

Abstract
Objective
To determine whether high CSF levels of neurogranin (Ng) predict longitudinal decline in memory and executive function during early-stage Alzheimer disease (AD).
Methods
Baseline levels of CSF Ng were studied in relation to cross-sectional and longitudinal cognitive performance over 8 years. Data were obtained from the Alzheimer's Disease Neuroimaging Initiative database, and participants with normal cognition (n = 111) and mild cognitive impairment (MCI) (n = 193) were included.
Results
High levels of CSF Ng were associated with poor baseline memory scores (β = −0.21, p < 0.0001). CSF Ng predicted both memory and executive function decline over time (β = −0.0313, p = 0.0068 and β = −0.0346, p = 0.0169, respectively) independently of age, sex, education, and APOE ε4 status. When the rate of decline by tertiles was examined, CSF Ng was a level-dependent predictor of memory function, whereby the group with highest levels of Ng showed the fastest rates of decline in both memory and executive function. When examined separately, elevated Ng was associated with cognitive decline in participants with MCI but not in those with normal cognition. The levels of CSF Ng were not associated with cognitive measures when tau and amyloid 42 (Aβ42) were controlled for in these analyses.
Conclusions
High CSF Ng associates with poor memory scores in participants with MCI cross-sectionally and with poor memory and executive function longitudinally. The association of Ng with cognitive measures disappears when tau and Aβ42 are included in the statistical models. Our findings suggest that CSF Ng may serve as a biomarker of cognition. Synaptic dysfunction contributes to cognitive impairment in early-stage AD.
Alzheimer disease (AD) is the most common neurodegenerative disorder. Memory loss is an early and predominant feature of AD.1 Patients with late-onset AD tend to display a pattern of disproportionately more impaired memory relative to executive function, yet about one-fifth of people with AD have been found to have executive predominance with executive function scores lower than memory scores.2
Over the last 2 decades, amyloid 42 (Aβ42), phosphorylated tau, and total tau (t-tau) in the CSF have been identified as important biomarkers of the pathophysiologic process underlying AD.3,4 However, the correlation of these biomarkers with the rate of cognitive deterioration is inconsistent. Searching for additional biomarkers associated with cognitive decline in AD is of critical importance in enabling earlier diagnosis, tracking mental status, and developing new treatments.
Neurogranin (Ng), a postsynaptic protein, is elevated in the CSF of patients with AD and mild cognitive impairment (MCI) compared to participants with normal cognition (NC).5–7 High CSF Ng levels correlate with poor Mini-Mental State Examination (MMSE) scores in these participants.8,9 Levels of Ng predict progression of MCI to AD.10 To study the role of Ng in specific cognitive domains, we examined whether high CSF Ng would predict cognitive performance in participants at risk for AD. We hypothesized that high CSF Ng would be a sensitive predictor of memory loss and executive dysfunction. In this study, baseline levels of CSF Ng were analyzed in relation to baseline memory, executive function, global cognition, and function scores in participants with NC and MCI. The longitudinal study was performed with Ng used as a predictor of change in memory, executive function, global cognition, and function scores over an 8-year period.
Methods
Alzheimer's Disease Neuroimaging Initiative study
Data used for this study were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (http://adni.loni.usc.edu). The ADNI was launched in 2003 as a public-private partnership with the primary objective of testing whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessments can be combined to measure the progression of MCI and early AD.
Standard protocol approvals, registrations, and patient consent
As stated in the ADNI procedures manual, ADNI centers obtain local Institutional Review Board protocol approval, and patient consents are obtained at the participants’ initial screening visit.
Participants
We included participants who met criteria for NC and MCI and had baseline CSF Ng samples and follow-up evaluations of memory and executive function. A total of 304 individuals were analyzed, 111 with NC and 193 with MCI. The numbers of participants present at the follow-up visits are summarized in table 1.
Table 1.
Demographic information of study participants

All participants were between 55 and 90 years old at the time of inclusion, had completed at least 6 years of education, were fluent in English or Spanish, and were free of any other neurologic diseases. Participants with NC had an MMSE score ≥24 and a Clinical Dementia Rating (CDR) score of zero. Participants with MCI had an MMSE score ≥24, CDR score of 0.5, objective memory loss based on delayed recall scores of the Wechsler Memory Scale Logical memory II, and absence of dementia.
Composite memory and executive function scores
ADNI created composite memory and executive function scores derived from tests used in its neuropsychological battery. The ADNI memory composite score (ADNI-Mem) was developed from the Rey Auditory Verbal Learning Test (2 versions), Alzheimer’s Disease Assessment Schedule-Cognition (ADAS-Cog, 3 versions), MMSE, and Logical Memory data.11 The executive function score (ADNI-EF) derives from Wechsler Adult Intelligence Scale-Revised Digit Symbol Substitution, Digit Span Backwards, Trails A and B, Category Fluency, and Clock Drawing.12 Both ADNI-Mem and ADNI-EF have been validated and described to be as good as or better than any of the composite parts.11,12 In our study, we included up to 8 years of follow-up scores.
