

Abstract
Abnormal Wnt activity is a major mechanism responsible for many diseases, including cancer. Previously, we reported that the anthelmintic drug Niclosamide (NIC) inhibits Wnt/β-catenin signaling and suppresses colon cancer cell growth. Although the pharmacokinetic properties of NIC are appropriate for use as an anthelmintic agent, its low solubility, low bioavailability and low systemic exposure limit its usefulness in treating systemic diseases. To overcome these limitations, we conjugated NIC to recombinant chimeric polypeptides (CPs), and the CP-NIC conjugate spontaneously self-assembled into sub-100 nm near-monodisperse nanoparticles. CP-NIC nanoparticles delivered intravenously act as a pro-drug of NIC to dramatically increase exposure of NIC compared to dosing with free NIC. CP-NIC improved anti-tumor activity compared to NIC in a xenograft model of human colon cancer. Because NIC has multiple biological activities, CP-NIC could be used for treatment of multiple diseases, including cancer, bacterial and viral infection, type II diabetes, NASH and NAFLD.
Graphical abstract
Attachment of the hydrophobic drug NIC (purple triangles) to CP (black chains) triggers self-assembly into cylindrical nanoparticles.
1. Introduction
Wnt ligand binding to Frizzled/LRP receptors recruits Dishevelled to prevent the APC/Axin/GSK3β-mediated phosphorylation, ubiquitination and destruction of β-catenin, allowing β-catenin to accumulate and enter the nucleus to regulate specific gene activity. The Wnt signalling pathway plays a key role in tissue development and homeostasis, but is also dysregulated in many diseases [1–3]. Specifically, in colorectal cancer (CRC), more than 80% of all sporadic and hereditary cancers exhibit hyperactivation of this pathway due to mutations in the adenomatous polyposis coli (APC) or β-catenin genes [1, 2]. Given the importance of Wnt signalling activity in promoting tumor formation and metastasis, therapies to target this pathway are medically needed. However, there is a lack of druggable Wnt signalling pathway drug targets downstream of APC and β-catenin, and because protein-protein interactions have traditionally been difficult to target with small drug-like molecules [1, 4], drug discovery targeting this pathway at the level of these proteins has been problematic.
Recently, we and others have begun to provide insights into mechanisms and chemical starting points to inhibit the Wnt pathway [5–19], although no drugs targeting this pathway have been approved to date [20]. Using a high-throughput drug screening approach, we found that Niclosamide (NIC), a drug approved by the FDA for human use as an anthelmintic, promotes Frizzled internalization [5]. We subsequently found that NIC down-regulates Dishevelled and β-catenin, and inhibits colon cancer cell growth in vitro and in vivo [5, 7]. These findings have since been confirmed by studies from other laboratories [21–23].
NIC is a multi-functional drug. It was used initially as an anthelmintic agent in livestock in the early 1960s before being approved by the FDA for use in humans in 1982 to treat tapeworm infections [24, 25]. NIC has been found to inhibit the proliferation of tumor cell lines from multiple tumor types, e.g. breast, lung, prostate, lung, ovary, blood and pancreas, in addition to colon cancer, over an IC50 range of 0.13 - 4 μM that overlaps with the IC50 of inhibition of Wnt/β-catenin signalling [26]. NIC also has anti-tumor activity in drug resistant cancers [7, 27, 28]. NIC has been reported to inhibit key oncogenic signalling pathways [26] in addition to Wnt [5, 7, 21, 22], including Notch [29], mTOR [30], NF-kB [31], and STAT-3 [32].
While the pharmacodynamic properties of NIC are appropriate for use in the gut lumen as an anthelmintic agent, its low solubility, low bioavailability and poor pharmacokinetic profile result in low plasma exposure when dosed orally [25, 33–36]. Efforts to improve its solubility have included the preparation of salt forms and of derivatives containing hydrophilic groups [25, 37, 38]. In the search for STAT-3 inhibitors with improved solubility to treat cancer, water-solubilizing amine groups were incorporated into NIC [39]. Recently, efforts to identify nanoparticle formulations of NIC for use in cancer therapy have been reported, but these formulations did not result in significant improvement of pharmacological properties [35, 40]. In one study in which the pharmacokinetic parameters of a nanocrystal formulation of NIC was evaluated in vivo, the nanocrystal formulation did not significantly change the plasma concentration vs. time profile when administered intravenously (i.v.) to rats, though an increased tissue concentration at 2 hours was noted [35].
Here, we have taken advantage of a new nanotechnology platform we have developed in which NIC is covalently conjugated to a genetically-encoded elastin-based chimeric polypeptide (CP)[41]. The CP consists of an elastin-like polypeptide (ELP), a disordered and highly water soluble recombinant peptide polymer[41], fused to a short (Cys-Gly-Gly)8 peptide segment that provides thiol reactive sites for chemical conjugation of chemotherapeutic drugs of interest. ELPs are biopolymers composed of a Val-Pro-Gly-Xaa-Gly pentapeptide repeat (where the “guest residue “Xaa” is any amino acid except Pro) derived from a structural motif found in mammalian elastin [41]. Attachment of multiple copies of a hydrophobic drug to the carboxyl terminus of the CP triggers self-assembly of the CP-drug conjugate into spherical nanoparticles. We showed previously that CP nanoparticles incorporating hydrophobic chemotherapeutics such as doxorubicin (DOX) and paclitaxel (PTX) show significantly better tumor regression compared to the free drug, and in the case of paclitaxel, lead to a better outcome in multiple mouse tumor models compared to Abraxane, a clinically approved nanoparticle formulation of paclitaxel [42–44].
While CP-drug nanoparticles have been used successfully to improve the pharmacokinetics and pharmacodynamics of drugs, the parent ELPs that CPs are derived from have presented a good safety profile in multiple clinical trials as fusions to peptide drugs such as glucagon-like peptide, vasoactive intestinal peptide-1 and insulin [41]. ELP drug carriers for treatment of diabetes and heart disease are successfully progressing toward clinical use, as clinical trials have demonstrated that ELPs are well tolerated in humans and do not induce a significant immune response in most individuals [41].
Here we show that conjugation of multiple copies of NIC to CP can trigger self-assembly of the conjugate into nanoparticles in aqueous solution. When injected intravenously, CP-NIC nanoparticles are better tolerated than free NIC, allowing use of a higher maximum tolerated dose in a form that leads to increased plasma concentration as a function of time and to longer duration of exposure, compared to free NIC. CP-NIC exhibits greater anti-cancer activity than NIC in a colon cancer xenograft model, with no observable adverse effects over two weeks of intravenous dosing. The ability of CP-NIC to increase the tissue exposure of NIC when dosed intravenously may provide a valuable preclinical research tool to study the effectiveness of NIC in preclinical models of cancer and other diseases known to be affected by NIC.
