morbidity and mortality in type 2 diabetes. Type 2 diabetes contributes to the development of heart failure through a variety of mechanisms, including disease-specific myocardial structural, functional and metabolic changes. This review will focus on the contemporary contributions of state of the art non-invasive technologies to our understanding of diabetic cardiomyopathy, including data on cardiac disease phenotype, cardiac energy metabolism and energetic deficiency, ectopic and visceral adiposity, diabetic liver disease, metabolic modulation strategies and cardiovascular outcomes with new classes of glucose-lowering therapies.
Introduction
Diabetes has reached epidemic proportions and is now among the top 10 causes of death worldwide (1). Type 2 diabetes (T2D) is associated with an increased risk of both heart failure (HF) and cardiovascular mortality even in the absence of coronary artery disease (2, 3). Cardiovascular disease is the leading cause of mortality in patients with diabetes, despite advances in treatment (4, 5). HF is a particularly common complication of diabetes (6, 7, 8), with poor outcomes and five-year survival rates of <25% (5). Poorer glycemic control (hazard ratio (HR) 1.32 per percentage point of HbA1c) is an important predictor of HF development (3).
T2D contributes to the development of HF through a variety of mechanisms, including disease-specific myocardial structural, functional and metabolic changes. The term diabetic cardiomyopathy is applied when cardiac structural and haemodynamic changes are not directly attributable to other confounding factors such as coronary artery disease and hypertension, in patients with diabetes (9). This clinical entity is currently poorly understood, but is clearly of significant clinical importance, given the robust association of diabetes with HF and increased cardiovascular mortality.
Myocardial structural changes in diabetes
Although the link between HF and diabetes had first been suggested by Leyden as early as 1881 (10), it was not until 1972 when Rubler described the evidence that myocardial damage exists in diabetes independently of other vascular diseases (11). They observed ventricular hypertrophy with diffuse fibrotic strands extending between bundles of muscle fibres and myofibrillar hypertrophy on histopathology in a series of post-mortem studies of four diabetic cases and coined the term ‘diabetic cardiomyopathy’.
In the last 2 decades, there has been an expansion in the armamentarium of non-invasive imaging technologies capable of providing detailed information about the structure of the heart in the health and disease. Patients with diabetes have been extensively phenotyped with a nuanced description of disease burden using these technologies, demonstrating the presence of hypertrophic response of the left ventricle (LV) independently of arterial blood pressure (12). However, the strong association among hypertension, and diabetes (13) is universally accepted, with a significant amount of overlap between the complications of diabetes and hypertension (14); making it difficult to distinguish the impact of diabetes from that of hypertension on the myocardial structural changes reported by many studies.
Several alterations in LV geometry have been demonstrated in patients with diabetes. One study has reported a 1% rise in HbA1c level was associated with a 3.0 g increase in LV mass in elderly subjects (15). Although an increased LV mass is independently associated with diabetes, often this increase was shown to be modest (16, 17). LV concentric remodelling represents the main structural characteristic of diabetic heart disease, precedes the development of clinical HF and was shown to be a strong predictor of adverse cardiovascular events (18). There is less evidence that diabetes itself can cause left ventricular dilatation and eccentric remodelling in the absence of CAD, obesity or hypertension (19). Further, LV concentric remodelling was shown to be more strongly predictive of cardiovascular mortality than eccentric remodelling (18).
Interstitial fibrosis has been implicated in the pathogenesis of LVH and has been identified in the more advanced stages of diabetic cardiomyopathy (11). The role of interstitial fibrosis in the pathogenesis of LVH in stable/early diabetic cardiomyopathy is much less clear, as abnormal myocyte hypertrophy rather than fibrosis appears to predominate in the early stages (20). Cardiovascular magnetic resonance (CMR) imaging native and post-contrast T1 mapping for extracellular volume (ECV) quantification allows for non-invasive quantification of myocardial extra cellular matrix expansion, and it was demonstrated that the ECV correlates closely with collagen proportionate area on histology samples obtained from patients with HF (21). Using this technique, two recent studies demonstrated no significant increase in ECV and native T1 mapping in patients with well-controlled T2D, suggesting the absence of significant extra cellular matrix expansion, even in the presence of LV concentric remodelling and diastolic dysfunction (22, 23). In a larger study of consecutive patients referred for CMR without amyloidosis, investigators showed higher median ECV in patients with diabetes (n = 231) than in those without diabetes (n = 945) (24). However, in this study, 85% of the patients with diabetes had diagnosed hypertension, which confounds the results.
Describing the myocardial structural changes detected in hypertensive heart disease in detail is beyond the scope of this review article. However, given the significant overlap with the diabetic cardiomyopathy phenotype, in summary hypertension results in increasing arterial stiffness and afterload, leading to remodelling of the myocardium due to cardiomyocyte hypertrophy, fibroblast stimulation and then increased collagen formation (3). In a cohort of well-controlled hypertensive patients, CMR T1 mapping revealed increased diffuse myocardial fibrosis, with small increases in T1 values which were only detected in patients with significant LV hypertrophy (25). Another study has shown concentric LV hypertrophy to be more prevalent than eccentric remodelling in hypertensive patients (26).
Myocardial functional changes in diabetes
Despite the link with HF on a population level (12), the majority of studies report that diabetes has little or no effect on global LV ejection fraction (LVEF), with the exception of the Strong Heart Study, which has demonstrated the presence of a mild reduction in LVEF (16). However, diabetes traditionally has been linked to diastolic dysfunction mainly based on echocardiography. Consequently, diastolic abnormalities have been suggested as the earliest functional effect of diabetic cardiomyopathy, with reported prevalence rates in asymptomatic, normotensive patients with T2D varying from 15 to as high as 75 per cent (27). The Strong Heart Study demonstrated that the extent and frequency of diastolic dysfunction was directly proportional to the HbA1c level (16).
The combination of pulsed tissue Doppler velocity of the medial mitral annulus (e′) with early passive transmitral inflow velocity (E) has been validated as a reliable index of left ventricular filling pressure. E/e′ ratio has been shown to be a useful prognostic biomarker in diabetic patients. Importantly, abnormality in E/e′ was shown to be associated with insulin resistance (28). From and coworkers in large study of 1760 diabetic patients with a tissue Doppler echocardiographic assessment showed that abnormalities in E/e’ in diabetic patients is associated with the subsequent development of HF and increased mortality independent of hypertension, coronary disease or other echocardiographic parameters (29).
The recent use of relatively less load dependent, sensitive measures of myocardial function with strain imaging by echocardiography and CMR has demonstrated the presence of subtle systolic dysfunction to be frequent as a marker of subclinical heart disease in diabetic patients. Both reduced longitudinal contractility and impaired systolic circumferential strain have been shown in diabetics (30). Although these subclinical abnormalities in contractility are widely considered to be a precursor to the onset of clinical HF in diabetes, prognostic data on the use of strain measures in diabetes is lacking and large longitudinal studies will need to assess this and better define the spectrum of diabetic heart disease.
Myocardial metabolic changes in diabetes
Myocardial energy metabolism in diabetes
Maintenance of adequate levels of cardiac high-energy phosphate metabolites, ATP, the energy source for contraction and phosphocreatine (PCr), the major energy storage compound, are of vital importance for normal heart function. Altered myocardial metabolism has been widely considered among the potential mechanisms leading to diabetic heart disease. In the normal heart, 60–90% of ATP synthesis is generated from fatty acids (FA), with a lesser proportion (10–40%) from glucose (31). In diabetes, insulin fails to suppress hormone-sensitive lipase in adipose tissue and very low-density lipoprotein secretion in the liver leading to high circulating FAs. This, in turn activates peroxisome proliferator activated receptor-α (PPARα), which upregulates myocardial FA uptake and metabolism while decreasing glucose transporter 4 (GLUT4) (32, 33). Hence, these systemic metabolic changes in diabetes modify metabolism in the heart, culminating in abnormal cardiac substrate utilisation, impaired cardiac efficiency and decreased energy generation (19, 34, 35, 36, 37). FA regulate glucose metabolism in the heart by activating pathways that lead to the attenuation of insulin signals, thereby inhibiting insulin-mediated glucose transport (38, 39, 40). Due to increased FA availability as a substrate and increased gene expression of FA oxidation enzymes via peroxisome PPARα activation, the β-oxidation increases. This increase in FA availability, and consequently, increased cardiac usage (36, 41, 42, 43, 44, 45, 46), is thought to result in a loss of metabolic flexibility, efficiency between substrate use and ATP production in the diabetic heart (46). The free energy yielded by hydrolysis of ATP is affected by the substrate oxidized (47) and this is lower when excess FA are used compared to glucose (48, 49, 50), resulting in mitochondrial inefficiency and lower ATP yield.