CSF analysis
CSF Ng levels were measured with antibody-based electrochemiluminescence technology (Meso Scale Discovery, Gaithersburg, MD) with Ng 7 (a monoclonal antibody with epitope at amino acids 53–64 of Ng)13 as the coating antibody and polyclonal Ng anti-rabbit (ab 23570, Upstate) as the detector antibody.5
Statistical analysis
We used t tests and χ2 tests to assess differences in demographic, clinical, genotype, and CSF biomarker variables between the NC and MCI groups. To examine the cross-sectional associations between CSF Ng levels and both baseline ADNI-Mem and ADNI-EF scores, various general linear regression models were constructed: model 1 was unadjusted; model 2 was adjusted for age, sex, and years of education; model 3 was adjusted for APOE ε4 status; and model 4 was adjusted for t-tau and Aβ42. To evaluate the relationship between baseline CSF Ng levels and rate of decline per year in ADNI-Mem and ADNI-EF scores, linear mixed models were fitted with the same adjustments as above. For these models, baseline CSF Ng was first assessed as a continuous variable and then categorized as tertiles to look for a gradient effect on the decline in memory and/or executive function. To determine whether clinical status had a modification effect, these analyses were conducted within each diagnostic group separately. The Pearson correlation was performed to evaluate the correlation between CSF Ng and the other biomarkers. The level of statistical significance was set at p < 0.05. All data analyses were performed with statistical software (SPSS, version 24, IBM, Armonk, NY; Stata, version 13, StataCorp LP, College Station, TX; or SAS, version 9.4, SAS Institute, Cary, NC).
Results
Demographic information
Within the ADNI dataset, participants with MCI (n = 193) and NC (n = 111) were analyzed (table 1). Compared with healthy controls, participants with MCI were more likely to be male and APOE ε4 carriers and had greater CDR Sum of Boxes scores, lower MMSE scores, and higher levels of CSF Ng, t-tau, and phosphorylated tau but lower levels of Aβ42. No significant difference was found in age or education attainment between the 2 groups (table 1).
Association of CSF Ng levels with memory scores in cross-sectional study
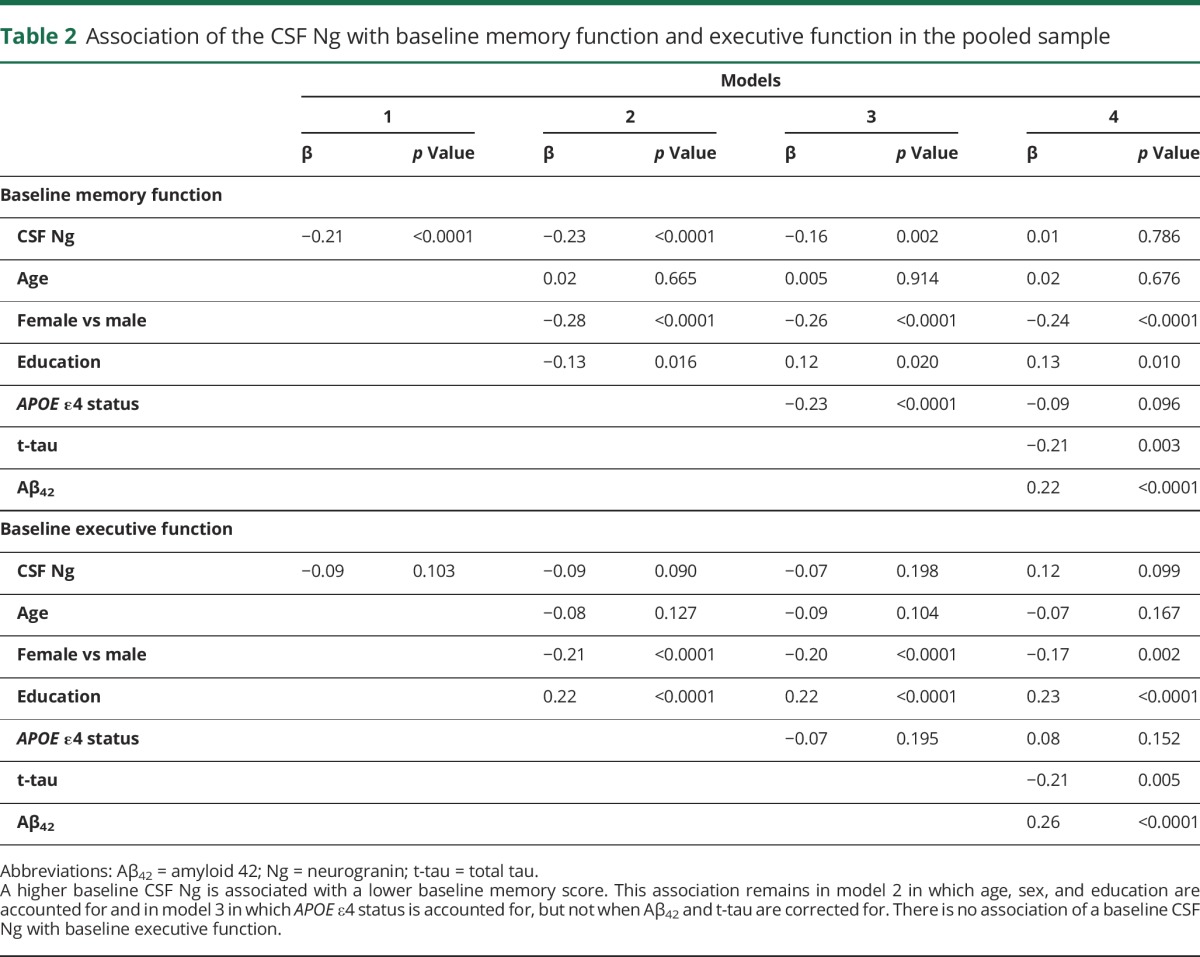