2. Experimental
Additional methods can be found in Supplementary Information.
2.1 Static and Dynamic Light Scattering
Dynamic light scattering (DLS) was used to measure the particle size at 25 °C and at 10 μM concentration (n=3) in PBS after filtration through an Anotop syringe filter with 0.22 μm size pores (Whatman; Florham Park, NJ) using a DynaPro Plate Reader (Wyatt Technology; Santa Barbara, CA). To obtain size histograms, regularization fits were used to determine the hydrodynamic radius (Rh) as weighted by the percent by mass. Static and dynamic light scattering (SLS/DLS) measurements were performed on an ALV/CGS-3 goniometer system (Langen, Germany). Samples for the goniometer were prepared in PBS and filtered through 0.22 μm Millex-GV filters into a 10 mm disposable borosilicate glass tube (Fisher). Simultaneous SLS and DLS measurements were obtained at 22°C for angles between 30°-150° at 5° increments, with measurements at each angle consisting of 3 runs for 15 seconds. The differential refractive index (dn/dc) was determined by measuring the refractive index at five different concentrations using an Abbemat 500 refractometer (Anton Paar, Graz, Austria). DLS data were analyzed by fitting the autocorrelation function to a biexponential decay using the HDRC software package (Germany). Rh was plotted against angle and extrapolated to zero. SLS data were analyzed by partial Zimm plots using ALV/Dynamic and Static FIT and PLOT software in order to determine the radius of gyration and molecular weight.
2.2 Cryogenic Transmission Electron Microscopy
Cryogenic transmission electron microscopy (cryo-TEM) was performed at Duke University’s Shared Materials Instrumentation Facility (Durham, NC). Lacey holey carbon grids (Ted Pella, Redding, CA) were glow discharged in a PELCO EasiGlow Cleaning System (Ted Pella, Redding, CA). A 3 μl drop of a sample was deposited onto the grid, blotted for 3 s with an offset of −3 mm, and vitrified in liquid ethane using the Vitrobot Mark III (FEI, Eindhoven, Netherlands). Prior to vitrification, the sample chamber was maintained at 22 °C and 100% relative humidity to prevent sample evaporation. Grids were transferred to a Gatan 626 cryoholder (Gatan, Pleasanton, CA) and imaged on a FEI Tecnai G2 Twin TEM (FEI, Eindhoven, Netherlands).
2.3 Atomic-force Microscopy (AFM)
Samples for AFM imaging were prepared by placing a drop of sample solution (~0.2 mg/ml) onto a freshly cleaved mica surfaces and incubating for 15 minutes. Then, the sample was gently rinsed with Milli-Q H2O and dried under a N2 stream. All AFM images were acquired with Tapping Mode under ambient conditions using a MultiMode AFM (Bruker). TappingMode silicon cantilever was used for all the AFM images (kF = 40 N/m, fres = 300 kHz).
2.4 Western blot
Western blots were performed following a procedure similar to that reported previously [7]. Briefly, HCT-116 cells were grown to about 80% confluency on poly-D-lysine coated six-well plates for 48 and then incubated with 2.5 μM NIC in DMSO, molar equivalent CP-NIC or DMSO control for 18 hours in growth medium. After treatment, the cytosolic fraction was isolated as described [5]. Immunoblot was used to detect the β-catenin, c-myc and cyclin D1 protein levels in cytosol, with β-actin immunoblots used for loading control.
2.5 Pharmacokinetic analysis of CP-NIC
CP-NIC was dissolved in PBS at a concentration of 40 mg/ml and injected i.v. in the tail vein of CD1 mice at a dose of 128 mg/kg of body weight. Blood samples were obtained at 30 min prior to the dosing and at 5, 10, 20 and 40 min, and 1.5, 4, 8, 12, and 24 h after drug administration. Quantification of free NIC in mouse plasma was done by LC/MS–MS using methods similar to those previously published [7].
2.6 In vitro cell proliferation
HCT116 human colon carcinoma cells were purchased from American Type Culture Collection (Manassas, VA, USA) and maintained in McCoy’s 5A medium supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals, Lawrenceville, GA, USA), 200 U/ml penicillin, and 50 ng/ml streptomycin (Invitrogen, Grand island, NY, USA). Cells were grown at 37oC in 5% CO2. The cells were plated at 5,000 cells per well into 96 well plates and treated with compounds (n=3) for 48 hours, at which point the cell proliferation was measured using the colorimetric MTS assay (Promega, Madison, WI, USA). Values were normalized as a percentage of DMSO treated cells. The dose response data were fit with sigmoidal dose-response using Graphpad Prism.
2.7 Dose Escalation and Tumor Inhibition
Prior to in vivo implantation, HCT116 cells were washed twice in Minimum Essential Media (MEM) (51200-038; Invitrogen; Carlsbad, CA). HCT116 cells were implanted in the right flank of male nude mice by subcutaneous injection of 1-2×106 cells in 50 μL. All animals were treated in accordance with National Institute of Health Guide for the Care and Use of Laboratory Animals under protocols approved by the Duke University Institutional Animal Care and Use Committee.
Male nude mice (6–8 weeks old) bearing subcutaneous HCT116 tumors were treated when the mice had a tumor volume of 75-100 mm3. Controls or drugs were administered by tail vein infusion (50 μL/min) of 500 μL. Dose escalation was performed with CP-NIC at 5, 10, 15, 20, and 25 mg/kg BW (BW: body weight). Mice were treated 3 days/week for 2 weeks with either 5 mg/kg BW unconjugated NIC, or 20 mg/kg BW CP-NIC, the maximum tolerated doses, respectively. Tumor dimensions and BW were measured 3-4 times a week, and the tumor volume was calculated according to Volume [mm3] = length × width × depth × 1/2.
Mice were monitored for BW loss, and euthanized upon exceeding 15% loss in BW or if their tumors grew to a volume greater than 1000 mm3. The maximum tolerated dose (MTD) was determined in mice with tumors. Cumulative survival curves were compared using Kaplan-Meier analysis, and the Sidak test, Tukey Test and Wilcoxon test were calculated using GraphPad Prism 6 software.
