

Abstract
Objective
To systematically review evidence regarding ataxia treatment.
Methods
A comprehensive systematic review was performed according to American Academy of Neurology methodology.
Conclusions
For patients with episodic ataxia type 2, 4-aminopyridine 15 mg/d probably reduces ataxia attack frequency over 3 months (1 Class I study). For patients with ataxia of mixed etiology, riluzole probably improves ataxia signs at 8 weeks (1 Class I study). For patients with Friedreich ataxia or spinocerebellar ataxia (SCA), riluzole probably improves ataxia signs at 12 months (1 Class I study). For patients with SCA type 3, valproic acid 1,200 mg/d possibly improves ataxia at 12 weeks. For patients with spinocerebellar degeneration, thyrotropin-releasing hormone possibly improves some ataxia signs over 10 to 14 days (1 Class II study). For patients with SCA type 3 who are ambulatory, lithium probably does not improve signs of ataxia over 48 weeks (1 Class I study). For patients with Friedreich ataxia, deferiprone possibly worsens ataxia signs over 6 months (1 Class II study). Data are insufficient to support or refute the use of numerous agents. For nonpharmacologic options, in patients with degenerative ataxias, 4-week inpatient rehabilitation probably improves ataxia and function (1 Class I study); transcranial magnetic stimulation possibly improves cerebellar motor signs at 21 days (1 Class II study). For patients with multiple sclerosis–associated ataxia, the addition of pressure splints possibly has no additional benefit compared with neuromuscular rehabilitation alone (1 Class II study). Data are insufficient to support or refute use of stochastic whole-body vibration therapy (1 Class III study).
The cerebellum is composed of the vermis, the hemispheres, and 3 cerebellar peduncles on each side, and contributes largely to balance and motor coordination. The causes of cerebellar dysfunction are numerous and include vitamin deficiencies, structural lesions (caused by tumors or trauma), infection, inflammation, toxins, neurodegeneration, genetics, stroke, multiple sclerosis (MS), and metabolic disorders. Motor signs resulting from cerebellar dysfunction may include some or all of the following: imbalance, impaired coordination, limb and body tremor, dysarthria, and oculomotor abnormalities. Other neurologic symptoms and signs may accompany cerebellar dysfunction, including dystonia, muscle weakness, oculomotor abnormalities, neuropathy, parkinsonism, spasticity, impaired visual acuity, and sensory impairment; these symptoms and signs are beyond the scope of this review. Mood, cognitive disorders, and autonomic dysfunction may also occur. Ataxia may result from cerebellar or sensory impairment.
There is currently no approved therapy to treat cerebellar motor dysfunction, and no pharmacologic or surgical treatment is routinely used. Various therapies have been studied in clinical trials for the past 40 years, although no consensus has been reached on their effectiveness. This comprehensive systematic review synthesizes the literature on the treatment of cerebellar motor dysfunction to answer the following questions:
For patients with cerebellar motor dysfunction, do pharmacologic therapies, compared with no (or alternative) treatments, improve motor symptoms with acceptable safety and tolerability?
For patients with cerebellar motor dysfunction, do surgical or other interventional therapies (e.g., physical training), compared with no (or alternative) treatments, improve motor symptoms with acceptable safety and tolerability?
For patients with cerebellar motor dysfunction, does transcranial magnetic stimulation (TMS) or transcranial direct current stimulation (tDCS), compared with no (or alternative) treatments, improve motor symptoms with acceptable safety and tolerability?
This article summarizes the systematic review findings and conclusions. Full text of the systematic review is available as a data supplement at http://npub.org/0t5ncn.
Description of the analytic process
This project used a hybrid of American Academy of Neurology (AAN) systematic review methodologic processes; the development panel used the AAN's 2004 process manual1 for the overall approach but held a public comment period and applied the updated classification of evidence scheme for therapeutic studies that had been approved and then published as an amendment to the 2011 manual.2 A description of the exact methodology followed, including the convening of the author panel, literature search strategy, and the process for reviewing evidence, is available in the full-length review. Articles authored by individuals participating in the systematic review were assessed by nonconflicted panel members. Conflicts of interest were assessed and judged to be balanced when the comprehensive systematic review was initiated and again at its conclusion. Although new conflicts appeared during the multiyear process, at least half of the panel was without conflict throughout the entirety of the process. Studies without an independent control group are considered Class IV under the updated classification of evidence scheme. Because many studies predate the determination of genotypes causing cerebellar motor dysfunction, the development panel retained the nosology used by the authors of each article. As the pathophysiology and neurochemistry of the ataxias may vary between types, the different diagnoses were considered separately wherever possible. Adverse events are discussed in the full-length review.
Analysis of evidence
Question 1: For patients with cerebellar dysfunction, do pharmacologic therapies, compared with no (or alternative) treatments, improve motor symptoms with acceptable safety and tolerability?