There was a significant association between baseline levels of CSF Ng and baseline memory scores in the pooled participants with NC and MCI (table 2). Increased CSF Ng was associated with lower baseline memory scores (β = −0.21, p < 0.0001). This association remained after adjustment for age, sex, and education in model 2 (β = −0.23, p < 0.0001) and after adjustment for APOE ε4 in model 3 (β = −0.16, p = 0.002). The levels of CSF Ng were not associated with baseline memory when tau and Aβ42 were controlled for in model 4 (β = 0.01, p = 0.786). There was no association between baseline CSF Ng and baseline executive function in both unadjusted and multivariable-adjusted models (table 2).
Table 2.
Association of the CSF Ng with baseline memory function and executive function in the pooled sample
Association of CSF Ng levels with memory and executive scores over a period of 8 years
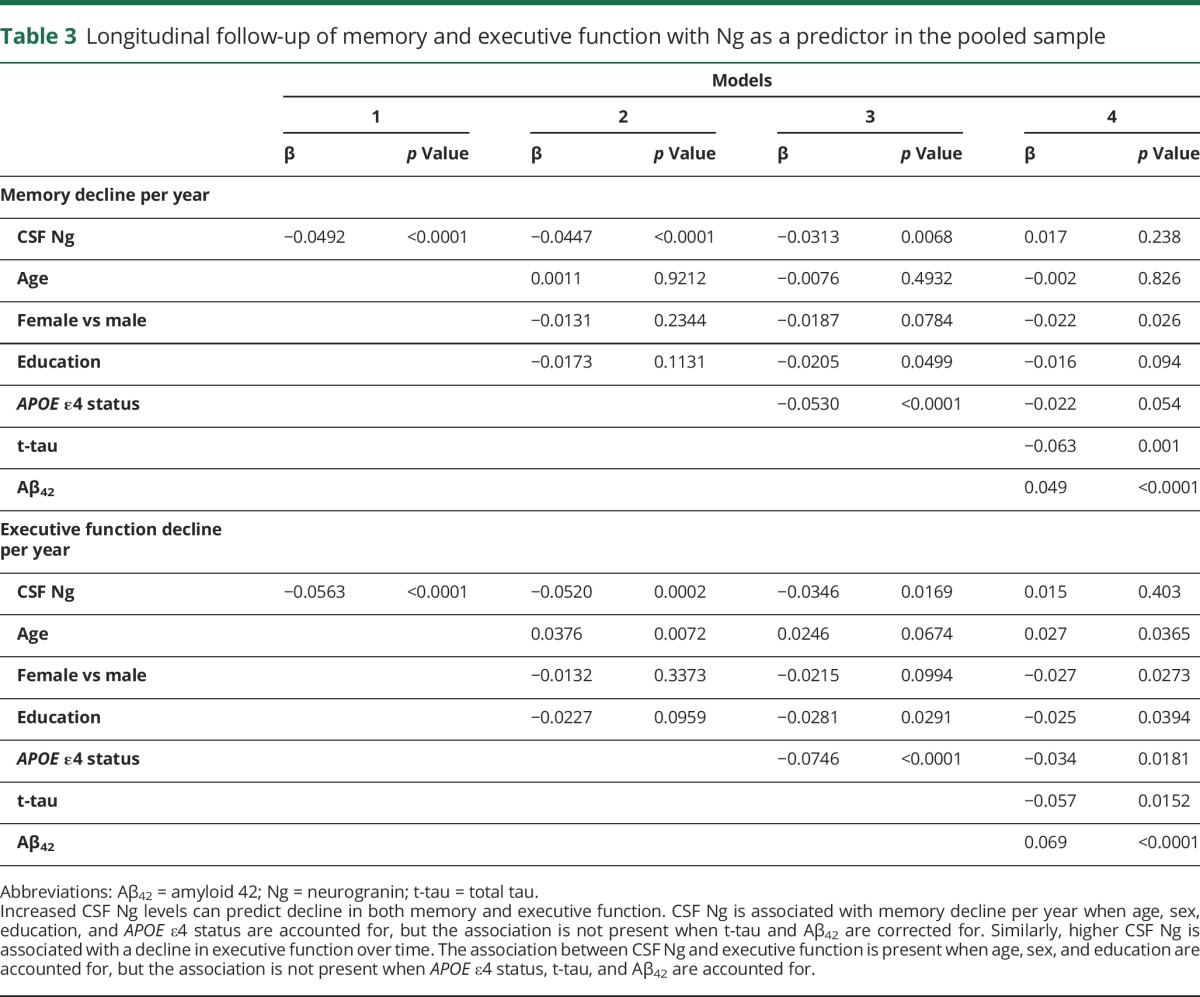
Longitudinal analysis of NC and MCI participants showed that baseline CSF Ng levels predicted decline in both memory and executive function over an 8-year period. Table 3 demonstrates that baseline CSF Ng was associated with memory decline per year (β = −0.049, p < 0.0001), and after accounting for age, sex, education, and APOE ε4 status, this association remained (β = −0.031, p = 0.0068). There was an association of baseline CSF Ng with executive function decline per year (β = −0.056, p < 0.0001), and this association remained after adjustment for age, sex, education, and APOE ε4 status in model 3 (β = −0.035, p = 0.0169). As shown in model 4, when CSF Aβ42 and tau levels were corrected for, CSF Ng was no longer a predictor of longitudinal memory or executive function decline (β = 0.017, p = 0.238 and β = 0.015, p = 0.403, respectively).
Table 3.
Longitudinal follow-up of memory and executive function with Ng as a predictor in the pooled sample
When the participants were divided into tertiles based on CSF Ng levels, there was a clear gradient relationship between baseline CSF Ng levels and the rate of memory decline (figure). The rate of memory decline could be differentially predicted by all CSF Ng tertiles. Tertiles 2 and 3 showed incrementally faster rates of decline in memory function compared to tertile 1. Executive function decline could be differentially predicted only by the top CSF Ng tertile (tertile 3). The rate of decline in executive function was not different between the tertile 2 and 1 groups (figure).