3. Results
3.1 Synthesis of CP-NIC conjugate
In our previous studies, we demonstrated that conjugation of multiple copies of doxorubicin or paclitaxel to CP can trigger self-assembly into nanoparticles, and that these drug-loaded nanoparticles show greatly improved anti-tumor efficacy compared to the parent drug [42]. Here we sought to explore the advantage of this nano-formulation technology to deliver a targeted therapeutic —NIC— to improve its pharmacodynamic properties (Figure 1). The amino acid sequence of CP is shown in Figure 1a. The CP was expressed from a plasmid-borne synthetic gene in E. coli and purified by inverse transition cycling (ITC), using temperature-dependent self-assembly phase shift [45–46]. Three rounds of ITC yielded 100 mg of purified monodisperse CP from 1 L of culture. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-MS) (Figure 2a, and Table 1) and SDS-PAGE analysis (Supplementary Figure 2) showed that the molecular weight of the CP is 62550 Da.
Fig 1.
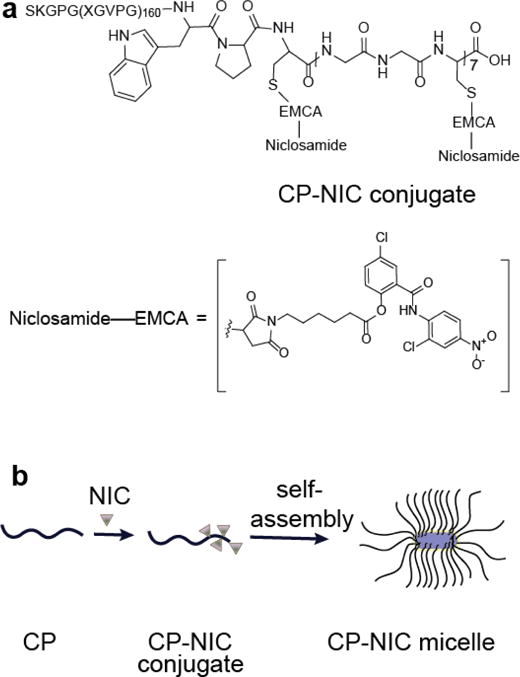
Structure of CP–NIC conjugate and schematic of the structure of CP-NIC nanoparticles. (a) The CP was synthesized by genetically encoded synthesis in E. coli, and conjugated to NIC at the multiple Cys residues at the C-terminal end of the CP. (b) Attachment of the hydrophobic drug NIC (triangles) triggers self-assembly of the CP into cylindrical nanoparticles with a drug-rich (purple) core surrounded by a hydrophilic polypeptide corona (black chains).
Fig 2.
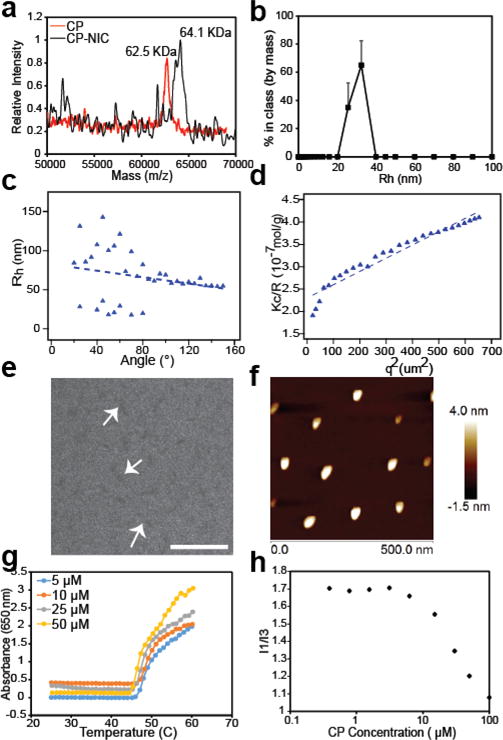
Characterization of CP–NIC nanoparticles. (a) MALDI-MS of CP and CP-NIC conjugate. (b) DLS measurement of CP-NIC conjugate (n=3). (c) Angular dependence of Rh of CP–NIC nanoparticles. (d) Partial Zimm plot of Kc/R vs q2 of CP-NIC conjugate. (e) Cryo-TEM image of CP-NIC nanoparticles. Scale 200 nm. (f) AFM images of CP-NIC conjugate. (g) Transition temperature (Tt), and (h) Critical aggregation concentration (CAC) of CP-NIC conjugate.
Table 1.
Physicochemical properties of the CP-NIC conjugate.
| CP-polypeptide sequence | SKGPG(XGVPG)160WPC(GGC)7 |
| Guest residues (X) | V:A:G [1:8:7] |
| Molecular weight of CP (KDa) | 62.5 |
| 1Drugs per CP | 4 |
| 2Rh (nm) | 30.1 ± 10.4 |
| 2Rg (nm) | 81.5 ± 5.8% |
| 3Z (chains per nanoparticle) | 90 |
| ρ | 1.65 |
| MW (g/mol) | 5.83 × 106 ± 3.9% |
| CMC (μM) | 3.1 |
Drug molecules calculated from MALDI MS.
Rh determined by DLS at 25 °C in PBS. Mean ± %PD (n=3).
Aggregation number (Z): Number of CP-NIC molecules per nanoparticle, as determined by SLS.
A terminal maleimide [42, 43] was added to NIC via a substituted hexanoic acid to enable conjugation of NIC to the polypeptide. Treatment of NIC with 6-Maleimidohexanoic acid and N,N’-dicyclohexylcarbodiimide(DCC) produced the 6-Maleimidohexanoic ester derivative of NIC (I), which was covalently attached to the Cys residues of the CP (Figure 1 a, b, Supplementary Figure 1). Purified CP-NIC conjugate (Supplementary Figure 2) has ~4 drug molecules per CP, as determined from the mass difference between the conjugate and the parent CP (Table 1) measured by MALDI-TOF MS (Figure 2a) [43]. The drug loading efficiency is 2 wt. % of the conjugate and is consistent with our previous CP-drug conjugates [42, 43]. In principle, up to 7 drug molecules can be conjugated per CP molecule. However, in our previous studies with CP-DOX and CP-PTX, where we conjugated DOX or PTX to the same CP, we found 4 and 2 drugs respectively per CP [42, 43], and the 2 wt. % loading suggests an average of 4 NIC molecules per CP. Because CP-NIC does not ionize well in MALDI, it is difficult to determine the poly-dispersity of CP-NIC from the peak width as the [M+H]+1 peak for the CP-NIC conjugate has relatively low intensity and the low signal-to noise precludes deconvolution of the peak into components that represent CP-NIC conjugates with distinct stoichiometry.