Medications with evidence of benefit
4-Aminopyridine
Ten patients with familial episodic ataxia type 2 (EA2) were administered 4-aminopyridine 15 mg/d in a Class I randomized, double-blind, placebo-controlled, crossover study.3 After 3 months of treatment, the median monthly attack frequency was 1.65 (interquartile range 1.00–4.78) compared with a median monthly attack frequency of 6.50 (interquartile range 2.33–13.75) with placebo (p = 0.03).
4-Aminopyridine conclusion
For patients with EA2, 4-aminopyridine 15 mg/d probably reduces the frequency of ataxia attacks over a 3-month period (1 Class I study).
Riluzole
Forty patients with ataxia of mixed etiology were administered riluzole 100 mg/d in a randomized, double-blind, placebo-controlled, single-center Class I study.4 A 5-point decrease in International Cooperative Ataxia Rating Scale (ICARS) score after 4 weeks was seen in 9 of 19 patients receiving riluzole vs 1 of 19 patients receiving placebo (odds ratio [OR] 16.2, 95% confidence interval [CI] 1.8–147.1). Four more patients receiving riluzole experienced a benefit after 8 weeks (OR 39.0, 95% CI 4.2–364.2). Absolute risk difference was 63.2% (95% CI 33.5%–79.9%) after 8 weeks. Riluzole treatment resulted in greater mean decreases in the ICARS total and subscale scores compared with placebo (mean difference in ICARS total change −7.05 [95% CI −9.74 to −4.68]; subscales described in full-length review). Whether these changes reflect clinically meaningful changes is unknown. Because of the small number of participants with each condition and the varied signs and physiology of each condition, this study could not inform disease-specific conclusions.
A follow-up, randomized, double-blind, placebo-controlled, Class I study investigated the benefit of riluzole 50 mg twice daily for 12 months in 60 patients with spinocerebellar ataxia (SCA) or Friedreich ataxia (FA).5 The primary endpoint was the proportion of patients with an improved Scale for the Assessment and Rating of Ataxia (SARA) score at 12 months, which was better in the riluzole group (OR 8.00, 95% CI 1.95–32.83), including after a post hoc logistic regression analysis adjusting for sex, age, and ataxia type (OR 9.76, 95% CI 2.08–45.80), in the 55 patients who received treatment. Mean difference in change in SARA score was also better in the riluzole group (−1.50, 95% CI −2.59 to −0.40, at 3 months; −2.68, 95% CI −3.98 to −1.39, at 12 months).
Riluzole conclusion
For patients with ataxia of various etiologies, riluzole 100 mg/d is probably effective for short-term treatment as measured by the ICARS at 8 weeks (1 Class I study). In patients with SCA or FA, riluzole 100 mg/d is probably effective for improving ataxia as measured by the SARA at 12 months (1 Class I study). Patients receiving riluzole require monitoring of liver enzymes.
Weak evidence
Valproic acid
In a Class II, randomized, double-blind, placebo-controlled study,6 patients with SCA type 3 (SCA3)/Machado–Joseph disease were randomized to receive high-dose valproic acid (VPA) (1,200 mg/d), low-dose VPA (800 mg/d), or placebo for 12 weeks. Mean change in SARA total score over 12 weeks was significantly greater in the 1,200-mg/d group (−2.05) compared with both the 800-mg/d (−1.58) and placebo (−0.75) groups (analysis of variance p = 0.021). The clinical importance of this difference in mean change is uncertain.
VPA conclusion
For patients with SCA3, VPA 1,200 mg/d is possibly effective for improving SARA total score at 12 weeks (1 Class II study).
Thyrotropin-releasing hormone
A randomized, double-blind, placebo-controlled, Class II study (predating genetic testing) of 254 patients with “spinocerebellar degeneration” (SCD) administered 0.5 and 2 mg of thyrotropin-releasing hormone (TRH), intramuscularly, once daily for 2 weeks.7 A higher percentage of patients with late-onset cerebellar cortical atrophy and olivopontocerebellar atrophy were rated as “markedly improved” or “moderately improved” at 2 weeks when treated with TRH compared with placebo (p < 0.05, exact value not reported). In the overall group, more patients treated with TRH had a higher “improvement ratio” for the signs of dysarthria, standing, and gait disorder (p < 0.05, exact value not reported). The article focused only on signs that improved. The clinical significance of these change scores is unknown.
TRH conclusion
For patients with SCD, TRH use possibly improves some signs of ataxia over 10 to 14 days (1 Class II study). The clinical significance of these changes is uncertain.
Medications with evidence against benefit
Moderate evidence
Lithium carbonate
A double-blind, randomized, placebo-controlled, Class I study evaluated lithium carbonate (dosed to serum target levels of 0.5–0.8 mEq/L) in 62 patients with SCA3 who were ambulatory.8 After 48 weeks of treatment, no difference was seen in mean scores on the primary endpoint, the Neurological Examination Score for Spinocerebellar Ataxia (NESSCA), as assessed by a generalized estimation equation using baseline measurements as covariates (NESSCA total score −0.38 points in the lithium group vs placebo, 95% CI −1.7 to 1.0). No difference was observed on the SARA total score (a secondary outcome) at 48 weeks (lithium effect vs placebo −0.96, 95% CI −2.38 to 0.46). Small but statistically significant changes were noted in certain secondary outcome measures when those receiving lithium were compared with the placebo group; the clinical relevance of these scales is not established. In further analysis,9 the treatment group had less worsening on the cerebellar NESSCA (range: 0–7 points) at 24 weeks (−0.81, 95% CI −1.18 to −0.44) and 48 weeks (−0.64, 95% CI −1.05 to −0.23).