Figure. Pattern of memory and executive function decline with Ng as a function in the pooled sample.

Three tertiles were created based on baseline CSF neurogranin (Ng) level. Tertile 1 comprises the lowest level CSF Ng; tertile 2 is intermediate; and tertile 3 includes the highest level of CSF Ng. Trajectories of memory (MEM) and executive function (EF) decline over time, showing that Ng is an effective longitudinal predictor for cognitive function over an 8-year period. (A) The rate of memory decline is fastest for the highest CSF Ng level, and the rate of decline is faster in the intermediate tertile than the lowest tertile. (B) For executive function, the highest level of CSF Ng predicts a faster rate of executive function decline, but the rates for the 2 lower tertiles are very similar.
Association of CSF Ng with global cognition and function
The baseline and longitudinal associations between CSF Ng and multiple global cognitive measures, including ADAS-Cog 11, ADAS-Cog 13, MMSE, and CDR Sum of Boxes, were studied (tables e-1–e-12, http://links.lww.com/WNL/A210). At baseline, CSF Ng is associated with ADAS-Cog 11 (β = 0.13, p = 0.02), ADAS-Cog 13 (β = 0.17, p < 0.01), and CDR Sum of Boxes scores (β = 0.12, p = 0.03). These associations remained when age, sex, and years of education were accounted for. However, only baseline ADAS-Cog 13 was associated with CSF Ng when APOE ε4 status was corrected for (β = 0.11, p = 0.04). In model 4, CSF Ng was not associated with any of these cognitive measures after adjustment for tau and Aβ42.
Longitudinal analysis revealed an association between CSF Ng and all of the cognitive scores analyzed: ADAS-Cog 11 (β = 4.77, p < 0.01), ADAS-Cog 13 (β = 5.42, p < 0.001), MMSE (β = −2.07, p < 0.001), and CDR Sum of Boxes (β = 1.215, p < 0.001). In models 2 and 3 in which demographic information and APOE ε4 status were corrected, the longitudinal associations remained significant. As with the baseline associations, CSF Ng was not significantly associated with longitudinal ADAS-Cog 11, ADAS-Cog 13, MMSE, or CDR Sum of Boxes scores when t-tau and Aβ42 were additionally corrected for in model 4.