3.2 Characterization of CP-NIC conjugate
Upon conjugation with NIC, the CP-NIC conjugate spontaneously self-assembled into near-monodisperse cylindrical micelles (Figure 1b). As NIC is a hydrophobic drug with a logD of 4.48 at pH 7, these results are consistent with our previous observation that conjugation of multiple copies of a hydrophobic small molecule drug with a logD > 1.5 to one end of a hydrophilic polypeptide (CP) impart sufficient amphiphilicity to trigger the self-assembly of CP into nanoparticles [47]. The radius of gyration (Rg), and hydrodynamic radius (Rh) of CP-NIC conjugate were determined by static and dynamic light scattering (SLS/DLS). The Rh of CP-NIC conjuagte measured by fixed angle DLS using a DynaProTM Plate Reader (Wyatt Technology; Santa Barbara, CA) at 25 °C. To obtain histograms, regularization fits were used to determine the hydrodynamic radius (Rh) as weighted by the percent by mass and the Rh of CP-NIC conjugate was calculated as 30 nm (Figure 2b, Figure S3, Figure S7). However, the Rh calculated from the SLS/DLS measurement with ALV instrument was 49.3 nm (Table 1). This discrepancy is likely due to the fact that the SLS and DLS measurement on the ALV instrument are carried out simultaneously in the angular range of 30°-150° at 5° increments, and the Rh was calculated from the inflection point at 0°, whereas the DLS measurement in the Wyatt instrument is at a fixed angle of 145°. Analysis of the partial Zimm plot obtained from SLS showed that the radius of gyration (Rg) of the CP-NIC nanoparticles was 81.5 nm, and that the aggregation number of the nanoparticles was 90 (Figure 2 b, c and Table 1). The experimentally determined form factor (ρ)—calculated as Rg/Rh—was 1.65, which is close to the theoretical value for cylindrical particles with high aspect ratio [48].
The size and rod-like morphology of the CP-NIC nanoparticles were confirmed by cryo-TEM, which allows for the direct visualization of self-assembled structures in a near-native, hydrated state (Figure 2e, Figure S4). Only the hydrophobic core of CP-NIC nanoparticles is visualized by cryo-TEM, due to the low electron density and high degree of hydration of the ELP chains in the corona of the nanoparticles. However, gaining better contrast in cryo-TEM is experimentally close to impossible. This is because contrast in cryo-TEM is generated from differences in electron density between the sample and the vitreous ice layer, and unfortunately, the contrast for our polypeptide materials in cryo-TEM is fundamentally limited by two factors. First, because they are composed of relatively light atoms, polypeptides have lower electron density than many synthetic polymers, and thus exhibit low contrast. Second, the hydrophobic core of our assembled nanoparticles likely remain quite hydrated [50–51], further limiting the difference in electron density between the core and the solvent. This is in contrast to synthetic diblock copolymers where the core-forming domain is typically much less hydrated, and hence provides good contrast in cryo-TEM. The core’s low electron density and its likely high degree of hydration combine to limit the contrast achievable by cryo-TEM.
It is possible to obtain greater contrast, and thus more easily interpretable images, through negative staining and conventional TEM, but this comes at the cost of potential major changes to the sample size and morphology during the sample preparation process. Our judgment is that despite the low contrast images, cryo-TEM, which captures images of the micelles in their near-native state, combined with light scattering is the best approach to characterize polypeptide self-assembly. The average length of the cylindrical nanoparticle determined by cryo-TEM (LTEM) was measured as 74±10 nm (n = 10), and the average diameter (DTEM) was measured as 12.5±3.5 nm. The micellar morphologies were further verified by atomic force microscopy (AFM) under ambient condition (Figure 2f, Figure S5). The AFM images show distinct particles with a rod or worm-like morphology. The observed width of the worm-like micelle is much larger than their heights, which is likely attributed to the spreading of the micelles on the mica surface during sample preparation and because of the tip–induced broadening effect inherent to AFM.
CPs are thermally responsive and display lower critical solution temperature (LCST) phase transition behaviour, in which the protein goes from a soluble state to an insoluble coacervate phase upon raising the solution temperature above its cloud point, also called the inverse transition temperature (Tt). We measured the thermal responsiveness of the CP-NIC nanoparticles as a function of the CP concentration in mouse serum to model the physiological milieu that the CP-NIC nanoparticles would be in after i.v. injection (Figure 2g). In serum, the Tt of the CP-NIC nanoparticles was independent of the CP-NIC concentration in the range of 5-50 μM (45 °C at 25 μM), which is in sharp contrast to unconjugated CP, where Tt varied significantly with concentration (ranging from 48 °C for 50 μm to 65 °C for 5 μM) (Figure S6). This result is consistent with our previous studies of other drug conjugates (e.g., CP-Paclitaxel and CP-Doxorubicin [42, 43]) that form nanoparticles, and suggests that the high local polypeptide concentration in the CP-NIC nanoparticles make the Tt nearly independent of its overall solution concentration [47].
CP-NIC nanoparticles were further characterized by fluorescence spectroscopy using pyrene as a probe of local hydrophobicity, which enables measurement of the critical aggregation concentration (CAC) of the self-assembled nanoparticles [49]. The ratio of the first fluorescence emission peak (I370-373) to the third peak (I381-384) was plotted over a range of CP concentrations (Figure 2f). The sigmoid of best fit was used to calculate the CAC, defined as the inflection point of the curve, giving the CAC of the CP-NIC nanoparticles of ~3 μM (Figure 2f).
3.3 In vitro anti-cancer efficacy
Having demonstrated that we can package NIC in the core of the CP-NIC nanoparticles, we next turned our attention to verify that this formulation retains activity of the drug. Prior SAR studies indicated that the ester attachment did not affect NIC activity [19]. We chose the human colon carcinoma HCT116 cell line to evaluate the in vitro cytotoxicity of the CP-NIC conjugate, as NIC has been proposed for clinical use in human colon carcinoma [7, 21]. After 72 h exposure to CP-NIC nanoparticles, HCT116 cell proliferation was significantly inhibited (Figure 3a). The IC50, defined as the concentration of NIC (or NIC equivalent for the CP-NIC nanoparticles) needed to inhibit the proliferation of cells by 50%, was found to be 0.94 μM for CP-NIC and 0.85 μM for free NIC.
Fig 3.