Lithium carbonate conclusion
For patients with SCA3 who are ambulatory, lithium probably does not improve ataxia over 48 weeks as measured by the NESSCA and SARA total scores (1 Class I study), although minimal clinically important differences on these scales have not been established and small changes cannot be excluded.
Weak evidence
Deferiprone
A Class II study described the administration of deferiprone (20, 40, and 60 mg/kg/d divided in 2 doses) over 6 months to 72 patients with FA who were ambulatory.10 The 60-mg/kg/d group was discontinued because of perceived/observed worsening of ataxia. Patients receiving 40 mg/d experienced significant worsening of ataxia compared with the placebo group, as measured by the Friedreich Ataxia Rating Scale (FARS) total score (difference in mean change 5.4, 95% CI 1.5–9.3) and the ICARS total score (difference in mean change 4.7, 95% CI 0.5–8.9). There were no significant differences between the group treated with 20 mg/kg/d and the placebo group (difference in FARS total score mean change −0.3, 95% CI −3.8 to 3.2; difference in ICARS total score mean change −0.6, 95% CI −4.5 to 3.3).
Deferiprone conclusion
For patients with FA, deferiprone 40 mg/kg/d possibly worsens ataxia signs over 6 months (1 Class II study).
Medications with conflicting results or insufficient evidence
Pharmacologic therapies for which no conclusions for or against use could be drawn are described in the table, with details provided in the full-length review. The idebenone literature is described briefly here because this treatment has been the subject of 3 randomized controlled trials. In the first study,11 there was no difference in ICARS change scores at 6 months by analysis of covariance (p = 0.17), but the intermediate- and high-dose groups had a greater mean change on the ICARS compared with the placebo group (difference in change vs placebo: low-dose 5 mg/kg −1.99 [95% CI −7.54 to 3.57], Bonferroni-adjusted p = 1.00; intermediate-dose 15 mg/kg −6.24 [95% CI −10.89 to −1.60], Bonferroni-adjusted p = 0.03; high-dose 45 mg/kg −7.76 [95% CI −12.56 to −2.96], Bonferroni-adjusted p = 0.010); the difference in mean change on the FARS between the treatment and control groups was not significant, but the CIs included the possibility of clinically important effects. The second study found no difference in improvement on the ICARS scores between groups, but did not have sufficient precision to exclude a clinically important effect.12 A random-effects meta-analysis of these 2 studies showed a greater mean change on the ICARS in the idebenone group, but with CIs that included the possibility of no effect (difference in mean score change −4.2, 95% CI −9.0 to 0.7, I2 = 38%). When data for the 45-mg/kg group in the first study were combined with those for the 30- to 54-mg/kg group in the second study, the difference in mean score change between idebenone and placebo was −4.5 (95% CI −11.0 to 2.0, I2 = 71%).
Table.
Pharmacologic agents for which no conclusions can be drawn
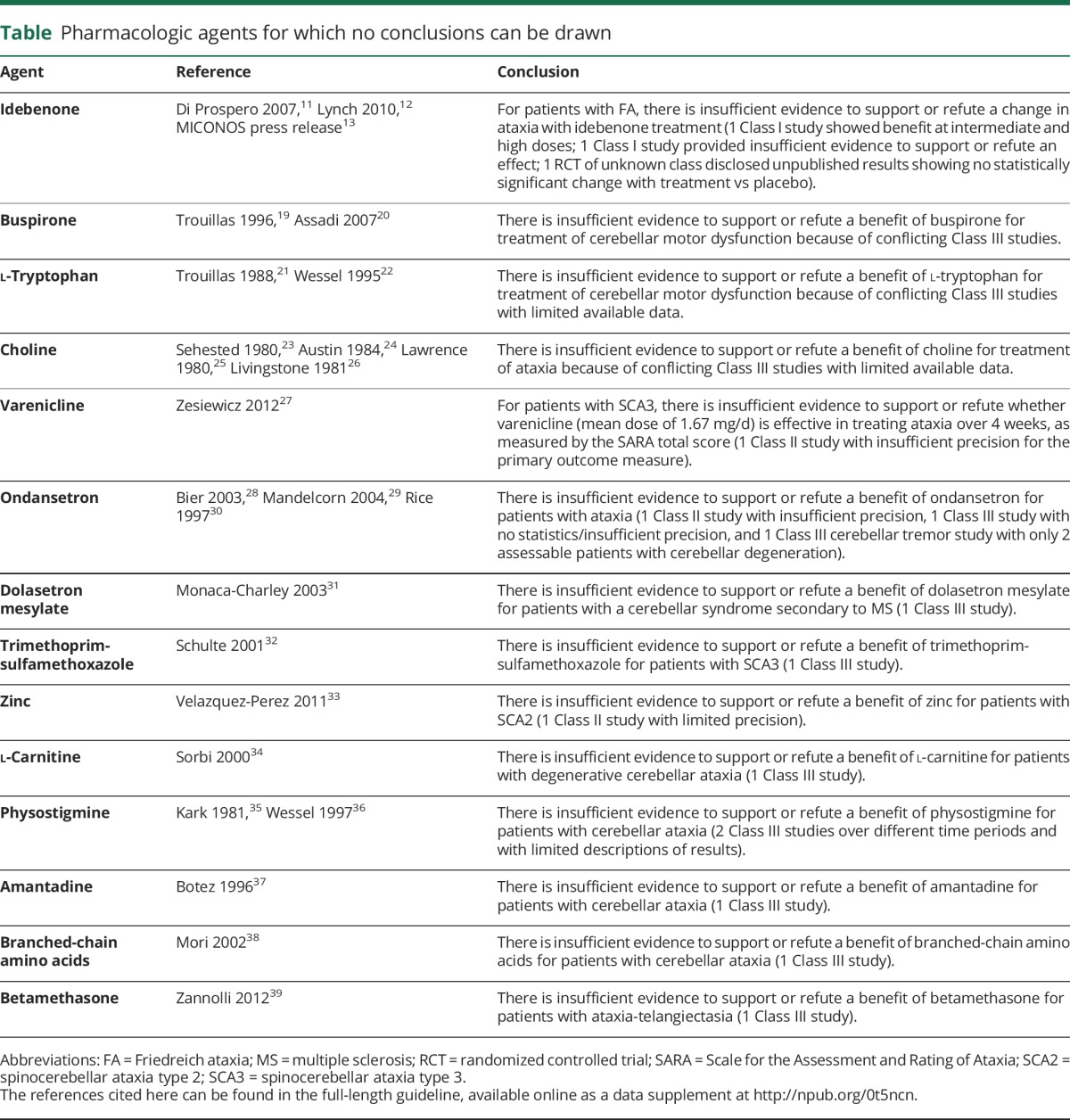