High levels of CSF Ng are associated with memory and executive function decline in participants with MCI but not those with NC over an 8-year period.
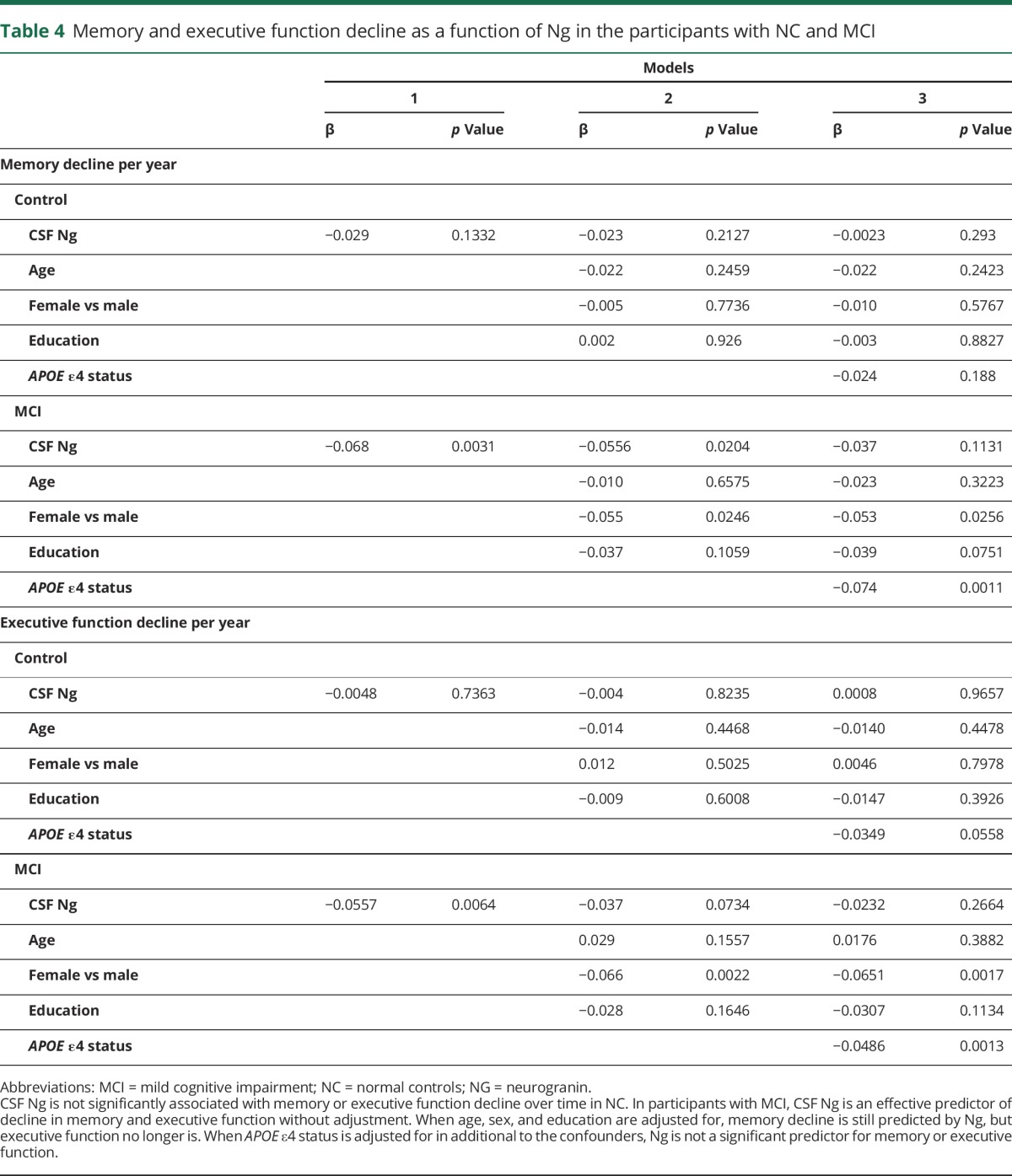
To explore the potential modification effect of baseline diagnostic status, the relationship between CSF Ng and memory and executive function decline was examined in participants with MCI and healthy controls separately. Table 4 reveals that baseline CSF Ng was not associated with longitudinal memory decline in healthy controls (β = −0.029, p = 0.133) but was associated with memory decline in participants with MCI (β = −0.068, p = 0.0031). The association in the MCI group remained after accounting for age, sex, and education (β = −0.056, p = 0.020) but not after accounting for APOE ε4 status (β = −0.037, p = 0.113). Baseline CSF Ng predicted a decline in executive function for participants with MCI (β = −0.056, p = 0.006) but not for healthy controls (β = −0.0048, p = 0.736). Baseline CSF Ng was not associated with executive function decline in the MCI group after adjustment for confounders.
