

 Ternary transition state analogue (TSA) complexes probing the isomerization of β-d-glucose 1-phosphate (G1P) into d-glucose 6-phosphate (G6P) catalyzed by catalytically active, fluorinated (5-fluorotryptophan), β-phosphoglucomutase (βPGM) have been observed directly by 19F NMR spectroscopy.
Ternary transition state analogue (TSA) complexes probing the isomerization of β-d-glucose 1-phosphate (G1P) into d-glucose 6-phosphate (G6P) catalyzed by catalytically active, fluorinated (5-fluorotryptophan), β-phosphoglucomutase (βPGM) have been observed directly by 19F NMR spectroscopy.
Abstract
Ternary transition state analogue (TSA) complexes probing the isomerization of β-d-glucose 1-phosphate (G1P) into d-glucose 6-phosphate (G6P) catalyzed by catalytically active, fluorinated (5-fluorotryptophan), β-phosphoglucomutase (βPGM) have been observed directly by 19F NMR spectroscopy. In these complexes MgF3– and AlF4– are surrogates for the transferring phosphate. However, the relevance of these metal fluorides as TSA complexes has been queried. The 1D 19F spectrum of a ternary TSA complex presented a molar equivalence between fluorinated enzyme, metal fluoride and non-isomerizable fluoromethylenephosphonate substrate analogue. Ring flips of the 5-fluoroindole ring remote from the active site were observed by both 19F NMR and X-ray crystallography, but did not perturb function. This data unequivocally demonstrates that the concentration of the metal fluoride complexes is equivalent to the concentration of enzyme and ligand in the TSA complex in aqueous solution.
Introduction
Phosphates are essential for cellular energy, metabolism, structure, and function, thus, enzymes that are able to transfer phosphate are crucial for cell survival.1,2 The haloacid dehalogenase (HAD) superfamily contain enzymes that catalyze phosphotransferase reactions with the use of Mg2+ as a cofactor and a conserved aspartic acid residue acting as a nucleophile.3 A member of this superfamily, β-phosphoglucomutase (βPGM), is able to concisely perform two different phosphotransfer reactions in the same active site while accelerating rates of conversion by a factor of 1021. βPGM catalyzes the conversion of β-glucose-1-phosphate (βG1P) into glucose-6-phosphate (G6P) via a ping-pong mechanism (Fig. 1A).4,5
Fig. 1. (A) Enzymatic mechanism of βPGM converting βG1P to G6P via a βG16BP intermediate. Conversion occurs through a bi bi ping-pong mechanism allowing the intermediate to reorient itself in the enzyme active site. (B) Inhibition of step 1 and 2 of the enzymatic reaction by the formation of previously reported TSA complexes.7,8 .

Studies on the enzyme structure of βPGM reveal that the protein is monomeric, containing a helical cap domain and an α/β core domain with its active site at the interface.4 Upon binding of βG1P, the structure of βPGM changes from its “cap-open” conformation to its “cap-closed” conformation by the rigid rotation of its cap domain.6 The phosphorylated aspartic acid residue (Asp8) transfers its phosphate to the 6-OH of βG1P producing a β-glucose-1,6-bisphosphate (βG16BP) intermediate causing the enzyme to convert to its “cap-open” conformation. βG16BP dissociates from the enzyme, repositioning itself to allow for dephosphorylation of the phosphate at C-1, before it rebinds to the enzyme prompting it to convert back into its “cap-closed” conformation. Recent attempts to further understand the two-step mechanism of βPGM have been made by using metal fluorides to form transition state analogue (TSA) complexes.7–9 Studies have shown that MgF3– and AlF4– are able to closely mimic the charge and geometry of the transferred phosphate such that they can form long-lived complexes with various substrate analogues.9,10 G6P complexes were successfully isolated and analyzed (Fig. 1B) as a representation of transition state 2 (TS2) of the enzymatic reaction, however, complexes with βG1P were inaccessible as a result of its turnover.8 To solve this problem, methylenephosphonate and α-fluoromethylenephosphonate analogues of βG1P were synthesized to enable formation of the TSA complexes (Fig. 1B), mimicking step 1 (TS1) of the enzymatic reaction.7 Crystallographic evidence of TSA complexes revealed that TSA complexes for step 1 involve direct contact of amino acid residues with the substrate, while TSA step 2 complexes involve two conserved water molecules bridging the substrate and amino acid residues.7 Although the ability of βPGM to readily form stable metal fluoride complexes provides a 19F spectroscopic probe to detect stable TSA complex formation, it lacks a direct enzymatic signal and questions whether metal fluoride TSA complexes are representative of the enzymatic reaction coordinate. Incorporating a spectroscopic probe into the enzyme would test this hypothesis. Further, it would offer the opportunity to screen and analyze inhibitors without the use of metal fluorides. The use of fluorine as a spectroscopic probe has monitored conformational changes and protein-ligand binding.11–23 Herein, we present the use of fluorine as a spectroscopic probe on βPGM to provide insight into ternary TSA complexes of phospho-transfer enzymes, and demonstrate that the concentration of the fluorinated enzyme directly correlates to the concentration of the metal fluoride complexes in aqueous solution.