A third double-blind, placebo-controlled trial investigating idebenone for use in FA ended in 2010, but it could not be included, as it is unpublished and available data are insufficient for classification of evidence and analysis. According to a press release,13 there was no difference in the mean change in ICARS score from baseline between the active arms and placebo, and a meta-analysis of the manufacturer's 3 studies showed no statistically significant mean change in ICARS scores between groups.
Because of these different findings, the lack of statistical precision, and the inability to incorporate a large unpublished randomized controlled trial, no conclusions could be drawn for or against idebenone use (table).
The manufacturer of idebenone is not currently pursuing approval of idebenone for the treatment of FA, and this medication is not routinely used for this indication in clinical practice. Idebenone is not approved for use within the United States.
Question 2: For patients with cerebellar dysfunction, do surgical or other interventional therapies (e.g., physical training), compared with no (or alternative) treatments, improve motor symptoms with acceptable safety and tolerability?
Pressure splints
A Class II study of patients with MS-associated ataxia randomized patients to receive neuromuscular rehabilitation only (control group, n = 13) or neuromuscular rehabilitation plus pressure splints (treatment group, n = 13) 3 times weekly for 4 weeks.14 No posttreatment differences were noted between groups for most gait parameters/equilibrium tests. Data were insufficient to calculate 95% CIs for between-group change scores.
Pressure splints conclusion
For patients with MS-associated ataxia, the addition of pressure splints to neuromuscular rehabilitation possibly has no additional benefit over neuromuscular rehabilitation alone (1 Class II study).
Physical and occupational therapy
Various therapy approaches have been evaluated to improve symptoms of ataxia; most studies in this area are rated Class IV. In a single Class I study, daily inpatient physical and occupational therapy for 4 weeks was compared with a wait-list control.15 At 4 weeks, patients with SCA type 6, SCA type 31, and idiopathic cerebellar ataxia receiving rehabilitation had a greater reduction in the SARA total score (mean difference −3.0, 95% CI −4.3 to −1.8) and a small but significant improvement in the Functional Independence Measure total score (mean difference 1.3, 95% CI 0.4–2.0).
Physical and occupational therapy conclusion
Four-week inpatient rehabilitation in patients with isolated degenerative ataxias probably improves ataxia and functional abilities as measured at 4 weeks (1 Class I study).
Stochastic vibration therapy
There is insufficient information to support or refute the use of stochastic whole-body vibration therapy in patients with SCAs (1 Class III study).16
Question 3: For patients with cerebellar dysfunction, does TMS or tDCS, compared with no (or alternative) treatments, improve motor symptoms with acceptable safety and tolerability?