Table 4.
Memory and executive function decline as a function of Ng in the participants with NC and MCI
Correlation of CSF Ng levels with t-tau and Aβ42 levels
To evaluate the relationship between CSF Ng and t-tau and Aβ42, a Pearson correlation test was performed. Increasing levels of CSF Ng were associated with increasing t-tau levels (r = 0.69, p < 0.0001). Conversely, increasing levels of CSF Ng were associated with decreasing Aβ42 levels (r = −0.33, p < 0.0001).
Discussion
In this study, cross-sectional analysis showed that high CSF Ng was significantly associated with lower memory scores in pooled participants with NC and MCI. Longitudinal analysis revealed that baseline CSF Ng was significantly associated with both memory and executive function decline in the pooled sample. In addition, Ng predicted memory, executive function, and global cognitive decline over an 8-year period. Finally, a significant association of CSF Ng with memory and executive function decline was associated only with individuals with MCI.
In AD, memory impairment is an initial and central presentation in most patients.1,14 Amnestic MCI has a higher likelihood of progression to AD compared to nonamnestic MCI.15 This is consistent with neuropathologic studies showing that the limbic system has degenerative changes in the early stage of AD.16 Although a clear, disproportionately severe episodic memory disorder was observed in patients with amnestic MCI, increasing evidence also shows that there is a dysexecutive form of MCI associated with AD.2,17,18 The molecular mechanism underlying memory and executive dysfunction remains unclear.
Ng, a postsynaptic protein, has been shown to be associated with synaptic plasticity.19 Deletion of the Ng gene in mice results in decreased short- and long-term plasticity and impaired spatial memory.19,20 Ng influences synaptic plasticity by regulating the level of calmodulin and the pattern of calcium/calmodulin–dependent postsynaptic signaling at dendritic spines.21 In AD, a marked reduction in Ng levels is found in the hippocampus and frontal cortex.22
In this study, high CSF Ng was significantly associated with poor memory scores in both cross-sectional and longitudinal analyses. This finding suggests that Ng plays an important role in memory dysfunction in AD. Given the fact that excitatory synapses terminating on dendritic spines are involved in the formation of new memories and are damaged in AD, changes of the levels of postsynaptic proteins in CSF in relation to memory function are to be expected.23,24
However, high CSF Ng levels were associated with worse executive function only in the longitudinal analysis, not the cross-sectional analysis. This finding raises several possibilities. First, the participants with MCI in this study may have had a relatively subtle deficit of executive function at baseline. Alternatively, the neuropsychological battery used in this study may have been too insensitive to detect dysexecutive function in MCI. Another possible explanation is that our MCI group is composed of a more amnestic phenotype than dysexecutive phenotype because our population has a high percentage of APOE ε4 carriers in the MCI group (53%). A previous study noted that APOE ε4 carriers were more common in the amnestic group than the dysexecutive phenotype.17 As a result, the association of memory dysfunction with CSF Ng was readily apparent. Finally, it is possible that there is a temporal relationship between involvement of memory and executive function in the pathophysiologic process of AD. Declining performance on episodic memory was found to occur 7 years before diagnosis, while declining performance on executive function was found to accelerate 2 to 3 years before diagnosis.25 Nevertheless, our findings provide evidence that Ng is involved in executive dysfunction in patients with MCI.
In this study, we further investigated the utility of CSF Ng as a maker for global cognition and functional status. We confirmed that high CSF Ng was associated with global cognitive impairment cross-sectionally and longitudinally. In addition, high CSF Ng was associated with poor functional scores longitudinally. These findings suggest that CSF Ng has potential to monitor the progress of this disease, serving as a biomarker for assessing the effect of treatment.
In this study, the association of Ng with memory, executive function, and global cognitive and functional measures was no longer present when tau and Aβ42 were controlled for in the cross-sectional and longitudinal analyses. It is suggested that tau and Aβ42 are the key factors involved in postsynaptic injury in patients with MCI. This finding is supported by our correlation analysis. A recent study also reports that the association of Ng with regional brain atrophy was found in individuals with Aβ pathology.26 Emerging evidence suggests that Aβ-induced synaptic dysfunction is dependent on NMDA receptor–mediated pathways, leading to dendritic spine loss.27–29 Tau pathology has been reported to contribute more to synapse degeneration and resultant dementia.30,31 The involvement of amyloid and tau pathology in synaptic damage requires further investigation.