Results and discussion
Expression of 19F-labeled βPGM
Incorporation of fluorine into the βPGM enzyme structure was accomplished by use of 5-fluorotryptophan (5FW). The βPGM structure contains two tryptophan residues, W24 and W216. Analysis of a previously recorded crystal structure of βPGM with a TSA complex (PDB ID code 4C4R)7 reveals that W24 is on the enzyme cap domain and W216 is on the enzyme core domain. W24 is within ∼3 Å of the active site, allowing the indole nitrogen to form a hydrogen bond with the substrate during catalysis, while W216 is on the outer surface of the core domain, ∼22 Å from the active site, and is not in direct contact with the substrate (Fig. S1†). Since the two tryptophan residues are located on different domains, we anticipated that these differences in environment would result in distinct 19F resonances by 19F NMR spectroscopy.
19F-labelling was incorporated into the wild-type βPGM as well as a W24F and W216F βPGM mutants. To substitute the natural tryptophan residues for 19F-tryptophan, two different methods were explored. The first protocol was developed by Crowley and coworkers24 who found that upon addition of 5-fluoroindole to cell media, E. coli BL21 cells were able to incorporate the fluorinated indole into tryptophan biosynthesis (method A). The second protocol involved the use of glyphosate to induce aromatic amino acid auxotrophy, followed by the addition of 5-fluorotryptophan, unlabeled phenylalanine, and unlabeled tyrosine (method B).24 Both methods did not show any major deleterious effect on protein overexpression or purification, however, the yields observed for the production of 5-fluorotryptophan βPGM (5FWβPGM) were ∼47% for method 1 and ∼85% for method B compared to the wild-type βPGM. Incorporation of 19F-labeled amino acids are known to inhibit bacterial growth to different degrees.21
Efficacy of 19F-labeling methods
To analyze the efficacy of both labelling methods, 1H-15N HSQC and LC-MS/MS were performed. Upon 15N-labeling of 5FWβPGM produced by method A, we expected the 1H-15N HSQC of the labeled protein to show cross peaks for all nitrogen resonances except the two 5-fluoroindole tryptophan side chains. Therefore, comparing the integrations of the cross peaks from the tryptophan backbone and tryptophan side chain resonances would provide a quantitative analysis of 5-fluoroindole incorporation (method A). 15N-labeling of 5FWβPGM produced by method B would result in an unlabeled side chain and backbone nitrogen for 5FW residues, which would be absent from the 1H-15N HSQC. Comparing the integration of the tryptophan backbone cross peak to a backbone residue close in proximity in the enzyme structure would, therefore provide a quantitative analysis of 5-fluorotryptophan incorporation. An HSQC spectra overlay of the wild-type 15N-labeled βPGM and the 15N-labeled βPGM from method A and B (Fig. S8†) revealed that the incorporation of both 19F-labeled tryptophan residues did not cause any structural perturbations in the enzyme tertiary structure, while suggesting high efficacy labelling with both methods. The HSQC spectra for both methods A and B showed an absence of the distinct Trp side chain cross peaks as seen in the 15N-labeled βPGM spectra which we interpreted as a high percent incorporation of 19F-labeled tryptophan (Fig. S9–11†). To more accurately quantify the effects of labeling, LC-MS/MS analysis of the peptides showed corresponding masses for fragments containing tryptophan (W24 and W216) and corresponding masses for fragments containing fluorotryptophan (5FW24 and 5FW216). To generate peptides, endoproteinase Lys-C was used to digest βPGM. A ratio between the total ion count of fragments containing 5FW24 : W24 and 5FW216 : W216 provided a quantitative result of each method. Method A showed ∼70% incorporation of 5-fluoroindole and method B showed ∼85% incorporation of 5-fluorotryptophan. We, therefore, chose to use method B to produce 5FWβPGM since both the levels of incorporation and quantity of protein production were highest.