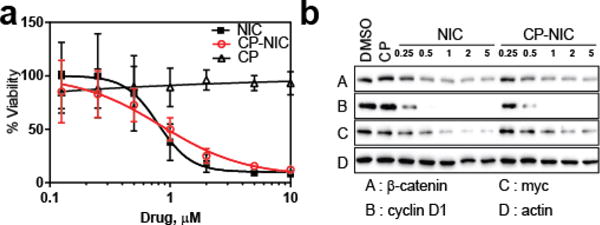
In vitro activity of CP–NIC nanoparticles. (a) Cell viability in the presence of the indicated doses of free NIC, CP–NIC (equivalent dose to free NIC), or unconjugated CP (equivalent to CP-NIC) in HCT-116 cells (n=3, mean ± 95%CI). (b) Inhibition of Wnt/β-catenin signalling measured as cytosolic β-catenin level, as well as Wnt-targets c-myc and cyclin D1, by the indicated doses of CP–NIC versus free NIC in HCT-116 cells.
We also determined the efficacy of CP-NIC to inhibit Wnt signalling in HCT116 cells. Wnt signalling was quantified as cytosolic β-catenin level by Western blot, an assay routinely used for Wnt signalling activity measurement [19]. Upon treatment of HCT116 cells with CP-NIC at doses ranging from 0.25 to 5 μM (NIC equivalent) for 18 h, β-catenin levels were significantly decreased in HCT116 cells (Fig 3b), similar to the inhibition observed for the same equivalent dose of free NIC [5]. Levels of the Wnt target proteins c-myc and cyclin D1 were also similarly reduced by treatment with NIC, as shown by western blotting. These data, together with data from the cell proliferation assay, clearly demonstrate that the CP-NIC nanoparticles inhibit the in vitro proliferation of HCT116 cells and the Wnt signalling pathway, and that conjugation of NIC to CP does not significantly decrease the activity of the drug.
3.4 Pharmacokinetic analysis of CP-NIC
To compare the plasma exposure to NIC from CP-NIC nanoparticles versus NIC, CP-NIC nanoparticles or NIC were administered intravenously and the plasma NIC concentration was measured as a function of time post-injection (Figure 4). We employed LCMS-MS analysis to determine the in vivo concentration of NIC as free drug liberated from CP-NIC nanoparticles. The mechanism of cleavage of NIC from the particle has not been defined, but is thought to involve protease cleavage and/or aqueous hydrolysis. The pharmacokinetic parameters were calculated using a non-compartment pharmacokinetic approach using the WinNonlin software, yielding a terminal half-life of NIC derived from the CP-NIC nanoparticles of 4.2 ± 1.34 h and a plasma AUC of 36.9 ± 7.34 μg/mL/h (Supplementary Table 1). In contrast, the terminal half-life and AUC for NIC in mice (treated at the same dose of unconjugated NIC) are only 1.0 ± 0.22 h and 3.3 ± 1.3 μg/mL/h respectively (Supplementary Table 1).
Fig 4.
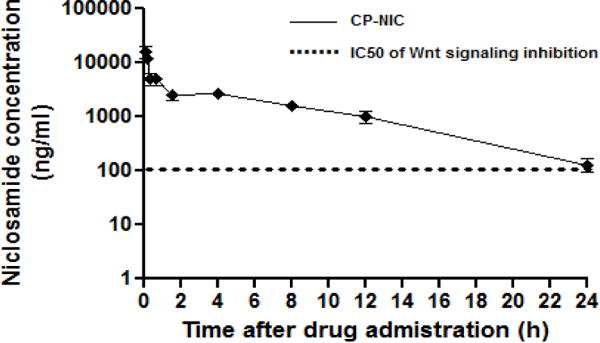
Plasma pharmacokinetics. CD1 mice were dosed with CP-NIC (i.v., 128mg/kg). Blood samples were obtained 0.5 h prior to dosing and at 10, 20 and 40 min, and, 1.5, 4, 8, 12, 24 hours after drug administration (n=4 per time point). Quantification of NIC in mouse plasma was done by LC/MS-MS and reported as ng/ml. A non-compartment model was fitted to the plasma NIC concentration, which yielded a terminal half-life of 4.2 h for CP–NIC (mean ± 95% CI, n=4). The dotted line denotes the IC50 of NIC inhibition of Wnt/β-catenin signaling in the Wnt-stimulated TOPFlash assay.
In fact, the plasma levels of NIC obtained by dosing CP-NIC at 128 mg CP-NIC Equiv/kg BW remained above the IC50 of inhibition of Wnt signalling by NIC in the TOPFlash assay for nearly 24 h (Figure 4), whereas the reported plasma levels of NIC dosed as a free drug solution at 200 mg/kg BW were only above the IC50 for Wnt inhibition for less than 1 h [7].
3.5 In vivo anti-tumor activity
To compare the therapeutic effect of CP–NIC nanoparticles versus free NIC, CP-NIC formulations were administered in a dose escalation study. The maximum deliverable dose (MDD) of CP–NIC due to solution viscosity was 20 mg NIC Equiv/kg BW (Figure S8). We believe that the maximum tolerated dose (MTD) of CP-NIC nanoparticles is greater than 20 mg/kg, but we were unable to increase the dose beyond this due to viscosity of the solution.
Next, we evaluated the tumor inhibition efficacy of the maximum deliverable/tolerated dose of CP-NIC versus free NIC in the HCT-116 cell xenograft model. Mice with HCT-116 tumors were treated every third day for two weeks intravenously with PBS, unconjugated NIC (5 mg/kg), or CP–NIC nanoparticles (20 mg/kg of NIC-equivalent) (Figure 5a). The 5 mg/kg dose of free NIC was chosen because in a pilot study with NIC formulated in a mixture of N-dimethylacetamide (DMA) and Polyethylene Glycol 400 (1:2 v/v), the LD50 of NIC in nude mice was found to be 5 mg/kg BW (unpublished data). Body-weight loss was also measured throughout the treatment of free NIC and CP-NIC conjugate. All treatments were tolerated for the period of the study (Figure S8). Ten days after the start of the treatment, CP–NIC treated mice had a mean tumor volume of 339 mm3 (n=8) versus 661 mm3 (n=8) for NIC-treated (Tukey; p=0.001), compared to 1111 mm3 (n=8) for PBS-treated controls (Tukey; p=0.0001). The CP–NIC formulation outperforms free drug in reducing growth in tumor volume, which correlated with extended animal survival (Figure 5b, Figure S9). The median survival time for mice treated with PBS (n = 8) was 13 days, and treatment with the free NIC (n= 8) slightly increased survival to 16 days (Kaplan–Meier, log-rank test, p<0.0001) (see Methods). Treatment with CP-NIC (n= 8) further increased survival to 26 days (Kaplan–Meier, log-rank test, p<0.0001). In this study the presumed mechanism of action of NIC released from nanoparticles is expected to be same as free NIC, which we have already characterized [7], so we did not further analyze samples. We did not treat mice with CP alone, because we have shown in a previous study that CP alone is indistinguishable from PBS vehicle [43]. These results demonstrate that treatment with CP-NIC nanoparticles improved the survival of mice bearing a subcutaneous HCT-116 cell tumor, compared to treatment with free NIC drug.