A double-blind, Class II study compared 21 daily TMS treatments over the cerebellum with sham treatments in 74 patients with various ataxias.17 The patients treated with TMS had a greater reduction in timed 10-m walk (−1.1 seconds, estimated 95% CI −2.3 to −0.005) and 10-m steps (−1.7, estimated 95% CI −3.4 to −0.007), and a greater improvement in the number of tandem steps (1.0, estimated 95% CI 0.3–1.7) and standing capacity (as assessed on a 0- to 6-point scale with lower scores indicating better function (−0.32, estimated 95% CI −0.6 to −0.001). The clinical significance of these differences is uncertain.
A Class III, randomized, double-blind, crossover study18 compared a single session of anodal cerebellar tDCS with sham stimulation separated by at least 1 week in 19 patients with various ataxias. The SARA score was better after tDCS treatment vs sham (mean difference 1.40, 95% CI 0.94–1.85), as was the ICARS (mean difference 4.37, 95% CI 3.27–5.47).
Conclusion
TMS over the cerebellum possibly improves cerebellar motor function at 21 days in patients with SCD and olivopontocerebellar atrophy (1 Class II study). There is insufficient evidence to support or refute use of a single session of anodal cerebellar tDCS for the treatment of ataxia (1 Class III study).
Discussion and suggestions for future research
Although studies of populations with rare diseases are challenging, rigorous study design is critical to assess the outcomes associated with new therapeutic options. This is true for both pharmacologic and nonpharmacologic studies. In addition to the studies described here, numerous Class IV studies were identified in the literature search. Under the 2011 AAN process, as amended, masked pretreatment and posttreatment study designs are insufficient to achieve Class III status.2 Only 2 rehabilitation studies were identified with a classification better than Class IV, and yet in practice, many clinicians find it helpful to refer patients with ataxia for therapy to help with daily function if not the ataxia itself. This review focused specifically on treatment of cerebellar motor dysfunction and ataxia; many of these conditions have associated signs and symptoms both within and outside the neurologic system that could potentially benefit from therapies not covered in this review. Dietary changes, including the use of a gluten-free diet to treat ataxia, were outside the scope of this systematic review. In addition, historical treatment approaches, such as the use of acetazolamide for the treatment of EA2, can have clinical value even in the absence of clinical trial evidence.
Future research in cerebellar motor dysfunction should analyze and document specific causes (genotype); define groups of diseases according to their mechanism of action (e.g., gain vs loss of function, toxicity); and utilize more precise outcome measures, including clinical and functional rating scales. More specific and potent candidate drugs for both symptomatic and disease-modifying studies are needed, as well as more sensitive clinical measures and biomarkers. Moreover, long-term studies to detect disease-modifying potential beyond symptomatic treatment should be conducted. Finally, the clinical trials must be adequately powered to detect a meaningful difference for each etiology.
Glossary
- AAN
American Academy of Neurology
- CI
confidence interval
- EA2
episodic ataxia type 2
- FA
Friedreich ataxia
- FARS
Friedreich Ataxia Rating Scale
- ICARS
International Cooperative Ataxia Rating Scale
- MS
multiple sclerosis
- NESSCA
Neurological Examination Score for Spinocerebellar Ataxia
- OR
odds ratio
- SARA
Scale for the Assessment and Rating of Ataxia
- SCA
spinocerebellar ataxia
- SCA3
spinocerebellar ataxia type 3
- SCD
spinocerebellar degeneration
- tDCS
transcranial direct current stimulation
- TMS
transcranial magnetic stimulation
- TRH
thyrotropin-releasing hormone
- VPA
valproic acid
Footnotes
Full text of guidelines at: NPub.org/0t5ncn
Disclaimer
Clinical practice guidelines, practice advisories, systematic reviews, and other guidance published by the American Academy of Neurology and its affiliates are assessments of current scientific and clinical information provided as an educational service. The information: (1) should not be considered inclusive of all proper treatments, methods of care, or as a statement of the standard of care; (2) is not continually updated and may not reflect the most recent evidence (new evidence may emerge between the time information is developed and when it is published or read); (3) addresses only the question(s) specifically identified; (4) does not mandate any particular course of medical care; and (5) is not intended to substitute for the independent professional judgment of the treating provider, as the information does not account for individual variation among patients. In all cases, the selected course of action should be considered by the treating provider in the context of treating the individual patient. Use of the information is voluntary. AAN provides this information on an “as is” basis, and makes no warranty, expressed or implied, regarding the information. AAN specifically disclaims any warranties of merchantability or fitness for a particular use or purpose. AAN assumes no responsibility for any injury or damage to persons or property arising out of or related to any use of this information or for any errors or omissions.