19F NMR analysis of 5FWβPGM and assignment of resonances
The three protein constructs, W24F, W216F and wild-type βPGM were induced using method B and the recombinant purified 5FW proteins isolated. For the wild-type 5FW24 5FW216 βPGM there were three major resonances present in the 1D 19F NMR spectrum at –120, –123.5, and –125 ppm (Fig. 2A). The resonance seen at –123.5 ppm was noticeably broad showing the presence of a second species. The 19F NMR spectrum for the 5FW216 W24F mutant showed two broad resonances at –123.5 ppm and –125 ppm (Fig. 2B). The occurrence of two resonances corresponding to one 5FW residue could be attributable to a 5-fluoroindole ring flip, or a core domain conformation not observed crystallographically (vide infra).25,26 For the 5FW24 W216F 19F NMR spectrum, a single resonance observed at –123.7 ppm that was readily assigned to the 5FW abutting the active site (Fig. 3A) and, therefore, be most likely to report upon the formation of TSA complexes. All three of the 5FW protein constructs were subjected to conditions to form TSA complexes for both TS1 and TS2 and with MgF3– or AlF4– (Fig. S12 and S13†). All of the 5FW protein constructs formed TSA complexes as observed by the characteristic three signals in MgF3– complexes, or the characteristic four signals in the AlF4– complexes in the 19F spectra. The chemical shifts for the metal fluoride complexes were consistent with previous literature reports for TSA1 and TSA2. Fig. 2C shows the formation of the W24F 5FW216 βPGM–MgF3––G6P TSA2 complex. This is the fluorinated βPGM where the 5FW is remote from the active site. That the two resonances present from the 5FW216 did not change intensity or chemical shift appreciably is suggestive that residue W216, being remote from the active site (∼22 Å), does not function effectively as a probe of TSA formation. It also indicates that the core domain where W216 is located, does not undergo a conformational change, observable by 19F NMR of the 5FW216, upon formation of a TSA complex. The formation of the W216F 5FW24-MgF3––G6P TSA2 complex is shown in Fig. 3B. In this instance, the singular peak at –123.7 ppm corresponding to the 5FW24 resonance adjacent to the active site had disappeared, and a new peak at –121.7 ppm had appeared, a change of 2 ppm. The intensity of the new peak at –121.7 was comparable to the intensity of the three peaks corresponding to the MgF3– resonances between –145 and –160 ppm. The appearance of the new peak at –121.7 ppm was indicative that all of the protein in aqueous solution was involved in the formation of the TSA2 complex with G6P. Next we looked at utilizing the 19F probe to monitor the formation of the TSA1 complex. It is not possible to form TSA1 complexes using the substrate βG1P since it is readily turned over by the enzyme to product. Our initial attempts to form MgF3– complexes using the non-isomerizable and non-hydrolysable analogues of βG1P namely, βG1CP and βG1CFSP,7 were confounded by the significantly less intense peaks arising from the MgF3– (Fig. S12 and S13†) in comparison to the intensity of the corresponding peaks in the W216F 5FW24-MgF3––G6P and other complexes. These results are consistent with previously reported data stating the weaker binding affinity of βG1CP versus G6P.7 Upon addition of AlCl3 to the solutions, however, distinct peaks of comparable intensity and consistent chemical shift to TSA1 complexes formed previously7 were observed, that were indicative of TSA1 formation (Fig. 3C and S12 and S13†). The W216F 5FW24-AlF4–-βG1CFSP TSA1 complex is particularly insightful since the substrate analogue also possesses a fluorine atom. Thus, in this spectrum, there are fluorine resonances from the singularly fluorinated protein 5FW24, from the AlF4–, that is an isoelectronic representative of the transferring phosphate, and from the fluorinated non-isomerizable and non-hydrolysable analogue. The peak at –215 ppm arises from the bound ligand, the four peaks at –160, –141, –138 and –131 arise from the AlF4–, and the signal at –122 from the 5FW24 in the TSA1 complex. All of these peaks had comparable integration, accepting that there are differing rates of relaxation for each type of fluorine. This unequivocally demonstrates that the AlF4– species is in a molar equivalence with the 5FW24 βPGM in aqueous solution. This corroborates the structure observed by X-ray crystallographic data,7,8 and dispels any notion that metal fluoride complexes are not representative of phosphoryl transfer and are merely bespoke artifacts.