Fig 5.
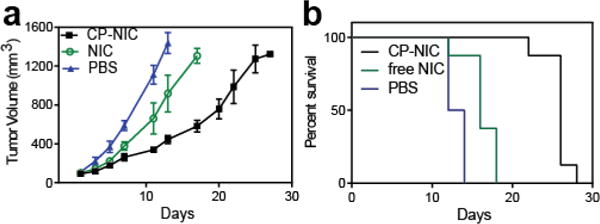
In vivo anti-tumor activity of CP–NIC nanoparticles. (a) and (b) tumor cells (HCT-116) were implanted in the right flank of male nude mice on day zero. When the tumor volume reached ~100 mm3, mice were treated intravenously every third day for two weeks with PBS (n=8), unconjugated NIC (5 mg/kg BW, n=8) or CP–NIC (20 mg NIC equiv/kg BW, n=8). (a) Tumor volume up to day 30 (mean ± 95% CI, n= 8). p > 0.0001 for CP–NIC, NIC or PBS treatment. At day 10, comparison was made among groups using the Tukey test. (b) Cumulative survival of mice (Kaplan–Meier).
4. Discussion
NIC, an FDA-approved anthelmintic drug, is also a potential cancer chemotherapeutic because it inhibits Wnt/β-catenin signalling, a signalling pathway that is implicated in tumor progression and metastasis in colon cancers [19].
Previously, we demonstrated that NIC exerted anti-proliferative effects in human colon cancer cell lines by inhibiting Wnt/β-catenin pathway activation, down-regulating Dvl2 and reducing downstream β-catenin signalling [5]. When administered orally in mice bearing a human colorectal cancer xenograft, NIC inhibited tumor growth [7]. Because NIC is poorly absorbed and metabolizes rapidly in vivo [7], we recently designed and synthesized a prodrug of NIC that metabolizes to NIC in vivo. Oral administration of this prodrug improved the plasma exposure and extended the duration of therapeutic exposure of NIC drug in mice [19]. In previous anti-tumor studies of NIC in vivo, NIC was formulated for oral administration in organic solvents such as 90% PEG300 and 10% 1-methyl-2-pyrrolidone that may generate additional solvent-related side effects [7]. Recently, NIC was encapsulated using the co-assembly of diblock copolymer (polystyrene-block-polyacrylic acid) in a micellar suspension of polyethylene glycol cetyl ether [52]. Treatment with these NIC-loaded nanocells showed anti-proliferative effects against MDA-MB-231 and MCF-7 cells in culture. However, the in vivo pharmacological properties and anti-tumor effect of this formulation have not been reported. In contrast to these studies, here we show that conjugation of multiple copies of NIC to a CP triggers self-assembly of the CP-NIC conjugate into cylindrical nanoparticles that are highly water soluble. These nanoparticles show comparable inhibition of β-catenin expression and growth of HCT-116 cells as free drug, and when administered intravenously, liberate NIC with better systemic NIC distribution and better anti-tumor in vivo efficacy compared to direct dosing with free NIC.
In an alternative approach, hydrophobic drugs can be physically encapsulated into polymeric micelles, such as poly(d,l-lactide-co-glycolide) (PLL-PLG), poly(ethylene glycol)–poly(γ-benzyl-l-glutamate) (PEG-PBLG) poly(ethylene glycol)–poly(d,l-lactide) (PEG-PLL), methoxy-poly(ethylene glycol)-co-poly(paclitaxel) (mPEG-PTX) and methoxy-poly(ethylene glycol)-co-poly trimethylene carbonate (paclitaxel) (mPEG-TMC-PTX) [53–56]. Drugs such as DOX, camptothecin and PTX respectively were encapsulated into these micelles ranging from ~1-12 % loading efficiency. Though our 2 wt. % loading of the CP-NIC conjugate is low, direct conjugation to the carrier enables precise control over the location of drug release in endosomes, as compared to physically encapsulated drugs, leading to excellent in vivo tumor regression efficacy of CP-drug conjugates [42, 43]. We also note that this issue of drug loading was recently addressed by Stenzel and coworkers, who demonstrated that a polymer micelle with low loading of cis-platinate showed better tumor cell uptake and greater tumor cytotoxicity than a similar micelle with high drug loading [57].
The CMC of the CP-NIC conjugate of ~3 μM is comparable to other polymer micelles, such as mPEG-polyPTX and mPEG-poly(TMC-PTX) that we recently reported, whose CMCs were 2 and 0.3 μM respectively [56]. Similarly, the CMC of a PEG-PLA copolymer was reported in the range of ~0.6-0.8 μM [55].
Beyond its efficacy, the CP-NIC nanoparticle formulation has several other useful characteristics compared to synthetic polymeric micelles and polymer conjugates that require complicated multistep procedures to synthesize. Because CPs are recombinant polypeptides, they can be synthesized in E. coli (or other expression systems) with high yield and purified readily using phase-shift coacervation, allowing complete control of their molecular weight and polydispersity. They are biodegradable and self-assemble in aqueous buffer into nearly monodisperse nanoparticles upon conjugation with NIC or other small hydrophobic drugs [58–59]. Attachment of hydrophobic drugs solely at the chain end ensures that the drug is sequestered within the nanoparticle core, unlike other nanoparticle drug carriers, such as dendrimers [60], metal nanoparticles [61] or carbon nanotubes [62] that expose the hydrophobic drugs at the nanoparticle-water interface. We believe that all these features make CP nanoparticles attractive for improving the delivery of the many hydrophobic chemotherapeutics that are clinically approved. Further, this nanoparticle method may also allow re-evaluation and eventual therapeutic use of drug candidates that have been discarded in drug development pipelines due to unfavourable physicochemical properties (such as very low water solubility) and poor bioavailability.