Myocardial energetic impairment in patients with type 2 diabetes
The relative concentration of PCr to ATP (PCr/ATP) is a sensitive index of the energetic state of the myocardium (31). Phosphorus magnetic resonance spectroscopy (31P-MRS) allows non-invasive assessment of the myocardial PCr/ATP. Decreased PCr/ATP is a predictor of mortality (31), linked to contractile dysfunction (51) and is a well-recognized complication of diabetes (30). This pre-existing energetic deficit in diabetic cardiomyopathy is exacerbated by exercise (30). Additionally, exercise PCr/ATP was shown to correlate with impaired myocardial perfusion and oxygenation, suggesting that, in diabetes, coronary microvascular dysfunction exacerbates derangement of cardiac energetics under conditions of increased workload (30).
Although significant correlations between myocardial systolic strain and PCr/ATP were demonstrated (30), the causal role of altered energetics in contractile dysfunction in diabetic hearts remains unclear, and additional research is therefore necessary to delineate the role of myocardial energetics in the development of cardiac dysfunction in patients with T2D.
Manipulation of substrate utilisation
Epidemiological data have shown an association between glycaemia and incident HF events in patients with or at risk of T2D (52, 53, 54, 55); as a result, major emphasis has been placed on the carbohydrate mechanism. Paradoxically, overall, glucose-lowering drugs or strategies increased the risk of HF compared with standard care (56). There is therefore a need for new and effective alternative therapeutic strategies to reduce the prevalence and incidence of HF in patients with T2D. As such substrate metabolism has become a potential target of pharmacological agents to improve the cardiac function. Myocardial utilization of the glucose and FAs are regulated by substrate availability, competition at the level of the mitochondria and also at the site of cellular entry (57) (Fig. 1). Thus, agents that affect mitochondrial substrate uptake or cellular substrate uptake have been developed. Table 1 includes a list of potential therapeutic strategies to restore the balance of fuel utilisation.
Figure 1.

Cardiac energy metabolism and the sites of action of the different metabolic modulators. These range from (A) mitochondrial carnitine shuttle (CPT inhibitors) and (B) β-fatty acid oxidation inhibitors (C) pyruvate dehydrogenase (PDH) activators. In practice, the latter two represent the clinically pertinent therapeutic targets. Group (A) includes perhexiline, etomoxir, oxfenicine and to a lesser extent amiodarone. Group (B) includes trimetazidine and ranolazine. Group (C) includes dichloroacetate. GLUT denotes glucose transporter, PCr phosphocreatine, Cr free creatine.
Table 1.
Potential therapeutic strategies to restore the balance of fuel utilisation in type 2 diabetes.
| Strategy/Agent | Effect |
|---|---|
| Pyruvate dehydrogenase kinase 1–4 inhibition | |
| Dichloroacetate | Increased flux through PDH Increased Krebs cycle flux Increased oxidative metabolism Pyruvate dehydrogenase activation |
| CPT-1 inhibition | |
| Perhexiline, Amiodarone, Etomoxir, Oxfenicine | Decreased fatty acid oxidation Increased glucose oxidation |
| Long-chain 3-ketoacyl-CoA thiolase inhibitors | |
| Trimetazidine, Ranolazine | Decreased fatty acid oxidation Increased glucose oxidation |
| γ-butyrobetaine hydroxylase partial inhibition | |
| Meldonium | Decreased l-carnitine synthesis Increased glucose oxidation |
| Carnitine acylcarnitine transferase activation | |
| l-propionylcarnitine l-carnitine | Increased fatty acid transport across mitochondrial membrane Increased glucose oxidation |
Supporting the hypothetical cardiovascular beneficial influences of reduced FA oxidation in animal models of T2D, trimetazidine was shown to ameliorate features of diabetic cardiomyopathy and reverse insulin resistance (58). Trimetazidine is a piperazine derivative with pleiotropic beneficial properties (e.g. anti-ischaemic, antioxidant and even anti-apoptotic roles have been reported) (59). It is licensed as an anti-anginal agent, which selectively inhibits long-chain 3-ketoacyl coenzyme A thiolase (the last enzyme involved in β-oxidation) activity, thereby modifying energy metabolism by partial inhibition of FA oxidation. Although there is a lack of large-scale clinical trials with metabolic modulators, there have been 16 randomised controlled clinical trials of patients with chronic HF suggesting the efficacy of trimetazidine. Trimetazidine was shown to reduce all-cause mortality, improve LVEF, reduce symptoms of HF and plasma B-type natriuretic peptide (BNP) levels (59). While studies reported beneficial effects of trimetazidine on clinical prognosis of diabetic patients with advanced ischaemic heart disease (60), whether or not these beneficial effects can prevent the development of HF in patients with T2D at an early stage has not been explored in clinical studies.
Perhexiline, 2-(2,2-dicyclohexylethyl) piperidine is another metabolic agent reducing FA metabolism through the inhibition of carnitine palmitoyltransferase, the enzyme responsible for mitochondrial uptake of long-chain FA. Perhexiline was also shown to improve LVEF, resting and peak stress myocardial function and skeletal muscle energetics, peak exercise oxygen consumption (VO2max) reduce HF symptoms in patients with chronic HF (61), and in a separate study, perhexiline improved myocardial energetics (62). Efficacy of perhexiline was also shown in patients with symptomatic hypertrophic cardiomyopathy (HCM), with improved myocardial energetics, diastolic function and exercise capacity (63). In patients with ischaemic cardiomyopathy, perhexiline was shown to have no effect on wall motion response to dobutamine stress and adverse effect on strain rate compared to placebo (64). Neither of these antianginal metabolic modulators showed any negative inotropic effect to reduce the cardiac workload, consequently, their beneficial cardiovascular effects are considered to be related to inhibition of FA uptake and a metabolic shift towards the use of glucose and lactate (61, 62).
L-carnitine plays a pivotal role in both FA and carbohydrate metabolism. It is responsible for transfer of long-chain FA into the mitochondrial matrix. Meldonium is also an antianginal drug, which partially inhibits γ-butyrobetaine hydroxylase, reducing L-carnitine biosynthesis and uptake and consequently leading to a shift away from FA metabolism towards glucose metabolism. In animal models of obesity and impaired glucose tolerance, meldonium reduced plasma insulin concentration and increased cardiac and hepatic PPAR-α activity (65).
There is evidence that increased FA utilisation may ‘paradoxically’ have beneficial effects on cardiovascular health in patients with T2D (66). In the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study, PPAR-α agonist fenofibrate treatment was associated with a statistically non-significant trend towards a reduction in the 5-year CVD risk of 14.5 to 13.1%, representing a proportional risk reduction of 11% (adjusted HR 0.89 (95% CI 0–21%), P = 0.052; absolute risk reduction 1.4%). These contradictory outcomes make it even more pertinent to delineate the precise metabolic changes that occur in patients with T2D. Rather than representing a paradox, this may indicate that it is the lack of metabolic flexibility, rather than specific substrate preference that predisposes the diabetic heart to injury (67). Further, PPAR-α activation may be beneficial with two significant advantages: (i) providing continued support to the muscle’s metabolic needs and (ii) avoiding accumulation of lipid byproducts that could be harmful to the cardiomyocyte (68). Additionally, pleiotropic benefits of these agents may be responsible for these beneficial effects (67).
Myocardial steatosis in patients with type 2 diabetes
Excess myocyte accumulation of lipids has emerged as an important contributor to the development of diabetic cardiomyopathy, particularly concentric LV remodelling (23). The discordance between the rates of FA availability and/or uptake with that of FA oxidation results in increased intracellular long-chain fatty acyl-CoA concentrations (19). Since cardiomyocytes are not specialised to store lipid, this finding suggests a deleterious effect and cellular lipid overloading underlies the concept of ‘lipotoxicity’ as a potential mechanism for impaired cardiac function (32). The excess long-chain fatty acyl-CoA is then diverted towards non-oxidative processes with the production of lipotoxic intermediates such as ceramide and diacyl-glycerol (32). These have been shown to activate signalling pathways affecting ATP production, insulin sensitivity, myo-cellular contractility and apoptosis (32, 69). Increased FA levels stimulate cardiac PPAR-α, resulting in upregulation of the lipid metabolising pathway, and PPAR-α-overexpressing mice show a phenotype similar to diabetes (32). This provides another potential mechanistic link between cardiac steatosis, lipotoxicity and concentric LV remodelling in diseases of upregulated FA metabolism such as diabetes.
Using proton (1H)-MRS, myocardial triglyceride content has been shown to be increased by 1.5- to 2.3-fold in T2D (23, 70). Importantly, myocardial steatosis has been shown to be modifiable (71, 72). Successful reduction of myocardial steatosis with GLP-1 agonists (71) and mineralocorticoid receptor blockers (72) have both been shown to reverse concentric LV remodelling. However, larger studies targeting myocardial lipid accumulation are needed to confirm these observations.