Fig. 2. 19F NMR spectra of 19F- labelled wild-type and W24F mutant βPGM. Diamonds (♦) are 5FW216 resonances and circles (•) are free MgF3– resonances (–155 ppm). Resonances at –119 are from free F–. Assignment of MgF3– and AlF4– resonances were previously determined by chemical shift.21,33 Assignment of MgF3– and AlF4– resonances were previously determined by chemical shift.21,33 (A) 5FWβPGM; (B) W24F, 5FW216 5FWβPGM; (C) W24F, 5FW216 5FWβPGM–MgF3–G6P TSA complex.

Fig. 3. 19F NMR spectra of the 5FWβPGM W216F mutant and its TSA1 and TSA2 complexes. Asterisks (*) are TSA 5FW24 resonances, squares (■) are apo 5FW24 resonances, circles (•) are free MgF3–, inverse triangles () are complexed G1CsFP, triangles (▲) are free G1CsFP. (A) W216F 5FWβPGM; (B) W216F 5FWβPGM–MgF3–G6P TSA complex; (C) W216F 5FWβPGM–AlF4-G1CsFP TSA complex.

Kinetic analysis of 5FWβPGM
Kinetic studies were performed for both wild-type and 19F-labeled enzyme (Table 1) to confirm kinetic competence of the fluorinated enzyme. α-Fructose-1,6-bisphosphate was used as an activator27 and the Michaelis–Menten equation was fitted to the steady-state velocity data to generate Km and kcat values. The Km of βG1P was 10.1 μM, which was comparable to previously recorded results (14.7 μM).28 This suggests that the incorporation of two fluorine atoms into the enzyme structure, one adjacent to the active site and one distal to the active site, did not have an effect on the binding affinity of the native substrate. The kcat value of the wild-type and 19F-labeled enzyme was slightly lower than reported (17 s–1)24 which could be a result of the incorporation of a His6-tag at the C-terminus of the enzyme sequence.
Table 1. Kinetic parameters for wild-type and 5FWβPGM with native substrate and competitive inhibitors.
| Wild-type βPGM | 5FWβPGM | |
| K m | 9.0 ± 0.7 μM | 10.1 ± 2.1 μM |
| k cat | 7.7 ± 0.1 s–1 | 3.8 ± 0.1 s–1 |
| IC50 (βG1CP) | 18 ± 3 μM | 13 ± 5 μM |
| IC50 (βG1CFsP) | 15 ± 2 μM | 11 ± 2 μM |
| K i(comp) (βG1CP) | 4.67 ± 0.04 μM | |
| K i(comp) (βG1CFsP) | 4.03 ± 0.03 μM |
Inhibition studies of phosphonates on 5FWβPGM
Although the formation of βG1CP and βG1CFSP TSA complexes have been extensively studied,7–9 the kinetic analysis of phosphonates as non-covalent inhibitors for βPGM has not been reported. Inhibition studies were performed without NH4F, as fluorine inhibition has already been reported in the low millimolar range.8 IC50 measurements of both ligands were performed with wild-type and 5FWβPGM, however, there was no significant discrepancy in values between the two enzymes (Table 1). For 5FWβPGM, βG1CP and βG1CFSP both show IC50 values in the micromolar range (13 ± 5 μM and 11 ± 2 μM respectively) (Fig. S2†). Time-dependent inhibition was not observed with either inhibitor. The mode of inhibition of these inhibitors was determined from double-reciprocal plots (Fig. 4) that showed both analogues as competitive inhibitors with Ki values of 4.67 ± 0.04 μM for βG1CP and 4.03 ± 0.03 μM for βG1CFSP. These values suggest that phosphonate compounds bind 5–6 times less strongly than the enzyme intermediate, βG16BP (Km = 0.72 ± 0.04 μM),28 whereas they show a slightly stronger binding affinity than the native substrate, βG1P.28 Nonetheless, these data are measured without fluorine in the solution and so it is not representative of the inhibition, or formation constant, of the MgF3– or AlF4– complexes with these compounds.