Conflict of interest
The American Academy of Neurology (AAN) is committed to producing independent, critical, and truthful comprehensive systematic reviews (SRs). Significant efforts are made to minimize the potential for conflicts of interest to influence the conclusions of this SR. To the extent possible, the AAN keeps separate those who have a financial stake in the success or failure of the products appraised in the SRs and the developers of the SRs. Conflict of interest forms were obtained from all authors and reviewed by an oversight committee prior to project initiation. AAN limits the participation of authors with substantial conflicts of interest. The AAN forbids commercial participation in, or funding of, systematic review projects. Drafts of the SR have been reviewed by at least 3 AAN committees, a network of neurologists, Neurology peer reviewers, and representatives from related fields. The AAN Guideline Author Conflict of Interest Policy can be viewed at www.aan.com. For complete information on this process, access the 2004 AAN process manual.1
Author contributions
Dr. Zesiewicz: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content, study supervision. Dr. Wilmot: acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Kuo: acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Perlman: acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Greenstein: analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Ying: analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Ashizawa: acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Subramony: acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Schmahmann: acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Figueroa: analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Mizusawa: analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Schöls: analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Ms. Shaw: analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Dubinsky: acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content. Dr. Armstrong: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content, study supervision. Dr. Gronseth: acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content, study supervision. Dr. Sullivan: study concept and design, acquisition of data, analysis or interpretation of data, drafting/revising the manuscript, critical revision of the manuscript for important intellectual content, study supervision.
Study funding
This comprehensive systematic review was developed with financial support from the American Academy of Neurology (AAN). Authors who serve or served as AAN subcommittee members or methodologists (T.A.Z., R.M.D., M.J.A., G.S.G., K.L.S.) were reimbursed by the AAN for expenses related to travel to subcommittee meetings where drafts of manuscripts were reviewed.
Disclosure
T. Zesiewicz has served as a clinical advisor for Steminent Biotherapeutics; has received travel reimbursement from the Department of Neurology at University of Southern Florida; has received travel reimbursement for a Biohaven Pharmaceuticals meeting; has served on the editorial board for Neurodegenerative Disease Management and Tremor and other Hyperkinetic Movements; has a patent for Methods of Treating Disease-Induced Ataxia and Non-Ataxic Imbalance (US Patent No. 9463190 B2); and has received research support for her division for approximately 20 clinical trials for Parkinson disease Friedreich ataxia, and spinocerebellar ataxias (SCAs). G. Wilmot has served on scientific advisory panels for Biohaven Pharmaceuticals and Santhera Pharmaceuticals, and has received financial or material research support or compensation from Friedreich's Ataxia Research Alliance, Reata Pharmaceuticals, and Shire. S.-H. Kuo and S. Perlman report no disclosures relevant to the manuscript. P. Greenstein has received an R21 grant award from the NIH to study the effect of transcranial magnetic stimulation on SCA (grant awarded in August of 2013; the study began in January 2014; no preliminary data yet available). S. Ying received a salary from Shire; received a salary during her employment with Pfizer Inc. and Takeda Pharmaceuticals Inc.; and received grant funding from the NIH. T. Ashizawa has nonfinancial competing interests with the Marigold Foundation, Myotonic Dystrophy Foundation, and the Muscular Dystrophy Association (MDA); receives honoraria from the NIH National Institute of Neurological Disorders and Stroke (NINDS) Neurological Sciences and Disorders B Study Section; received travel reimbursement from the MDA Medical Advisory Committee and the National Ataxia Foundation for the Ataxia Investigator Meeting; serves as an editor for PLoS ONE; has a patent (US Patent No. 6855497) on a DNA test for SCA type 10 (SCA10); receives funding for an NINDS research grant award R01NSNS083564; participates in a clinical trial of BHV-4157 (NCT02960893); and has received royalty payments from Baylor College of Medicine for a DNA test for SCA10 (US Patent No. 6855497). S. Subramony has received compensation for a lecture from Athena Diagnostics in October 2013; and has received travel reimbursement or honoraria from Reata Pharmaceuticals, ISIS Pharmaceuticals (now Ionis Pharmaceuticals), the NIH, the National Ataxia Foundation, and the Friedreich's Ataxia Research Alliance. J. Schmahmann serves as a consultant to Ataxion, Biogen, Biohaven, Pfizer, and Takeda, and receives grant support from the National Ataxia Foundation and the A-T Children's Project. K. Figueroa, H. Mizusawa, L. Schöls, and J. Shaw report no disclosures relevant to the manuscript. R. Dubinsky serves on the scientific advisory board for Allergan Pharmaceuticals; has received travel funding from the American Academy of Neurology (AAN), Allergan Pharmaceuticals, and the Huntington Study Group; serves as Level of Evidence associate editor for the AAN; receives honoraria from Allergan Pharmaceuticals; serves on the speakers bureau for Allergan Pharmaceuticals; and is involved with the commercial entity Allergan Pharmaceuticals and the government entities the NIH and the Agency for Healthcare Research and Quality. His spouse holds stock in Abbott Laboratories. M. Armstrong serves on the Level of Evidence editorial board for Neurology (not compensated financially) and is an AAN evidence-based methodologist. G. Gronseth serves as an associate editor (level of evidence review) for Neurology, serves on the editorial advisory board for Neurology Now, and is compensated by the AAN for methodologic activities. K. Sullivan has received research support from the Georgia Governor's Office of Highway Safety and has a patent for Methods of Treating Disease-Induced Ataxia and Non-Ataxic Imbalance (US Patent No. 9463190 B2). Go to Neurology.org/N for full disclosures.