5. Conclusion
We report the development and preclinical testing of NIC-loaded polypeptide nanoparticles that were synthesized by conjugating NIC to recombinant chimeric polypeptides (CPs) to generate CP-NIC. The CP-NIC nanoparticles exhibit Wnt signalling inhibition similar to that of free NIC in colon cancer cells. Notably, CP-NIC nanoparticles can behave as a pro-drug to significantly increase the plasma exposure of NIC as compared to free NIC after intravenous administration, and enhanced the efficacy of the drug in reducing tumor growth of human colon cancer xenografts in mice. To our knowledge, this is the first report of NIC-loaded nanoparticles that increase plasma exposure to NIC, extend its duration of exposure and improve its in vivo efficacy, thereby overcoming critical barriers to the clinical translation of NIC to treat cancer [26]. Moreover, this approach also enables the study of NIC in vivo in other diseases for which NIC has demonstrated biological activity. Thus, the findings described here may provide a breakthrough to treat diseases ranging from cancer, viral infection, and metabolic diseases in which Wnt signalling is implicated.
Supplementary Material
Acknowledgments
The authors thank Isaac J. Weitzhandler for performing the cryo-TEM imaging and Lei Tang for performing the AFM imaging. This work was funded in part by 5R01 CA172570 (WC), BC123280 (WC), and Clinical Oncology Research Center Development Grant 5K12-CA100639-08 (RAM, MC). Wei Chen is a V foundation Scholar and an American Cancer Society Research Scholar. A.C. thanks the NIH for support of this work through grants NIH R01 EB-00188 and R01 EB-007205.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Barker N, Clevers H. Nat Rev Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 2.Coombs GS, Covey TM, Virshup DM. Current Drug Targets. 2008;9:513–531. doi: 10.2174/138945008784911796. [DOI] [PubMed] [Google Scholar]
- 3.Besancon R, Valsesia-Wittmann S, Puisieux A, de Fromentel CC, Maguer-Satta V. Curr Med Chem. 2009;16:394–416. doi: 10.2174/092986709787315531. [DOI] [PubMed] [Google Scholar]
- 4.Meireles LMC, Mustata G. Curr Top Med Chem. 2011;11:248–57. doi: 10.2174/156802611794072632. [DOI] [PubMed] [Google Scholar]
- 5.Chen MY, Wang JB, Lu JY, Bond MC, Ren XR, Lyerly HK, et al. Biochemistry. 2009;48:10267–10274. doi: 10.1021/bi9009677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mook RA, Jr, Chen M, Lu J, Barak LS, Lyerly HK, Chen W. Bioorg Med Chem Lett. 2013;23:2187–2191. doi: 10.1016/j.bmcl.2013.01.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osada T, Chen MY, Yang XY, Spasojevic I, Vandeusen JB, Hsu D D, et al. Cancer Res. 2011;71:4172–4182. doi: 10.1158/0008-5472.CAN-10-3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang SMA, Mishina YM, Liu SM, Cheung A, Stegmeier F, Michaud GA, et al. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 9.Chen BZ, Dodge ME, Tang W, Lu JM, Ma ZQ, Fan CW, et al. Nat Chem Biol. 2009;5:100–107. doi: 10.1038/nchembio.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thorne CA, Hanson AJ, Schneider J, Tahinci E, Orton D, Cselenyi CS, et al. Nat Chem Biol. 2010;6:829–836. doi: 10.1038/nchembio.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, et al. Proc Natl Acad Sci USA. 2004;101:12682–12687. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiselyov AS, Tkachenko SE, Balakin KV, Ivachtchenko AV. Expert Opin Ther Targets. 2007;11:1087–1101. doi: 10.1517/14728222.11.8.1087. [DOI] [PubMed] [Google Scholar]
- 13.Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, et al. Cancer Cell. 2004;5:91–102. doi: 10.1016/s1535-6108(03)00334-9. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Pan S, Hsieh MH, Ng N, Sun F, Wang T, et al. Proc Natl Acad Sci USA. 2013;110:20224–20229. doi: 10.1073/pnas.1314239110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johannes JW, Almeida L, Barlaam B, Boriack-Sjodin PA, Casella R, Croft RA, et al. ACS Med Chem Lett. 2015;6:254–259. doi: 10.1021/ml5003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.James RG, Davidson KC, Bosch KA, Biechele TL, Robin NC, Taylor RJ, et al. Plos One. 2012;7 doi: 10.1371/journal.pone.0050457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park S, Lee JH, Lee JS, Song GY, Oh S. Bulletin of the Korean Chemical Society. 2013;34:1286–1289. [Google Scholar]
- 18.Mallinger A, Crumpler S, Pichowicz M, Waalboer D, Stubbs M, Adeniji-Popoola O, et al. J Med Chem, and references within. 2015 [Google Scholar]
- 19.Mook RA, Jr, Wang J, Ren XR, Chen M, Spasojevic I, Barak LS, et al. Bioorg Med Chem Lett. 2015;23:5829–5838. doi: 10.1016/j.bmc.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sebio A, Kahn M, Lenz H-J. Expert Opin Ther Targets. 2014;18:611–615. doi: 10.1517/14728222.2014.906580. [DOI] [PubMed] [Google Scholar]
- 21.Sack U, Walther W, Scudiero D, Selby M, Kobelt D, Lemm M, et al. J Natl Cancer Inst. 2011;103:1018–1036. doi: 10.1093/jnci/djr190. [DOI] [PubMed] [Google Scholar]
- 22.Lu W, Lin C, Roberts MJ, Waud WR, Piazza GA, Li Y. Plos One. 2011;6:e29290. doi: 10.1371/journal.pone.0029290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arend RC, Londono-Joshi AI, Samant RS, Li Y, Conner M, Hidalgo B, et al. Gynecologic oncology. 2014;134:112–120. doi: 10.1016/j.ygyno.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 24.Pearson RD, Hewlett EL. Ann Intern Med. 1985;102:550–551. doi: 10.7326/0003-4819-102-4-550. [DOI] [PubMed] [Google Scholar]