Fig. 4. Double-reciprocal plots for the inhibition of 50 nM βPGM with varying concentrations of βG1CP (A) and βG1CFSP (B), along with 2 mM MgCl2, 0.5 mM NADP+, 5 U mL–1 G6PDH, 150 μM fructose-1,6-bisphosphate, and βG1P in 50 mM HEPES pH 7.2. ▲ 0 μM, × 2 μM, ◆ 5 μM, ■ 10 μM, 20 μM.

X-ray structural analysis of 5FWβPGM and evidence of 5-fluoroindole ring flip
In order to better determine the origin of the observed resonances, and confirm that the labelled amino acids did not perturb protein function, the structures of 5FWβPGM apo-protein and the TSA complex with MgF3– and G6P were determined. Initially, crystals of the 5FWβPGM–MgF3––G6P complex were obtained from already established conditions.29 Clear density was visible for both 5FW24 and 216 (Fig. 5) showing that the labelled amino acid was incorporated and was in a similar conformation to that observed for the native protein. As the fluorine resonances indicated possible multiple conformations, the structures of the apo-protein and a new crystal form of the 5FWβPGM–MgF3––G6P complex, where crystal contacts are different in the region of W216, were also determined. In the latter, the two molecules in the asymmetric unit show two alternative orientations for 5FW216 (Fig. S19†). The orientations appear to be equally populated and it is likely that in solution both orientations are possible. This accounts for the two resonances observed for 5FW216 (Fig. 2B). In the apo-protein structure, 2 conformations were also observed for 5FW24 (Fig. S20†) but with the conformation not previously observed at very low occupancy (<10%), this could account for the slight line broadening observed for the apo-protein resonance (Fig. 3A). In all closed conformation structures 5FW24 is in the same conformation as the native protein (Fig. 6), confirming that the labelled protein can act as a reporter of the state of the enzyme without interfering with activity.
Fig. 5. Density for 5FW216 after molecular replacement before the inclusion of the modified amino acid in the model (2MFo-Fc blue mesh contoured at 1σ, Fo-Fc green mesh contoured at 3σ). Clear positive density is observed for the fluorine atom, confirming the presence of the label.
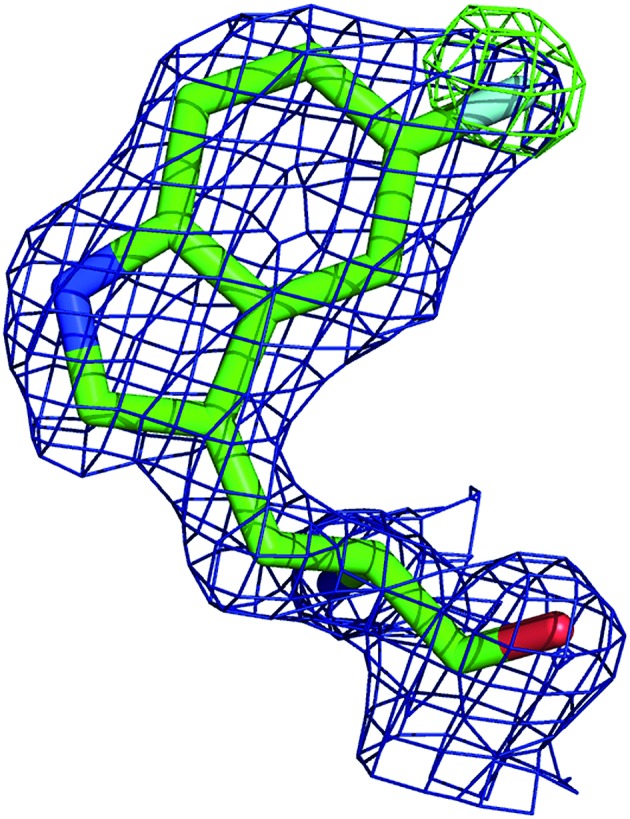
Fig. 6. Comparison of the conformation of labelled and unlabelled W24 in the βPGM–MgF3––G6P complex. 5FW24 βPGM is shown in magenta and βPGM in gold. The main amino acid side chains involved in ligand binding are shown. There is no discernible difference between the labelled and unlabelled proteins.