References
- 1.American Academy of Neurology. Clinical Practice Guidelines Process Manual. 2004 ed. St. Paul, MN: The American Academy of Neurology; 2004. [Google Scholar]
- 2.American Academy of Neurology. Clinical Practice Guideline Process Manual. 2011 ed. St. Paul, MN: The American Academy of Neurology; 2011. [Google Scholar]
- 3.Strupp M, Kalla R, Claassen J, et al. A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias. Neurology 2011;77:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ristori G, Romano S, Visconti A, et al. Riluzole in cerebellar ataxia: a randomized, double-blind, placebo-controlled pilot trial. Neurology 2010;74:839–845. [DOI] [PubMed] [Google Scholar]
- 5.Romano S, Coarelli G, Marcotulli C, et al. Riluzole in patients with hereditary cerebellar ataxia: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2015;14:985–991. [DOI] [PubMed] [Google Scholar]
- 6.Lei LF, Yang GP, Wang JL, et al. Safety and efficacy of valproic acid treatment in SCA3/MJD patients. Parkinsonism Relat Disord 2016;26:55–61. [DOI] [PubMed] [Google Scholar]
- 7.Sobue I, Takayanagi T, Nakanishi T, et al. Controlled trial of thyrotropin releasing hormone tartrate in ataxia of spinocerebellar degenerations. J Neurol Sci 1983;61:235–248. [DOI] [PubMed] [Google Scholar]
- 8.Saute JA, de Castilhos RM, Monte TL, et al. A randomized, phase 2 clinical trial of lithium carbonate in Machado-Joseph disease. Mov Disord 2014;29:568–573. [DOI] [PubMed] [Google Scholar]
- 9.Saute JA, Rieder CR, Castilhos RM, et al. Planning future clinical trials in Machado Joseph disease: lessons from a phase 2 trial. J Neurol Sci 2015;358:72–76. [DOI] [PubMed] [Google Scholar]
- 10.Pandolfo M, Arpa J, Delatycki MB, et al. Deferiprone in Friedreich ataxia: a 6-month randomized controlled trial. Ann Neurol 2014;76:509–521. [DOI] [PubMed] [Google Scholar]
- 11.Di Prospero NA, Baker A, Jeffries N, Fischbeck KH. Neurological effects of high-dose idebenone in patients with Friedreich's ataxia: a randomised, placebo-controlled trial. Lancet Neurol 2007;6:878–886. [DOI] [PubMed] [Google Scholar]
- 12.Lynch DR, Perlman SL, Meier T. A phase 3, double-blind, placebo-controlled trial of idebenone in Friedreich ataxia. Arch Neurol 2010;67:941–947. [DOI] [PubMed] [Google Scholar]
- 13.Santhera's MICONOS Trial with Catena in Friedreich's Ataxia Misses Primary Endpoint. Ataxia Canada. Available at: http://lacaf.org/en/santhera-trial-with-catena-in-friedreich-ataxia-misses-primary-endpoint. Accessed June 13, 2016. [Google Scholar]
- 14.Armutlu K, Karabudak R, Nurlu G. Physiotherapy approaches in the treatment of ataxic multiple sclerosis: a pilot study. Neurorehabil Neural Repair 2001;15:203–211. [DOI] [PubMed] [Google Scholar]
- 15.Miyai I, Ito M, Hattori N, et al. Cerebellar ataxia rehabilitation trial in degenerative cerebellar diseases. Neurorehabil Neural Repair 2012;265:515–522. [DOI] [PubMed] [Google Scholar]
- 16.Kaut O, Jacobi H, Coch C, et al. A randomized pilot study of stochastic vibration therapy in spinocerebellar ataxia. Cerebellum 2014;13:237–242. [DOI] [PubMed] [Google Scholar]
- 17.Shiga Y, Tsuda T, Itoyama Y, et al. Transcranial magnetic stimulation alleviates truncal ataxia in spinocerebellar degeneration. J Neurol Neurosurg Psychiatry 2002;72:124–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benussi A, Koch G, Cotelli M, Padovani A, Borroni B. Cerebellar transcranial direct current stimulation in patients with ataxia: a double-blind, randomized, sham-controlled study. Mov Disord 2015;30:1701–1705. [DOI] [PubMed] [Google Scholar]
- 19.Trouillas P , Xie J, Adeleine P. Treatment of cerebellar ataxia with buspirone: a double-blind study. Lancet 1996;348:759. [DOI] [PubMed] [Google Scholar]
- 20.Assadi M, Campellone J, Janson CG, Veloski JJ, Schwartzman RJ, Leone P. Treatment of spinocerebellar ataxia with buspirone. J Neurol Sci 2007;260:143–146. [DOI] [PubMed] [Google Scholar]