Ectopic and visceral adiposity and insulin resistance in patients with type 2 diabetes
Accumulating evidence suggests that: (i) the distribution of excess fat is an important determinant of cardiovascular risk; (ii) ectopic and visceral adiposity confer a much higher risk than subcutaneous adiposity (73, 74) and (iii) abnormal distribution of excess fat may also play a role in the pathogenesis of cardiomyopathy process associated with diabetes and obesity (75). Computed tomography (CT), MRI, ultrasonography and 1H-MRS have all been used to quantify adipose tissue amount or lipid content within an organ and to examine the association of various fat depots with both systemic and local manifestations of disease (70, 76, 77, 78, 79, 80). Recently, using these techniques, it was demonstrated that, irrespective of body mass index, diabetes is related to significant abnormalities in cardiac function, energetics and cardiac and hepatic steatosis (81). However, obese patients with T2D showed a greater propensity for ectopic fat deposition that is associated with cardiac contractile dysfunction and fibroinflammatory liver disease than lean T2D patients (81) (Fig. 2).
Figure 2.
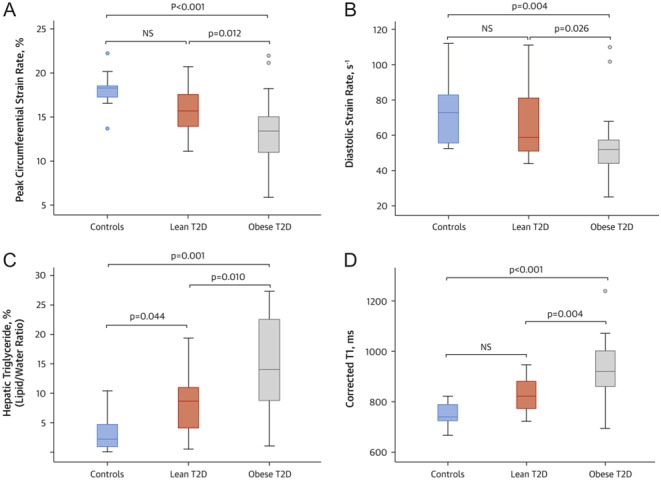
Differences in cardiac function, hepatic steatosis, and hepatic cT1 among the study cohorts. (A) Peak circumferential systolic strain; (B) diastolic strain rate; (C) hepatic triglyceride content (%) and (D) hepatic corrected T1 map (ms). The dots indicate values outside the interquartile range. Reproduced with permission from Levelt et al. (83). Copyright© The American College of Cardiology.
Epicardial adipose tissue (EAT), which is a form of visceral fat, has no anatomical barriers with the myocardium, and, by secreting proinflammatory adipokines and cytokines through paracrine/autocrine signalling pathways, EAT may play a significant role in diabetic heart disease. Supporting this theory, an inverse correlation of EAT volumes with cardiac systolic strain was demonstrated (81). Similarly, excess liver fat, which is a form of ectopic fat, has been shown to be accompanied by cardiac structural and functional changes (82).
Ectopic and visceral adiposity or ‘acquired lipodystrophy’ is linked to insulin resistance and diabetes (83). Multiple studies support the concept that insulin resistance is prompted, and sustained by, dysregulated fat tissue (84, 85, 86). It is possible that the insulin resistance may be responsible for the increased cardiovascular risk that is linked to ectopic and visceral adiposity. Additionally, there is evidence for a strong association between insulin resistance and non-ischaemic HF (87). There are many molecular mechanisms that may contribute to the association between insulin resistance and non-ischaemic cardiomyopathy (87). These include metabolic inefficiency (19), impaired vascular function (88), inflammation, mitogenic actions of insulin on myocardium leading to changes of left ventricular geometry (89). However, there are differing opinions whether this relationship is of protective or pathological nature (90, 91, 92). Although it has been demonstrated that insulin resistance and ectopic adiposity are associated with an even greater cardiovascular risk (93, 94), Nolan and coworkers recently argued that insulin resistance protects critical tissues, such as the heart, from nutrient-induced damage (92). It has been proposed that insulin resistance is an antioxidant defence mechanism (90). Consequently, lately, there has been something of a paradigm shift in the consensus regarding the nature of the role of insulin resistance in diabetes associated cardiovascular risk, when the traditional thinking had regarded insulin resistance as a primary etiological factor in the development of non-ischaemic HF. This is based on the evidence that impairment of mitochondrial oxidative capacity, which follows increased reactive oxygen species (ROS) production in muscles of diet-induced diabetic mice (95) and inhibition of mitochondrial ROS production reverses insulin resistance (96, 97). This novel perspective might offer answers to why some previous research therapeutically targeting impaired insulin sensitivity resulted in deleterious effects such as PPARs, including the development of HF in patients with diabetes (90, 98).
Novel glucose-lowering therapies and improved cardiovascular outcomes
Recently new classes of glucose-lowering therapies, such as glucagon-like peptide-1 (GLP-1) analogues (99) and inhibitors of sodium–glucose cotransporter 2 (7) have shown exciting results with improved glycaemic control as well as reduced cardiovascular mortality in patients with T2D. However, these recent trials were designed to assess the specific effects of these novel drugs on clinical outcomes, and therefore, the mechanisms behind the observed cardiovascular benefits are speculative. It would be interesting to see if the potential beneficial effects of these novel therapies on cardiovascular health will be reflected by the changes measured with non-invasive imaging techniques.
The biologic action of GLP-1 is focused on the intake, absorption, retention and disposal of energy-rich substrates (100). In normal physiology, endogenous GLP-1 is implicated in the control of appetite and satiety, not surprisingly therefore GLP-1 is currently under intensive investigation as a potential primary mediator of beneficial metabolic effects after bariatric surgery, with its eating-inhibitory, antiobesity and antidiabetes effects (101). The principal determinants of the levels of active plasma GLP-1 include enzymatic inactivation by dipeptidyl peptidase 4 (DPP-4) and neutral endopeptidase and renal clearance (102).
The incretin-based drugs include dipeptidyl peptidase 4 (DPP-4) inhibitors and GLP-1 analogues. The GLP-1 analogues exert their effect via the incretin system, specifically targeting the receptor for the incretin hormone GLP-1, which is partly responsible for augmenting glucose-dependent insulin secretion in response to nutrient intake (the ‘incretin effect’). The predominant actions of exogenously administered GLP-1 regulate blood glucose via inhibition of appetite, glucagon secretion and gastric emptying and stimulation of insulin secretion (103).
GLP-1 receptors are also expressed in the heart, and administration of GLP-1 improves cardiovascular function in the setting of experimental cardiac injury (104). The actions of GLP-1 on the heart may be directly through generation of cAMP in cardiomyocytes and/or indirectly by improvement of the metabolic environment through control of blood glucose, insulin and Fas (105). The Liraglutide Effect and Action in Diabetes: Evaluation of Cardiovascular Outcome Results (LEADER) trial showed that death from cardiovascular causes occurred in fewer patients in the GLP-1 analogue liraglutide group compared to the placebo group in patients with T2D and high cardiovascular risk. Similarly, in high-risk T2D patients, the rate of cardiovascular death, nonfatal myocardial infarction or nonfatal stroke was significantly lower for semaglutide than for placebo (106). Exenatide is an exendin-4-based GLP-1 receptor agonist which is a once-weekly, injectable, extended-release formulation drug. The Exenatide Study of Cardiovascular Event Lowering (EXSCEL) assessed the long-term cardiovascular safety and efficacy of exenatide, in patients with T2D who had a wide range of cardiovascular risk (107). The results of this study showed that exenatide was non-inferior to placebo with respect to cardiovascular safety, but it was not superior to placebo with respect to efficacy. The risk of death from any cause was 6.9% in the exenatide group and 7.9% in the placebo group (hazard ratio, 0.86; 95% CI, 0.77–0.97); this difference was not statistically significant. Furthermore, the Functional Impact of GLP-1 for Heart Failure Treatment (FIGHT) study which was a multicentre, double-blind, placebo-controlled randomized clinical trial of patients with established HF and reduced LVEF has demonstrated that liraglutide does not improve post-hospitalization clinical stability in patients with advanced HF and reduced LVEF despite prior studies indicating that GLP-1 therapy might ameliorate mechanisms of myocardial insulin resistance reported in patients with severe cardiomyopathies (108). No favourable effects of liraglutide on secondary end points based on echocardiographic measures, 6-minute walk distance or quality of life scores were shown. The negative outcome in this study was speculated to be potentially associated with the promotion of glucose-dependent insulin secretion with GLP1 agonists. This is also providing extra support for the argument put forward recently by Nolan and coworkers that insulin resistance protects critical tissues, such as the heart, from nutrient-induced damage (92) that enhancing endogenous insulin secretion is disadvantageous in the setting of HF, and myocardial insulin resistance in HF models might be an adaptive mechanism in patients with advanced HF.