Finally, in this study, we show that Ng was associated with memory and executive scores over time only in patients with MCI. It is suggested that elevated CSF Ng levels are especially related to the pathologic process of cognitive impairment in the early stage of the disease. Our findings are consistent with a recent study showing that individuals with progressive MCI have elevated CSF Ng levels that are associated with accelerated deterioration in the ADAS-Cog subscale.9 In the NC subgroup, there were no significant associations between cognitive function and Ng. Ng may therefore serve as a useful marker in conjunction with Aβ42 and t-tau to distinguish between MCI and NC. Ng has also been reported to be a useful biomarker for differentiating NC from MCI.8
This study has limitations. First, as with any longitudinal study of the elderly, attrition rate is a concern. We conducted an association study at the 36-month follow-up when dropout rate is less of an issue than at the 96-month follow-up (tables e-1–e-12, http://links.lww.com/WNL/A210). We also performed an analysis in the participants who completed the 96-month follow-up. We found that Ng also predicts memory and executive function decline longitudinally then. Nevertheless, we are aware that high dropout rates can cause the remaining participants to constitute a biased sample. The data should be interpreted with caution. Second, we examined the association of Ng only with memory, executive function, ADAS-Cog 11 and 13, MMSE, and CDR Sum of Boxes scores. It would be interesting to examine a more complete cognitive profile, including visuospatial and language domains, to see the timing and degree of association between CSF Ng and additional measures. Third, the findings relating Ng to cognitive measures disappear when tau and Aβ42 are included in the statistical models. It would be important to replicate these findings with other statistical models. The interactions and common pathways between Ng and Aβ42 and t-tau warrant further exploration. Fourth, data are also needed to clarify the temporal course of CSF Ng across the spectrum of MCI and AD and whether the rate of CSF Ng change parallels declines in cognition. Finally, because the ADNI cohort is a selected convenience sample of volunteers, sample selection bias should be taken into consideration in the interpretation of the data.
Glossary
- Aβ42
amyloid 42
- AD
Alzheimer disease
- ADAS-Cog
Alzheimer’s Disease Assessment Schedule-Cognition
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- ADNI-EF
ADNI executive function
- ADNI-Mem
ADNI memory
- CDR
Clinical Dementia Rating
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- NC
normal cognition
- NG
neurogranin
- t-tau
total tau
Footnotes
CME Course: NPub.org/cmelist
Author contributions
Dr. Sun had full access to all of the data in the study, initiated the study concept, was involved in study design, interpreted data, and edited the manuscript. Data analysis and drafting of the manuscript: Drs. Headley and DeLeon-benedetti. Study design, statistical analysis, and interpretation of data: Dr. Dong. Data analysis: Dr. Camargo. Acquisition, analysis, or interpretation of data: Drs. Blennow and Zetterberg. Interpretation of data and critical revision of the manuscript to enhance intellectual content: Drs. Levin, Blennow, Zetterberg, Loewenstein, Rundek, and Wright.
Study funding
This study was supported by the Swedish Alzheimer Foundation, the Brain Foundation, Sweden, and the Torsten Söderberg Foundation. Data collection and sharing for this project were funded by the ADNI (NIH grant U01 AG024904) and Department of Defense ADNI (award W81XWH-12-2-0012). ADNI is funded by the National Institute on Aging, by the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc; Biogen; Bristol-Myers Squibb Co; CereSpir, Inc; Eisai Inc; Elan Pharmaceuticals, Inc; Eli Lilly and Co; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc; Fujirebio; GE Healthcare; IXICO Ltd; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co, Inc; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corp; Pfizer Inc; Piramal Imaging; Servier; Takeda Pharmaceutical Co; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the NIH (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Disclosure
A. Headley, A. DeLeon-benedetti, C. Dong, B. Levin, D. Loewenstein, C. Camargo, and T. Rundek report no disclosures relevant to the manuscript. H. Zetterberg is a cofounder of Brain Biomarker Solutions in Gothenburg AB, a GU Holding–based platform company at the University of Gothenburg. K. Blennow has served as a consultant for Eli Lilly, Novartis, and Roche Diagnostics; has served on Advisory Boards for Amgen and IBL International; has given lectures for Fujirebio Europe and Lundbeck; and is a cofounder of Brain Biomarker Solutions in Gothenburg AB, a GU Holding–based platform company at the University of Gothenburg, Sweden. C. Wright and X. Sun report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Tarawneh R, Holtzman DM. The clinical problem of symptomatic Alzheimer disease and mild cognitive impairment. Cold Spring Harb Perspect Med 2012;2:a006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mez J, Mukherjee S, Thornton T, et al. The executive prominent/memory prominent spectrum in Alzheimer's disease is highly heritable. Neurobiol Aging 2016;41:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010;6:131–144. [DOI] [PubMed] [Google Scholar]