- 21.Trouillas P, Brudon F, Adeleine P. Improvement of cerebellar ataxia with levorotatory form of 5-hydroxytryptophan. Arch Neurol 1988;45:1217–1222. [DOI] [PubMed] [Google Scholar]
- 22.Wessel K, Hermsdorfer J, Deger K, et al. Double-blind crossover study with levorotatory form of hydroxytryptophan in patients with degenerative cerebellar diseases. Arch Neurol 1995;52:451–455. [DOI] [PubMed] [Google Scholar]
- 23.Sehested P, Lund HI, Kristensen O. Oral choline in cerebellar ataxia. Acta Neurol Scand 1980;62:124–126. [DOI] [PubMed] [Google Scholar]
- 24.Austin CA, Mundy KI, Dorey S. Low dose choline chloride in cerebellar degeneration. J Neurol Neurosurg Psychiatry 1984;47:1038–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawrence CM, Millac P, Stout GS, Ward JW. The use of choline chloride in ataxic disorders. J Neurol Neurosurg Psychiatry 1980;43:452–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Livingstone IR, Mastaglia FL, Pennington RJT, Skilbeck C. Choline chloride in the treatment of cerebellar and spinocerebellar ataxia. J Neurol Sci 1981;50:161–174. [DOI] [PubMed] [Google Scholar]
- 27.Zesiewicz TA, Greenstein PE, Sullivan KL, et al. A randomized trial of varenicline (Chantix) for the treatment of spinocerebellar ataxia type 3. Neurology 2012;78:545–550. [DOI] [PubMed] [Google Scholar]
- 28.Bier JC, Dethy S, Hildebrand J, et al. Effects of the oral form of ondansetron on cerebellar dysfunction: a multi-center double-blind study. J Neurol 2003;250:693–697. [DOI] [PubMed] [Google Scholar]
- 29.Mandelcorn J, Cullen N, Bayley MT, et al. A preliminary study of the efficacy of ondansetron in the treatment of ataxia, poor balance and incoordination from brain injury. Brain Inj 2004;18:1025–1039. [DOI] [PubMed] [Google Scholar]
- 30.Rice GP, Lesaux J, Vandervoort P, Macewan L, Ebers GC. Ondansetron, a 5-HT3 antagonist, improves cerebellar tremor. J Neurol Neurosurg Psychiatry 1997;62:282–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Monaca-Charley C, Stojkovic T, Duhamel A, De Seze J, Ferriby D, Vermersch P. Double-blind crossover study with dolasetron mesilate, a 5-HT3 receptor antagonist in cerebellar syndrome secondary to multiple sclerosis. J Neurol 2003;250:1190–1194. [DOI] [PubMed] [Google Scholar]
- 32.Schulte T, Mattern R, Berger K, et al. Double-blind crossover trial of trimethoprim-sulfamethoxazole in spinocerebellar ataxia type 3/Machado-Joseph disease. Arch Neurol 2001;58:1451–1457. [DOI] [PubMed] [Google Scholar]
- 33.Velazquez-Perez L, Rodriguez-Chanfrau J, García-Rodríguez JC, et al. Oral zinc sulphate supplementation for six months in SCA2 patients: a randomized, double-blind, placebo-controlled trial. Neurochem Res 2011;36:1793–1800. [DOI] [PubMed] [Google Scholar]
- 34.Sorbi S, Forleo P, Fani C, Piacentini S. Double-blind, crossover, placebo-controlled clinical trial with L-acetylcarnitine in patients with degenerative cerebellar ataxia. Clin Neuropharmacol 2000;23:114–118. [DOI] [PubMed] [Google Scholar]
- 35.Kark RA, Budelli MM, Wachsner R. Double-blind, triple-crossover trial of low doses of oral physostigmine in inherited ataxias. Neurology 1981;31:288–292. [DOI] [PubMed] [Google Scholar]
- 36.Wessel K, Langenberger K, Nitschke MF, Kömpf D. Double-blind crossover study with physostigmine in patients with degenerative cerebellar diseases. Arch Neurol 1997;54:397–400. [DOI] [PubMed] [Google Scholar]
- 37.Botez MI, Botez-Marquard T, Elie R, Pedraza OL, Goyette K, Lalonde R. Amantadine hydrochloride treatment in heredodegenerative ataxias: a double blind study. J Neurol Neurosurg Psychiatry 1996;61:259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mori M, Adachi Y, Mori N, et al. Double-blind crossover study of branched-chain amino acid therapy in patients with spinocerebellar degeneration. J Neurol Sci 2002;195:149–152. [DOI] [PubMed] [Google Scholar]
- 39.Zannolli R, Buoni S, Betti G, et al. A randomized trial of oral betamethasone to reduce ataxia symptoms in ataxia telangiectasia. Mov Disord 2012;27:1312–1316. [DOI] [PubMed] [Google Scholar]