Inhibitors of DPP-4 reduce the breakdown of endogenous GLP-1. Unlike with GLP-1 analogues, there has been considerable speculation about the potential beneficial effects of DPP-4 inhibitors on the cardiovascular system. The results of multicentre observational studies of DPP4 inhibitors showed no beneficial results on their effect on HF admissions: (i) SAVOR–TIMI 53 trial (109) showed 27% increase in the risk of hospitalization for HF in patients with T2D assigned to saxagliptin compared to those who received placebo, and the drug did not provide any cardioprotective benefit; (ii) cardiovascular outcomes studies EXAMINE trial (110) of alogliptin vs standard care and TECOS trial (111) of sitagliptin both showed no increase in the overall risk of hospitalization for HF among patients randomly assigned to alogliptin and sitagliptin, vs standard care respectively. Meta-analysis of several large cohorts of patients with T2D, the use of incretin-based drugs, as compared with combinations of oral antidiabetic drugs, was not associated with an increased risk of hospitalization for HF (112).
The incretin-based drugs were also shown to reduce the occurrence and degree of hepatic steatosis independent of their action on body weight in an experimental study (113) and also in a small clinical phase 2 study in overweight patients with nonalcoholic steatohepatitis (114).
Finally, inhibitors of sodium–glucose cotransporter 2 reduce rates of hyperglycemia in patients with T2D by decreasing renal glucose reabsorption, thereby increasing urinary glucose excretion (115). EMPA-REG OUTCOME trial showed that patients with T2D at high risk for cardiovascular events who received empagliflozin, a selective inhibitor of sodium–glucose cotransporter 2, had significantly lower rates of the primary composite cardiovascular outcome and of death from any cause than did those in the placebo group when the study drugs were added to standard care with almost immediate beneficial effect despite a modest improvement in glycaemic control, with approximately 0.4% reduction in glycated haemoglobin of over 94 weeks (7). Canagliflozin is another SGLT2 inhibitor. The Canagliflozin Cardiovascular Assessment Study (CANVAS) Program, comprising two sister trials, was designed to assess the cardiovascular safety and efficacy of canagliflozin. The trial program showed that T2D patients with high risk of cardiovascular disease treated with canagliflozin had a significantly lower risk of death from cardiovascular causes, nonfatal myocardial infarction or nonfatal stroke than those who received placebo, but they were at a greater risk of amputation (116). The CVD-REAL Study was a retrospective registry study designed to evaluate the association of outcomes of hospitalization for HF and all-cause death in patients with T2D treated with SGLT-2 inhibitors vs other glucose-lowering drugs. Consistent with the EMPA-REG OUTCOME, CVD-REAL Study showed treatment with SGLT-2 inhibitors was associated with 39% relative risk reduction in HF hospitalization, a 51% reduction in all-cause mortality. These beneficial effects, particularly relevant to HF admissions, appeared to be class related. Importantly, overwhelming majority (87%) of patients included in the study had no established CVD, suggesting that lower risk patients may derive similar benefits with SGLT-2 inhibitors, as those with higher risk. However, this study similar to others did not address the mechanisms linking use of SGLT-2 inhibitors and associated cardiovascular benefits (117).
As a result, the reasons for the beneficial cardiovascular effects are not yet clear, however recently suggested theories include: (i) SGLT2 inhibitor induced plasma volume contraction (5% increase in haematocrit in conjunction with a 35% relative risk reduction in hospital admission for HF on empaliflozin arm) (118); (ii) Restoring cellular energy homeostasis by activation of AMPK (119, 120); (iii) SGLT2 inhibitor induced mild ketosis (121, 122). This may improve myocardial/renal metabolic efficiency and function, given that the ketone body oxidation yields more ATP per oxygen consumption than palmitate, therefore, being more ‘energy-efficient’ (49).
Diabetic cardiomyopathy in type 1 diabetes
Relatively little research has taken place comparing the underlying mechanisms and clinical features of diabetic cardiomyopathy in type 1 vs type 2 diabetes. Although high prevalence of subclinical myocardial dysfunction has been reported in the early stage of type 1 diabetes (T1D), clinical presentations of HF is relatively rare in this type of diabetes compared to T2D (123). In a longitudinal observational study of a relatively large cohort of T1D patients without a previous history of heart disease only 17 patients out of 462 (3.7%) were shown to develop HF during a 12-year follow-up period (124). Those patients who developed HF were reported to be older with a longer duration of diabetes (35 ± 9 years), and had higher blood pressure, and higher prevalence of albuminuria and retinopathy compared to those without HF.
Similar to T2D (125), cardiomyocyte hypertrophy has been reported for different animal models of T1D (126), however significant reduction in the cardiomyocyte cross sectional area was also observed in a model of T1D (127). Fewer studies of T1D have shown an increase in LV mass compared to T2D. This may be due to the younger age and lower incidence of hypertension in T1D patients investigated in most studies. Significant LV dysfunction has also been detected by tissue Doppler and speckle tracking echocardiography techniques in T1D patients (128). Myocardial metabolic remodelling studies have been scarce in T1D. A single study demonstrated a significant reduction in myocardial energetics at rest independently of myocardial perfusion reserve changes, similarly to T2D (129). To our knowledge no studies to date have evaluated the role of myocardial steatosis in T1D.
Conclusions
Science has progressed significantly in its understanding of disease mechanisms in type 2 diabetes, and significant advances have been made in characterizing the metabolic phenotype in the diabetic heart and in defining the relationship among the myocardial metabolic remodelling, structural and functional changes. However, the fundamental question of whether or not a primary alteration in substrate utilisation in diabetes is responsible for cardiac dysfunction remains uncertain. The ability to manipulate cardiac metabolism is a promising therapeutic target which may shed light on this question. The mechanisms behind the observed cardiovascular mortality benefits of new classes of glucose-lowering therapies also remain to be shown. In search of treatment and prevention of diabetes-associated HF, the road ahead still appears long, but promises significant advances.
Declaration of interest
All authors have read and understood European Journal of Endocrinology policy on declaration of interests and declare that we have no competing interests.
Funding
This work was supported by the Wellcome Trust (grant number 207726/Z/17/Z).
Authors contribution statement
All authors made appropriate contributions according to the ICMJE guidance, and as such have read and approved the final manuscript. All authors take public responsibility for appropriate portions of the manuscript content; and agree to be accountable in ensuring that questions related to the accuracy or integrity of the work are appropriately investigated and resolved. E L, G S, S N and G M each contributed to drafting of manuscript and revisions.