- 4.Blennow K, Zetterberg H. The past and the future of Alzheimer's disease CSF biomarkers: a journey toward validated biochemical tests covering the whole spectrum of molecular events. Front Neurosci 2015;9:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kvartsberg H, Duits FH, Ingelsson M, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer's disease. Alzheimers Dement 2015;11:1180–1190. [DOI] [PubMed] [Google Scholar]
- 6.Sun X, Dong C, Levin B, et al. APOE epsilon4 carriers may undergo synaptic damage conferring risk of Alzheimer's disease. Alzheimers Dement 2016;12:1159–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorsell A, Bjerke M, Gobom J, et al. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer's disease. Brain Res 2010;1362:13–22. [DOI] [PubMed] [Google Scholar]
- 8.Kester MI, Teunissen CE, Crimmins DL, et al. Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol 2015;72:1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Portelius E, Zetterberg H, Skillback T, et al. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer's disease. Brain 2015;138:3373–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tarawneh R, D'Angelo G, Crimmins D, et al. Diagnostic and prognostic utility of the synaptic marker neurogranin in Alzheimer disease. JAMA Neurol 2016;73:561–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crane PK, Carle A, Gibbons LE, et al. Development and assessment of a composite score for memory in the Alzheimer's Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav 2012;6:502–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gibbons LE, Carle AC, Mackin RS, et al. A composite score for executive functioning, validated in Alzheimer's Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav 2012;6:517–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Vos A, Jacobs D, Struyfs H, et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer's disease. Alzheimers Dement 2015;11:1461–1469. [DOI] [PubMed] [Google Scholar]
- 14.Lambon Ralph MA, Patterson K, Graham N, Dawson K, Hodges JR. Homogeneity and heterogeneity in mild cognitive impairment and Alzheimer's disease: a cross-sectional and longitudinal study of 55 cases. Brain 2003;126:2350–2362. [DOI] [PubMed] [Google Scholar]
- 15.Knopman DS, Boeve BF, Petersen RC. Essentials of the proper diagnoses of mild cognitive impairment, dementia, and major subtypes of dementia. Mayo Clin Proc 2003;78:1290–1308. [DOI] [PubMed] [Google Scholar]
- 16.Scott SA, DeKosky ST, Scheff SW. Volumetric atrophy of the amygdala in Alzheimer's disease: quantitative serial reconstruction. Neurology 1991;41:351–356. [DOI] [PubMed] [Google Scholar]
- 17.Dickerson BC, Wolk DA; Alzheimer's Disease Neuroimaging Initiative. Dysexecutive versus amnesic phenotypes of very mild Alzheimer's disease are associated with distinct clinical, genetic and cortical thinning characteristics. J Neurol Neurosurg Psychiatry 2011;82:45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson JK, Vogt BA, Kim R, Cotman CW, Head E. Isolated executive impairment and associated frontal neuropathology. Dement Geriatr Cogn Disord 2004;17:360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyakawa T, Yared E, Pak JH, Huang FL, Huang KP, Crawley JN. Neurogranin null mutant mice display performance deficits on spatial learning tasks with anxiety related components. Hippocampus 2001;11:763–775. [DOI] [PubMed] [Google Scholar]
- 20.Kubota Y, Putkey JA, Waxham MN. Neurogranin controls the spatiotemporal pattern of postsynaptic Ca2+/CaM signaling. Biophys J 2007;93:3848–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhong L, Cherry T, Bies CE, Florence MA, Gerges NZ. Neurogranin enhances synaptic strength through its interaction with calmodulin. EMBO J 2009;28:3027–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davidsson P, Blennow K. Neurochemical dissection of synaptic pathology in Alzheimer's disease. Int Psychogeriatr 1998;10:11–23. [DOI] [PubMed] [Google Scholar]
- 23.Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K. Spine growth precedes synapse formation in the adult neocortex in vivo. Nat Neurosci 2006;9:1117–1124. [DOI] [PubMed] [Google Scholar]
- 24.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science 2002;298:789–791. [DOI] [PubMed] [Google Scholar]
- 25.Grober E, Hall CB, Lipton RB, Zonderman AB, Resnick SM, Kawas C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer's disease. J Int Neuropsychol Soc 2008;14:266–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pereira JB, Westman E, Hansson O; Alzheimer's Disease Neuroimaging Initiative. Association between cerebrospinal fluid and plasma neurodegeneration biomarkers with brain atrophy in Alzheimer's disease. Neurobiol Aging 2017;58:14–29. [DOI] [PubMed] [Google Scholar]
- 27.Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci 2010;13:812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Selkoe DJ. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res 2008;192:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tu S, Okamoto S, Lipton SA, Xu H. Oligomeric Abeta-induced synaptic dysfunction in Alzheimer's disease. Mol Neurodegener 2014;9:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spires-Jones TL, Hyman BT. The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron 2014;82:756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hyman BT. Amyloid-dependent and amyloid-independent stages of Alzheimer disease. Arch Neurol 2011;68:1062–1064. [DOI] [PubMed] [Google Scholar]