- 25.Andrews P, Thyssen J, Lorke D. Pharmac Ther. 1983;19:245–295. doi: 10.1016/0163-7258(82)90064-x. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Li PK, Roberts MJ, Arend RC, Samant RS, Buchsbaum DJ. Cancer Lett. 2014;349:8–14. doi: 10.1016/j.canlet.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, et al. Clin Cancer Res. 2014;20:3198–210. doi: 10.1158/1078-0432.CCR-13-3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li R, Hu Z, Sun SY, Chen ZG, Owonikoko TK, Sica GL, et al. Molecular cancer therapeutics. 2013;12:2200–2212. doi: 10.1158/1535-7163.MCT-13-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang AM, Ku HH, Liang YC, Chen YC, Hwu YM, Yeh TS. J Cell Biochem. 2009;106:682–92. doi: 10.1002/jcb.22065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fonseca BD, Diering GH, Bidinosti MA, Dalal K, Alain T, Balgi AD, et al. J Biol Chem. 2012;287:17530–17545. doi: 10.1074/jbc.M112.359638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin Y, Lu Z, Ding K, Li J, Du X, Chen C, et al. Cancer Res. 2010;70:2516–2527. doi: 10.1158/0008-5472.CAN-09-3950. [DOI] [PubMed] [Google Scholar]
- 32.Ren X, Duan I, He Q, Zhang Z, Wu D, Pan J, Pei D, Ding K. ACS Med Chem Lett. 2010;1:434–459. doi: 10.1021/ml100146z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang YW, Yeh TK, Lin KT, Chen WC, Yao HT, Lan SJ, et al. Yaowu Shipin Fenxi. 2006;14:329–333. [Google Scholar]
- 34.World_Health_Organization. Niclsoamide WHO Specifications and Evaluations for Public Health Pesticides. Geneva: WHO; 2002. http://www.who.int/whopes/quality/en/Niclosamide.pdf. [Google Scholar]
- 35.Ye Y, Zhang X, Zhang T, Wang H, Wu B. Drug Dev Ind Pharm. 2014:1–9. doi: 10.3109/03639045.2014.954585. [DOI] [PubMed] [Google Scholar]
- 36.Kasim NA, Whitehouse M, Ramachandran C, Bermejo M, Lennernäs H, Hussain AS, et al. Mol Pharm. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 37.Ding K, Pei D, Zhou J. Application: CN. CN: Guangzhou Institute of Biomedicine and Health, Chinese Academy of Sciences, Peop Rep China; 2010. p. 8. [Google Scholar]
- 38.Grifasi F, Chierotti MR, Gaglioti K, Gobetto R, Maini L, Braga D, et al. Crystal Growth & Design. 2015;15:1939–48. [Google Scholar]
- 39.Chen H, Yang Z, Ding C, Chu L, Zhang Y, Terry K, et al. ACS Med Chem Lett. 2013;4:180–185. doi: 10.1021/ml3003082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhushan B, Dubey P, Kumar SU, Sachdev A, Matai I, Gopinath P. RSC Advances. 2015;5:12078–12086. doi: 10.1016/j.cis.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 41.MacEwan SR, Chilkoti A. J Control Release. 2014;190:314–330. doi: 10.1016/j.jconrel.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Nat Mater. 2009;8:993–999. doi: 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bhattacharyya J, Bellucci JJ, Weitzhandler I, McDaniel JR, Spasojevic I, Li X, et al. Nat Commun. 2015;6:7939. doi: 10.1038/ncomms8939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bhattacharyya J, Weitzhandler I, Ho SB, McDaniel JR, Li X, Tang L, Liu J, Dewhirst M, Chilkoti A. Adv Funct Mater. 2017 doi: 10.1002/adfm.201605421. early view. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meyer DE, Chilkoti A. Nature Biotechnol. 1999;17:1112–1115. doi: 10.1038/15100. [DOI] [PubMed] [Google Scholar]
- 46.Bellucci JJ, Amiram M, Bhattacharyya J, McCafferty D, Chilkoti A. Angew Chem Int Ed (English) 2013;52:3703–3708. doi: 10.1002/anie.201208292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McDaniel JR, Bhattacharyya J, Vargo KB, Hassouneh W, Hammer DA, Chilkoti A. Angew Chem Int Ed (English) 2013;52:1683–87. doi: 10.1002/anie.201200899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Massey J, Power KN, Manners I, Winnik MA. J Am Chem Soc. 1998;120:9533–9540. [Google Scholar]
- 49.Kalyanasundaram KT. J Am Chem Soc. 1977:5. [Google Scholar]
- 50.McDaniel JR, Weitzhandler I, Prevost S, Vargo KB, Appavou MS, Hammer DA DA, et al. Nano Lett. 2014;14:6590–6598. doi: 10.1021/nl503221p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garanger E, MacEwan SR, Sandre O, Brulet A, Bataille L, Chilkoti A, Lecommandoux S. Macromolecules. 2015;48:6617–6627. [Google Scholar]
- 52.Misra SK, Jensen TW, Pan D. Nanoscale. 2015;7:7127–7132. doi: 10.1039/c5nr00403a. [DOI] [PubMed] [Google Scholar]
- 53.Ma Y, Boese SE, Luo Z, Nitin N, Gill HS. Biomed Microdevices. 2015;17:44. doi: 10.1007/s10544-015-9944-y. [DOI] [PubMed] [Google Scholar]
- 54.Thambi T, Yoon HY, Kim K, Kwon IC, Yoo CK, Park JH. Bioconjug Chem. 2011;22:1924–1931. doi: 10.1021/bc2000963. [DOI] [PubMed] [Google Scholar]
- 55.Alibolandi M, Ramezani M, Abnous K, Sadeghi F, Hadizadeh F. J Nanopart Res. 2015;17:1–16. [Google Scholar]
- 56.Liu J J, Pang Y, Bhattacharyya J, Liu W, Weitzhandler I, Li X, Chilkoti A. Adv Healthc Mater. 2016;5:1868–1873. doi: 10.1002/adhm.201600230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Callari M, De Souza PL, Rawal A, Stenzel MH. Angew Chem Int Ed. 2017;56:8441–8445. doi: 10.1002/anie.201701471. [DOI] [PubMed] [Google Scholar]
- 58.Liu W, Dreher MR, Furgeson DY, Peixoto KV, Yuan H, Zalutsky MR, et al. J Control Release. 2006;116:170–178. doi: 10.1016/j.jconrel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 59.Shamji MF, Betre H, Kraus VB, ChenJ J, Chilkoti A A, Pichika R, et al. Arthritis and rheumatism. 2007;56:3650–3661. doi: 10.1002/art.22952. [DOI] [PubMed] [Google Scholar]
- 60.Lee CC, MacKay JA, Frechet JM, Szoka FC. Nature Biotechnol. 2005;23:1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- 61.Huang X, Jain PK, El-Sayed IH, El-Sayed MA. Nanomedicine (London, England) 2007;2:681–693. doi: 10.2217/17435889.2.5.681. [DOI] [PubMed] [Google Scholar]
- 62.Martin CR, Kohli P. Nat Rev Drug Discov. 2003;2:29–37. doi: 10.1038/nrd988. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.