References
- 1.WHO. Fact sheet no 310; the top 10 causes of death. updated May 2014. Geneva: World Health Organization. (available at: http://www.who.int/mediacentre/factsheets/fs310/en/) [Google Scholar]
- 2.Garcia MJ, McNamara PM, Gordon T, Kannel WB. Morbidity and mortality in diabetics in the Framingham population. Sixteen year follow-up study. Diabetes 1974. 23 105–111. ( 10.2337/diab.23.2.105) [DOI] [PubMed] [Google Scholar]
- 3.Nichols GA, Gullion CM, Koro CE, Ephross SA, Brown JB. The incidence of congestive heart failure in type 2 diabetes: an update. Diabetes Care 2004. 27 1879–1884. ( 10.2337/diacare.27.8.1879) [DOI] [PubMed] [Google Scholar]
- 4.Morrish JN, Wang S-L, Stevens KL, Fuller HJ, Keen H. Mortality and causes of death in the WHO multinational study of vascular disease in diabetes. Diabetologia 2001. 44 S14–S21. ( 10.1007/PL00002934) [DOI] [PubMed] [Google Scholar]
- 5.Tancredi M, Rosengren A, Svensson A-M, Kosiborod M, Pivodic A, Gudbjörnsdottir S, Wedel H, Clements M, Dahlqvist S, Lind M. et al Excess mortality among persons with type 2 diabetes. New England Journal of Medicine 2015. 373 1720–1732. ( 10.1056/NEJMoa1504347) [DOI] [PubMed] [Google Scholar]
- 6.Cavender MA, Steg PG, Smith SC, Eagle K, Ohman EM, Goto S, Kuder J, Im K, Wilson PW, Bhatt DL. et al Impact of diabetes mellitus on hospitalization for heart failure, cardiovascular events, and death. Circulation 2015. 132 923. [DOI] [PubMed] [Google Scholar]
- 7.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ. et al Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. New England Journal of Medicine 2015. 373 2117–2128. [DOI] [PubMed] [Google Scholar]
- 8.Richard MC, Brook A, Adil R, Ben NM, Peysh AP, Guy G, Gale CP, Batin PD, Ajjan R, Kearney L. et al Diabetes mellitus is associated with adverse prognosis in chronic heart failure of ischaemic and non-ischaemic aetiology. Diabetes and Vascular Disease Research 2013. 10 330–336. ( 10.1177/1479164112471064) [DOI] [PubMed] [Google Scholar]
- 9.Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Reviews in Endocrine and Metabolic Disorders 2010. 11 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leyden E. Asthma and diabetes mellitus. Zeutschr Klin Med 1881. 3 358–364. [Google Scholar]
- 11.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. American Journal of Cardiology 1972. 30 595–602. ( 10.1016/0002-9149(72)90595-4) [DOI] [PubMed] [Google Scholar]
- 12.Kannel WB, McGee DL. Diabetes and cardiovascular disease: the framingham study. JAMA 1979. 241 2035–2038. ( 10.1001/jama.1979.03290450033020) [DOI] [PubMed] [Google Scholar]
- 13.Hansson L, Zanchetti A, Carruthers SG, Dahlöf B, Elmfeldt D, Julius S, Ménard J, Rahn KH, Wedel H, Westerling S. Effects of intensive blood-pressure lowering and low-dose aspirin in patients with hypertension: principal results of the Hypertension Optimal Treatment (HOT) randomised trial. Lancet 1998. 351 1755–1762. ( 10.1016/S0140-6736(98)04311-6) [DOI] [PubMed] [Google Scholar]
- 14.Long AN, Dagogo-Jack S. The comorbidities of diabetes and hypertension: mechanisms and approach to target organ protection. Journal of Clinical Hypertension 2011. 13 244–251. ( 10.1111/j.1751-7176.2011.00434.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skali H, Shah A, Gupta DK, Cheng S, Claggett B, Liu J, Bello N, Aguilar D, Vardeny O, Matsushita K. et al Cardiac structure and function across the glycemic spectrum in elderly men and women free of prevalent heart disease: the atherosclerosis risk in the community study. Circulation: Heart Failure 2015. 8 448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK, Fabsitz RR, Robbins D, Rhoades ER, Howard BV. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation 2000. 101 2271–2276. ( 10.1161/01.CIR.101.19.2271) [DOI] [PubMed] [Google Scholar]
- 17.De Marco M, de Simone G, Roman MJ, Chinali M, Lee ET, Calhoun D, Howard BV, Devereux RB. Cardiac geometry and function in diabetic or prediabetic adolescents and young adults. Diabetes Care 2011. 34 2300–2305. ( 10.2337/dc11-0191) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bluemke DA, Kronmal RA, Lima JAC, Liu K, Olson J, Burke GL, Folsom AR. The relationship of left ventricular mass and geometry to incident cardiovascular events: the MESA study. Journal of the American College of Cardiology 2008. 52 2148–2155. ( 10.1016/j.jacc.2008.09.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taegtmeyer H, McNulty P, Young ME. Adaptation and maladaptation of the heart in diabetes: part I: general concepts. Circulation 2002. 105 1727–1733. ( 10.1161/01.CIR.0000012466.50373.E8) [DOI] [PubMed] [Google Scholar]
- 20.van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, Ijsselmuiden AJ, Schalkwijk CG, Bronzwaer JG, Diamant M. et al Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 2008. 117 43–51. ( 10.1161/CIRCULATIONAHA.107.728550) [DOI] [PubMed] [Google Scholar]
- 21.Liu S, Han J, Nacif M, Jones J, Kawel N, Kellman P, Sibley CT, Bluemke DA. Diffuse myocardial fibrosis evaluation using cardiac magnetic resonance T1 mapping: sample size considerations for clinical trials. Journal of Cardiovascular Magnetic Resonance 2012. 14 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khan JN, Wilmot EG, Leggate M, Singh A, Yates T, Nimmo M, Khunti K, Horsfield MA, Biglands J, Clarysse P. et al Subclinical diastolic dysfunction in young adults with Type 2 diabetes mellitus: a multiparametric contrast-enhanced cardiovascular magnetic resonance pilot study assessing potential mechanisms. European Heart Journal – Cardiovascular Imaging 2014. 15 1263–1269. ( 10.1093/ehjci/jeu121) [DOI] [PubMed] [Google Scholar]
- 23.Levelt E, Mahmod M, Piechnik SK, Ariga R, Francis JM, Rodgers CT, Clarke WT, Sabharwal N, Schneider JE, Karamitsos TD. et al Relationship between left ventricular structural and metabolic remodelling in type 2 diabetes mellitus. Diabetes 2016. 65 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong TC, Piehler KM, Kang IA, Kadakkal A, Kellman P, Schwartzman DS, Mulukutla SR, Simon MA, Shroff SG, Kuller LH. et al Myocardial extracellular volume fraction quantified by cardiovascular magnetic resonance is increased in diabetes and associated with mortality and incident heart failure admission. European Heart Journal 2014. 35 657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Treibel TA, Zemrak F, Sado DM, Banypersad SM, White SK, Maestrini V, Barison A, Patel V, Herrey AS, Davies C. et al Extracellular volume quantification in isolated hypertension – changes at the detectable limits? Journal of Cardiovascular Magnetic Resonance 2015. 17 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodrigues JCL, Amadu AM, Dastidar AG, Szantho GV, Lyen SM, Godsave C, Ratcliffe LE, Burchell AE, Hart EC, Hamilton MC. et al Comprehensive characterisation of hypertensive heart disease left ventricular phenotypes. Heart 2016. 102 1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boyer JK, Thanigaraj S, Schechtman KB, Pérez JE. Prevalence of ventricular diastolic dysfunction in asymptomatic, normotensive patients with diabetes mellitus. American Journal of Cardiology 2004. 93 870–875. ( 10.1016/j.amjcard.2003.12.026) [DOI] [PubMed] [Google Scholar]
- 28.Di Bonito P, Moio N, Cavuto L, Covino G, Murena E, Scilla C, Turco S, Capaldo B, Sibilio G. Early detection of diabetic cardiomyopathy: usefulness of tissue Doppler imaging. Diabetic Medicine 2005. 22 1720–1725. ( 10.1111/j.1464-5491.2005.01685.x) [DOI] [PubMed] [Google Scholar]
- 29.From AM, Scott CG, Chen HH. The development of heart failure in patients with diabetes mellitus and preclinical diastolic dysfunction: a population based study. Journal of the American College of Cardiology 2010. 55 300–305. ( 10.1016/j.jacc.2009.12.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levelt E, Rodgers CT, Clarke WT, Mahmod M, Ariga R, Francis JM, Liu A, Wijesurendra RS, Dass S, Sabharwal N. et al Cardiac energetics, oxygenation, and perfusion during increased workload in patients with type 2 diabetes mellitus. European Heart Journal 2015. 37 3461–3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neubauer S. The failing heart – an engine out of fuel. New England Journal of Medicine 2007. 356 1140–1151. ( 10.1056/NEJMra063052) [DOI] [PubMed] [Google Scholar]
- 32.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD. et al The cardiac phenotype induced by PPARα overexpression mimics that caused by diabetes mellitus. Journal of Clinical Investigation 2002. 109 121–130. ( 10.1172/Jbib214080) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gulick T, Cresci S, Caira T, Moore DD, Kelly DP. The peroxisome proliferator-activated receptor regulates mitochondrial fatty acid oxidative enzyme gene expression. PNAS 1994. 91 11012–11016. ( 10.1073/pnas.91.23.11012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, Radda GK, Neubauer S, Clarke K. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation 2003. 107 3040–3046. ( 10.1161/01.CIR.0000072789.89096.10) [DOI] [PubMed] [Google Scholar]
- 35.Wright JJ, Kim J, Buchanan J, Boudina S, Sena S, Bakirtzi K, Ilkun O, Theobald HA, Cooksey RC, Kandror KV. et al Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovascular Research 2009. 82 351–360. ( 10.1093/cvr/cvp017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.How O-J, Aasum E, Severson DL, Chan WYA, Essop MF, Larsen TS. Increased myocardial oxygen consumption reduces cardiac efficiency in diabetic mice. Diabetes 2006. 55 466–473. ( 10.2337/diabetes.55.02.06.db05-1164) [DOI] [PubMed] [Google Scholar]
- 37.Taegtmeyer H, Hems R, Krebs HA. Utilization of energy-providing substrates in the isolated working rat heart. Biochemical Journal 1980. 186 701–711. ( 10.1042/bj1860701) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J-Y, Nolte LA, Hansen PA, Han D-H, Ferguson K, Thompson PA, Holloszy JO. High-fat diet-induced muscle insulin resistance: relationship to visceral fat mass. American Journal of Physiology – Regulatory, Integrative and Comparative Physiology 2000. 279 R2057–R2065. ( 10.1152/ajpregu.2000.279.6.R2057) [DOI] [PubMed] [Google Scholar]
- 39.Storlien LH, James DE, Burleigh KM, Chisholm DJ, Kraegen EW. Fat feeding causes widespread in vivo insulin resistance, decreased energy expenditure, and obesity in rats. American Journal of Physiology – Endocrinology and Metabolism 1986. 251 E576–E583. ( 10.1152/ajpendo.1986.251.5.E576) [DOI] [PubMed] [Google Scholar]
- 40.Grundleger ML, Thenen SW. Decreased insulin binding, glucose transport, and glucose metabolism in soleus muscle of rats fed a high fat diet. Diabetes 1982. 31 232–237. ( 10.2337/diab.31.3.232) [DOI] [PubMed] [Google Scholar]
- 41.Lopaschuk G. Metabolic abnormalities in the diabetic heart. Heart Failure Reviews 2002. 7 149–159. ( 10.1023/A:1015328625394) [DOI] [PubMed] [Google Scholar]
- 42.Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiological Reviews 2010. 90 207–258. ( 10.1152/physrev.00015.2009) [DOI] [PubMed] [Google Scholar]
- 43.An D, Rodrigues B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. American Journal of Physiology – Heart and Circulatory Physiology 2006. 291 H1489–H1506. ( 10.1152/ajpheart.00278.2006) [DOI] [PubMed] [Google Scholar]
- 44.Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochimica et Biophysica Acta (BBA) – Molecular and Cell Biology of Lipids 2005. 1734 112–126. ( 10.1016/j.bbalip.2005.03.005) [DOI] [PubMed] [Google Scholar]
- 45.Aasum E, Hafstad AD, Severson DL, Larsen TS. Age-dependent changes in metabolism, contractile function, and ischemic sensitivity in hearts from db/db mice. Diabetes 2003. 52 434–441. ( 10.2337/diabetes.52.2.434) [DOI] [PubMed] [Google Scholar]
- 46.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 2004. 53 2366–2374. ( 10.2337/diabetes.53.9.2366) [DOI] [PubMed] [Google Scholar]
- 47.Rider OJ, Cox P, Tyler D, Clarke K, Neubauer S. Myocardial substrate metabolism in obesity. International Journal of Obesity 2013. 37 972–979. ( 10.1038/ijo.2012.170) [DOI] [PubMed] [Google Scholar]
- 48.Veech RL. The determination of the redox states and phosphorylation potential in living tissues and their relationship to metabolic control of disease phenotypes. Biochemistry and Molecular Biology Education 2006. 34 168–179. ( 10.1002/bmb.2006.49403403168) [DOI] [PubMed] [Google Scholar]
- 49.Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins, Leukotrienes, and Essential Fatty Acids 2004. 70 309–319. ( 10.1016/j.plefa.2003.09.007) [DOI] [PubMed] [Google Scholar]
- 50.Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. American Journal of Physiology-Endocrinology and Metabolism 2009. 297 E578–E591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemical energy to support cardiac function. Circulation Research 2004. 95 135–145. ( 10.1161/01.RES.0000137170.41939.d9) [DOI] [PubMed] [Google Scholar]
- 52.He J, Ogden LG, Bazzano LA, Vupputuri S, Loria C, Whelton PK. Risk factors for congestive heart failure in us men and women: Nhanes i epidemiologic follow-up study. Archives of Internal Medicine 2001. 161 996–1002. ( 10.1001/archinte.161.7.996) [DOI] [PubMed] [Google Scholar]
- 53.Matsushita K, Blecker S, Pazin-Filho A, Bertoni A, Chang PP, Coresh J, Selvin E. The association of hemoglobin A1c with incident heart failure among people without diabetes: the atherosclerosis risk in communities study. Diabetes 2010. 59 2020–2026. ( 10.2337/db10-0165) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y, Hu G, Yuan Z, Chen L. Glycosylated hemoglobin in relationship to cardiovascular outcomes and death in patients with type 2 diabetes: a systematic review and meta-analysis. PLoS ONE 2012. 7 e42551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nielson C, Lange T. Blood glucose and heart failure in nondiabetic patients. Diabetes Care 2005. 28 607. [DOI] [PubMed] [Google Scholar]
- 56.Udell JA, Cavender MA, Bhatt DL, Chatterjee S, Farkouh ME, Scirica BM. Glucose-lowering drugs or strategies and cardiovascular outcomes in patients with or at risk for type 2 diabetes: a meta-analysis of randomised controlled trials. Lancet Diabetes and Endocrinology 2015. 3 356–366. [DOI] [PubMed] [Google Scholar]
- 57.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus Lancet 1963. 281 785–789. ( 10.1016/S0140-6736(63)91500-9) [DOI] [PubMed] [Google Scholar]
- 58.Zhang L, Ding W-Y, Wang Z-H, Tang M-X, Wang F, Li Y, Zhong M, Zhang Y, Zhang W. Early administration of trimetazidine attenuates diabetic cardiomyopathy in rats by alleviating fibrosis, reducing apoptosis and enhancing autophagy. Journal of Translational Medicine 2016. 14 109 ( 10.1186/s12967-016-0849-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang L, Lu Y, Jiang H, Zhang L, Sun A, Zou Y, Ge J. Additional use of trimetazidine in patients with chronic heart failure: a meta-analysis. Journal of the American College of Cardiology 2012. 59 913–922. ( 10.1016/j.jacc.2011.11.027) [DOI] [PubMed] [Google Scholar]
- 60.Zhao P, Zhang J, Yin X-G, Maharaj P, Narraindoo S, Cui L-Q, Tang YS. The effect of trimetazidine on cardiac function in diabetic patients with idiopathic dilated cardiomyopathy. Life Sciences 2013. 92 633–638. ( 10.1016/j.lfs.2012.03.015) [DOI] [PubMed] [Google Scholar]
- 61.Lee L, Campbell R, Scheuermann-Freestone M, Taylor R, Gunaruwan P, Williams L, Ashrafian H, Horowitz J, Fraser AG, Clarke K. et al Metabolic modulation with perhexiline in chronic heart failure. Circulation 2005. 112 3280. [DOI] [PubMed] [Google Scholar]
- 62.Beadle RM, Williams LK, Kuehl M, Bowater S, Abozguia K, Leyva F, Yousef Z, Wagenmakers AJ, Thies F, Horowitz J. et al Improvement in cardiac energetics by perhexiline in heart failure due to dilatedcardiomyopathy. JACC: Heart Failure 2015. 3 202–211. [DOI] [PubMed] [Google Scholar]
- 63.Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, Ahmed I, Maher AR, Kaur K, Taylor J, Henning A. et al Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation 2010. 122 1562. [DOI] [PubMed] [Google Scholar]
- 64.Bansal M, Chan J, Leano R, Pillans P, Horowitz J, Marwick TH. Effects of perhexiline on myocardial deformation in patients with ischaemic left ventricular dysfunction. International Journal of Cardiology 2010. 139 107–112. [DOI] [PubMed] [Google Scholar]
- 65.Liepinsh E, Skapare E, Svalbe B, Makrecka M, Cirule H, Dambrova M. Anti-diabetic effects of mildronate alone or in combination with metformin in obese Zucker rats. European Journal of Pharmacology 2011. 658 277–283. [DOI] [PubMed] [Google Scholar]
- 66.Scott R, O’Brien R, Fulcher G, Pardy C, d’Emden M, Tse D, Taskinen MR, Ehnholm C, Keech A. & Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study Investigators. Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care 2009. 32 493–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ashrafian H, Neubauer S. Metabolic modulation in heart failure: high time for a definitive clinical trial. Heart 2011. 97 267–268. ( 10.1136/hrt.2010.214932) [DOI] [PubMed] [Google Scholar]
- 68.Kassiotis C, Rajabi M, Taegtmeyer H. Metabolic reserve of the heart: the forgotten link between contraction and coronary flow. Progress in Cardiovascular Diseases 2008. 51 74–88. ( 10.1016/j.pcad.2007.11.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bielawska AE, Shapiro JP, Jiang L, Melkonyan HS, Piot C, Wolfe CL, Tomei LD, Hannun YA, Umansky SR. Ceramide is involved in triggering of cardiomyocyte apoptosis induced by ischemia and reperfusion. American Journal of Pathology 1997. 151 1257–1263. [PMC free article] [PubMed] [Google Scholar]
- 70.McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, Levine BD, Raskin P, Victor RG, Szczepaniak LS. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation 2007. 116 1170–1175. ( 10.1161/CIRCULATIONAHA.106.645614) [DOI] [PubMed] [Google Scholar]
- 71.Monji A, Mitsui T, Bando YK, Aoyama M, Shigeta T, Murohara T. Glucagon-like peptide-1 receptor activation reverses cardiac remodeling via normalizing cardiac steatosis and oxidative stress in type 2 diabetes. American Journal of Physiology – Heart and Circulatory Physiology 2013. 305 H295–H304. ( 10.1152/ajpheart.00990.2012) [DOI] [PubMed] [Google Scholar]
- 72.Ramírez E, Klett-Mingo M, Ares-Carrasco S, Picatoste B, Ferrarini A, Rupérez FJ, Caro-Vadillo A, Barbas C, Egido J, Tuñón J. et al Eplerenone attenuated cardiac steatosis, apoptosis and diastolic dysfunction in experimental type-II diabetes. Cardiovascular Diabetology 2013. 12 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Okura T, Nakata Y, Yamabuki K, Tanaka K. Regional body composition changes exhibit opposing effects on coronary heart disease risk factors. Arteriosclerosis, Thrombosis, and Vascular Biology 2004. 24 923–929. ( 10.1161/01.ATV.0000125702.26272.f6) [DOI] [PubMed] [Google Scholar]
- 74.Fantuzzi G, Mazzone T. Adipose tissue and atherosclerosis: exploring the connection. Arteriosclerosis, Thrombosis, and Vascular Biology 2007. 27 996–1003. ( 10.1161/ATVBAHA.106.131755) [DOI] [PubMed] [Google Scholar]
- 75.Montani JP, Carroll JF, Dwyer TM, Antic V, Yang Z, Dulloo AG. Ectopic fat storage in heart, blood vessels and kidneys in the pathogenesis of cardiovascular diseases. International Journal of Obesity and Related Metabolic Disorders 2004. 28 S58–S65. [DOI] [PubMed] [Google Scholar]
- 76.Fox CS, Massaro JM, Hoffmann U, Pou KM, Maurovich-Horvat P, Liu C-Y, Vasan RS, Murabito JM, Meigs JB, Cupples LA. et al Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation 2007. 116 39–48. ( 10.1161/CIRCULATIONAHA.106.675355) [DOI] [PubMed] [Google Scholar]
- 77.Fox CS, Gona P, Hoffmann U, Porter SA, Salton CJ, Massaro JM, Levy D, Larson MG, D’Agostino RB, Sr, O’Donnell CJ. et al Pericardial fat, intrathoracic fat, and measures of left ventricular structure and function: the Framingham Heart Study. Circulation 2009. 119 1586–1591. ( 10.1161/CIRCULATIONAHA.108.828970) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fox CS, Massaro JM, Schlett CL, Lehman SJ, Meigs JB, O’Donnell CJ, Hoffmann U, Murabito JM. Periaortic fat deposition is associated with peripheral arterial disease: the Framingham Heart Study. Circulation: Cardiovascular Imaging 2010. 3 515–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rijzewijk LJ, van der Meer RW, Smit JWA, Diamant M, Bax JJ, Hammer S, Romijn JA, de Roos A, Lamb HJ. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. Journal of the American College of Cardiology 2008. 52 1793–1799. ( 10.1016/j.jacc.2008.07.062) [DOI] [PubMed] [Google Scholar]
- 80.Rijzewijk LJ, Jonker JT, van der Meer RW, Lubberink M, de Jong HW, Romijn JA, Bax JJ, de Roos A, Heine RJ, Twisk JW. et al Effects of hepatic triglyceride content on myocardial metabolism in type 2 diabetes. Journal of the American College of Cardiology 2010. 56 225–233. ( 10.1016/j.jacc.2010.02.049) [DOI] [PubMed] [Google Scholar]
- 81.Levelt E, Pavlides M, Banerjee R, Mahmod M, Kelly C, Sellwood J, Ariga R, Thomas S, Francis J, Rodgers C. et al Ectopic and visceral fat deposition in lean and obese patients with type 2 diabetes. Journal of the American College of Cardiology 2016. 68 53–63. ( 10.1016/j.jacc.2016.03.597) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Petta S, Argano C, Colomba D, Cammà C, Di Marco V, Cabibi D, Tuttolomondo A, Marchesini G, Pinto A, Licata G. et al Epicardial fat, cardiac geometry and cardiac function in patients with non-alcoholic fatty liver disease: association with the severity of liver disease. Journal of Hepatology 2015. 62 928–933. ( 10.1016/j.jhep.2014.11.030) [DOI] [PubMed] [Google Scholar]
- 83.Heilbronn L, Smith SR, Ravussin E. Failure of fat cell proliferation, mitochondrial function and fat oxidation results in ectopic fat storage, insulin resistance and type II diabetes mellitus. International Journal of Obesity and Related Metabolic Disorders 2004. 28 S12–S21. [DOI] [PubMed] [Google Scholar]
- 84.Nelson MD, Victor RG, Szczepaniak EW, Simha V, Garg A, Szczepaniak LS. Cardiac steatosis and left ventricular hypertrophy in patients with generalized lipodystrophy as determined by magnetic resonance spectroscopy and imaging. American Journal of Cardiology 2013. 112 1019–1024. ( 10.1016/j.amjcard.2013.05.036) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Johannsen DL, Tchoukalova Y, Tam CS, Covington JD, Xie W, Schwarz J-M, Bajpeyi S, Ravussin E. Effect of 8 weeks of overfeeding on ectopic fat deposition and insulin sensitivity: testing the “adipose tissue expandability” hypothesis. Diabetes Care 2014. 37 2789–2797. ( 10.2337/dc14-0761) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Reitman ML. Metabolic lessons from genetically lean mice. Annual Review of Nutrition 2002. 22 459–482. ( 10.1146/annurev.nutr.22.010402.102849) [DOI] [PubMed] [Google Scholar]
- 87.Witteles RM, Fowler MB. Insulin-resistant cardiomyopathy: clinical evidence, mechanisms, and treatment options. Journal of the American College of Cardiology 2008. 51 93–102. ( 10.1016/j.jacc.2007.10.021) [DOI] [PubMed] [Google Scholar]
- 88.Zhang Q-J, Holland WL, Wilson L, Tanner JM, Kearns D, Cahoon JM, Pettey D, Losee J, Duncan B, Gale D. et al Ceramide mediates vascular dysfunction in diet-induced obesity by PP2A-mediated dephosphorylation of the eNOS-Akt complex. Diabetes 2012. 61 1848–1859. ( 10.2337/db11-1399) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circulation Research 2006. 98 596–605. ( 10.1161/01.RES.0000207406.94146.c2) [DOI] [PubMed] [Google Scholar]
- 90.Taegtmeyer H, Beauloye C, Harmancey R, Hue L. Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. American Journal of Physiology – Heart and Circulatory Physiology 2013. 305 H1693–H1697. ( 10.1152/ajpheart.00854.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nolan CJ, Ruderman NB, Prentki M. Intensive insulin for type 2 diabetes: the risk of causing harm. Lancet Diabetes and Endocrinology 2013. 1 9–10. [DOI] [PubMed] [Google Scholar]
- 92.Nolan CJ, Ruderman NB, Kahn SE, Pedersen O, Prentki M. Insulin resistance as a physiological defense against metabolic stress: implications for the management of subsets of type 2 diabetes. Diabetes 2015. 64 673–686. ( 10.2337/db14-0694) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Thakur ML, Sharma S, Kumar A, Bhatt SP, Luthra K, Guleria R, Pandey RM, Vikram NK. Nonalcoholic fatty liver disease is associated with subclinical atherosclerosis independent of obesity and metabolic syndrome in Asian Indians. Atherosclerosis 2012. 223 507–511. ( 10.1016/j.atherosclerosis.2012.06.005) [DOI] [PubMed] [Google Scholar]
- 94.Mazurek T, Zhang L, Zalewski A, Mannion JD, Diehl JT, Arafat H, Sarov-Blat L, O’Brien S, Keiper EA, Johnson AG. et al Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 2003. 108 2460–2466. ( 10.1161/01.CIR.0000099542.57313.C5) [DOI] [PubMed] [Google Scholar]
- 95.Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin M-A, Morio B, Vidal H, Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. Journal of Clinical Investigation 2008. 118 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hoehn KL, Salmon AB, Hohnen-Behrens C, Turner N, Hoy AJ, Maghzal GJ, Stocker R, Van Remmen H, Kraegen EW, Cooney GJ. et al Insulin resistance is a cellular antioxidant defense mechanism. PNAS 2009. 106 17787–17792. ( 10.1073/pnas.0902380106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sakai K, Matsumoto K, Nishikawa T, Suefuji M, Nakamaru K, Hirashima Y, Kawashima J, Shirotani T, Ichinose K, Brownlee M. et al Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochemical and Biophysical Research Communications 2003. 300 216–222. ( 10.1016/S0006-291X(02)02832-2) [DOI] [PubMed] [Google Scholar]
- 98.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine 2007. 356 2457–2471. ( 10.1056/NEJMoa072761) [DOI] [PubMed] [Google Scholar]
- 99.Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JFE, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS. et al Liraglutide and cardiovascular outcomes in type 2 diabetes. New England Journal of Medicine 2016. 375 311–322. ( 10.1056/NEJMoa1603827) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Drucker DJ, Nauck MA. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006. 368 1696–1705. ( 10.1016/S0140-6736(06)69705-5) [DOI] [PubMed] [Google Scholar]
- 101.Lutz TA, Osto E. Glucagon-like peptide-1, glucagon-like peptide-2, and lipid metabolism. Current Opinion in Lipidology 2016. 27 257–263. ( 10.1097/MOL.0000000000000293) [DOI] [PubMed] [Google Scholar]
- 102.Drucker DJ. Biologic actions and therapeutic potential of the proglucagon-derived peptides. Nature Clinical Practice Endocrinology and Metabolism 2005. 1 22–31. ( 10.1038/ncpendmebib17) [DOI] [PubMed] [Google Scholar]
- 103.Nauck MA, Heimesaat MM, Behle K, Holst JJ, Nauck MS, Ritzel R, Hüfner M, Schmiegel WH. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. Journal of Clinical Endocrinology and Metabolism 2002. 87 1239–1246. [DOI] [PubMed] [Google Scholar]
- 104.Nikolaidis LA, Elahi D, Hentosz T, Doverspike A, Huerbin R, Zourelias L, Stolarski C, Shen YT, Shannon RP. Recombinant glucagon-like peptide-1 increases myocardial glucose uptake and improves left ventricular performance in conscious dogs with pacing-induced dilated cardiomyopathy. Circulation 2004. 110 955. [DOI] [PubMed] [Google Scholar]
- 105.Bose AK, Mocanu MM, Carr RD, Brand CL, Yellon DM. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes 2004. 54 146. [DOI] [PubMed] [Google Scholar]
- 106.Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, Lingvay I, Rosenstock J, Seufert J, Warren ML. et al Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. New England Journal of Medicine 2016. 375 1834–1844. ( 10.1056/NEJMoa1607141) [DOI] [PubMed] [Google Scholar]
- 107.Holman RR, Bethel MA, Mentz RJ, Thompson VP, Lokhnygina Y, Buse JB, Chan JC, Choi J, Gustavson SM, Iqbal N. et al Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. New England Journal of Medicine 2017. 377 1228–1239. ( 10.1056/NEJMoa1612917) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Margulies KB, Hernandez AF, Redfield MM, Givertz MM, Oliveira GH, Cole R, Mann DL, Whellan DJ, Kiernan MS, Felker GM. et al Effects of liraglutide on clinical stability among patients with advanced heart failure and reduced ejection fraction: a randomized clinical trial. JAMA 2016. 316 500–508. ( 10.1001/jama.2016.10260) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, Ohman P, Frederich R, Wiviott SD, Hoffman EB. et al Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. New England Journal of Medicine 2013. 369 1317–1326. ( 10.1056/NEJMoa1307684) [DOI] [PubMed] [Google Scholar]
- 110.White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, Perez AT, Fleck PR, Mehta CR, Kupfer S. et al Alogliptin after acute coronary syndrome in patients with type 2 diabetes. New England Journal of Medicine 2013. 369 1327–1335. ( 10.1056/NEJMoa1305889) [DOI] [PubMed] [Google Scholar]
- 111.Green JB, Bethel MA, Armstrong PW, Buse JB, Engel SS, Garg J, Josse R, Kaufman KD, Koglin J, Korn S. et al Effect of sitagliptin on cardiovascular outcomes in type 2 diabetes. New England Journal of Medicine 2015. 373 232–242. [DOI] [PubMed] [Google Scholar]
- 112.Filion KB, Azoulay L, Platt RW, Dahl M, Dormuth CR, Clemens KK, Hu N, Paterson JM, Targownik L, Turin TC. et al A multicenter observational study of incretin-based drugs and heart failure. New England Journal of Medicine 2016. 374 1145–1154. ( 10.1056/NEJMoa1506115) [DOI] [PubMed] [Google Scholar]
- 113.Abdesselam I, Pepino P, Troalen T, Macia M, Ancel P, Masi B, Fourny N, Gaborit B, Giannesini B, Kober F. et al Time course of cardiometabolic alterations in a high fat high sucrose diet mice model and improvement after GLP-1 analog treatment using multimodal cardiovascular magnetic resonance. Journal of Cardiovascular Magnetic Resonance 2015. 17 95 ( 10.1186/s12968-015-0198-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Armstrong MJ, Hull D, Guo K, Barton D, Hazlehurst JM, Gathercole LL, Nasiri M, Yu J, Gough SC, Newsome PN. et al Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. Journal of Hepatology 64 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gallo LA, Wright EM, Vallon V. Probing SGLT2 as a therapeutic target for diabetes: basic physiology and consequences. Diabetes and Vascular Disease Research 2015. 12 78–89. ( 10.1177/1479164114561992) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR. et al Canagliflozin and cardiovascular and renal events in type 2 diabetes. New England Journal of Medicine 2017. 377 644–657. ( 10.1056/NEJMoa1611925) [DOI] [PubMed] [Google Scholar]
- 117.Kosiborod M, Cavender MA, Fu AZ, Wilding JP, Khunti K, Holl RW, Norhammar A, Birkeland KI, Jørgensen ME, Thuresson M. et al Lower risk of heart failure and death in patients initiated on SGLT-2 inhibitors versus other glucose-lowering drugs: the CVD-REAL Study. Circulation 2017. 136 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ceriello A, Genovese S, Mannucci E, Gronda E. Understanding EMPA-REG OUTCOME. Lancet Diabetes and Endocrinology 2015. 3 929–930. [DOI] [PubMed] [Google Scholar]
- 119.Hawley SA, Ford RJ, Smith BK, Gowans GJ, Mancini SJ, Pitt RD, Day EA, Salt IP, Steinberg GR, Hardie DG. The Na+/Glucose cotransporter inhibitor canagliflozin activates AMPK by inhibiting mitochondrial function and increasing cellular AMP levels. Diabetes 2016. 65 2784–2794. ( 10.2337/db16-0058) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hardie DG. AMPK – sensing energy while talking to other signaling pathways. Cell Metabolism 2014. 20 939–952. ( 10.1016/j.cmet.2014.09.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ferrannini E, Baldi S, Frascerra S, Astiarraga B, Heise T, Bizzotto R, Mari A, Pieber TR, Muscelli E. Shift to fatty substrate utilization in response to sodium-glucose cotransporter 2 inhibition in subjects without diabetes and patients with type 2 diabetes. Diabetes 2016. 65 1190–1195. ( 10.2337/db15-1356) [DOI] [PubMed] [Google Scholar]
- 122.Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME Study? A unifying hypothesis. Diabetes Care 2016. 39 1115–1122. ( 10.2337/dc16-0542) [DOI] [PubMed] [Google Scholar]
- 123.Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Failure Reviews 2013. 18 149–166. ( 10.1007/s10741-012-9313-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Torffvit O, Lövestam-Adrian M, Agardh E, Agardh CD. Nephropathy, but not retinopathy, is associated with the development of heart disease in Type 1 diabetes: a 12-year observation study of 462 patients. Diabetic Medicine 2005. 22 723–729. ( 10.1111/j.1464-5491.2005.01476.x) [DOI] [PubMed] [Google Scholar]
- 125.Sakata S, Lebeche D, Sakata Y, Sakata N, Chemaly ER, Liang L, Nakajima-Takenaka C, Tsuji T, Konishi N, del Monte F. et al Transcoronary gene transfer of SERCA2a increases coronary blood flow and decreases cardiomyocyte size in a Type 2 diabetic rat model. American Journal of Physiology – Heart and Circulatory Physiology. 2007. 292 H1204. [DOI] [PubMed] [Google Scholar]
- 126.Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T. Deficiency of rac1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes 2010. 59 2033–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Basu R, Oudit GY, Wang X, Zhang L, Ussher JR, Lopaschuk GD, Kassiri Z. Type 1 diabetic cardiomyopathy in the Akita (Ins2WT/C96Y) mouse model is characterized by lipotoxicity and diastolic dysfunction with preserved systolic function. American Journal of Physiology – Heart and Circulatory Physiology 2009. 297 H2096. [DOI] [PubMed] [Google Scholar]
- 128.Shivu GN, Abozguia K, Phan TT, Ahmed I, Weaver R, Narendran P, Stevens M, Frenneaux M. Increased left ventricular torsion in uncomplicated type 1 diabetic patients: the role of coronary microvascular function. Diabetes Care 2009. 32 1710–1712. ( 10.2337/dbib9-0408) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shivu GN, Phan TT, Abozguia K, Ahmed I, Wagenmakers A, Henning A, Narendran P, Stevens M, Frenneaux M. Relationship between coronary microvascular dysfunction and cardiac energetics impairment in type 1 diabetes mellitus. Circulation 2010. 121 1209–1215. ( 10.1161/CIRCULATIONAHA.109.873273) [DOI] [PubMed] [Google Scholar]