Conclusion
In this study, incorporation of 5-fluorotryptophan into the βPGM enzyme structure has been successfully completed with high efficiency and does not perturb catalytic efficiency or protein structure as determined by X-ray crystallography and 19F NMR spectroscopy. 19F NMR studies of 5FWβPGM W216F with metal fluoride TSA complexes enabled convenient evaluation of TSA complex formation for phosphoryl transfer enzymes by facilitating observation of protein, metal fluoride species, and ligand in a single NMR spectrum at anticipated molar ratios. The discovery of 5FW24, as a resonance in the βPGM enzyme structure that reports TSA complexation, has enabled us to demonstrate that the metal fluoride complexes are representative of phosphoryl transfer in aqueous solution. This will permit screening and analysis of inhibitors that may bind to the phosphoryl transfer site and perturb the formation of metal fluoride complexes.
Experimental
Preparation of βPGM-His mutant
The His6-Tag mutant was prepared from the pET22b(+)_pgmB template8 and commercial oligonucleotide primers, incorporating a silent mutation for SacI (underlined) by removing the TAA stop codon: 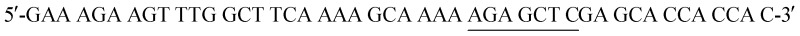 and
and 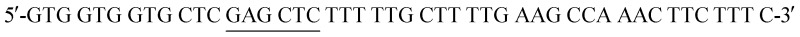 . Mutant preparation was performed according to the quikchange lightning site-directed mutagenesis kit. Confirmation of the mutation was accomplished by digesting plasmids with SacI and PstI (Fig. S3†). The purified plasmid was transformed into E. coli BL21(DE3) and the protein was expressed according to literature procedure.30 Protein purification was performed using a HiTrap 5 mL Chelating HP NTA-Ni2+ affinity column, eluting at a flow rate of 2.5 mL min–1 with a step-wise gradient of 250 mM and 25 mM imidazole buffer. Fractions were pooled and buffer exchanged into 50 mM HEPES buffer pH 7.2, providing 30.6 g L–1 of pure protein. Enzyme concentrations were determined using spectrophotometric analysis (ε280 = 19 940 M–1 cm–1).
. Mutant preparation was performed according to the quikchange lightning site-directed mutagenesis kit. Confirmation of the mutation was accomplished by digesting plasmids with SacI and PstI (Fig. S3†). The purified plasmid was transformed into E. coli BL21(DE3) and the protein was expressed according to literature procedure.30 Protein purification was performed using a HiTrap 5 mL Chelating HP NTA-Ni2+ affinity column, eluting at a flow rate of 2.5 mL min–1 with a step-wise gradient of 250 mM and 25 mM imidazole buffer. Fractions were pooled and buffer exchanged into 50 mM HEPES buffer pH 7.2, providing 30.6 g L–1 of pure protein. Enzyme concentrations were determined using spectrophotometric analysis (ε280 = 19 940 M–1 cm–1).
Expression and purification of 5FWβPGM
pET22b(+)_pgmB-His was expressed in E. coli BL21(DE3) and grown in minimal media according to the published Crowley methods.24 25 mL LBAmp was inoculated with 25 μL E. coli BL21(DE3) pET-22b(+)_pgmB-His glycerol stock and incubated at 37 °C with shaking (250 rpm) overnight. 3.3 mL of the overnight culture was used to inoculate 6 × 330 mL of LBAmp in 1 L flasks. Bacterial cultures were grown at 37 °C with shaking (250 rpm) for 2–3 h until an OD600 of ∼0.6–0.8 was reached. The bacterial cultures were centrifuged at 3750 rpm for 5 min and the supernatant was discarded. The cell pellet was resuspended in 12 mL of minimal growth media (12 g L–1 NaH2PO4, 6 g L–1 K2HPO4, 1 g L–1 NH4Cl, 2 g L–1d-glucose, 4 mL L–1 1 M MgSO4, and 1.8 mL L–1 1 mM FeSO4) and distributed evenly between 6 × 330 mL minimal media in 1 L flasks. Bacterial cultures were incubated at 37 °C with shaking (250 rpm) for 30 min. In 19F-labelling method B, 3 × 330 mL bacterial cultures were supplemented with 1 mL of sterile l-phenylalanine, l-tyrosine, 5-fluoro-d/l-tryptophan, and glyphosate solutions for final concentrations of 60 mg L–1, 60 mg L–1, 120 mg L–1, and 1 g L–1 respectively. In 19F-labelling method A, 3 × 330 mL bacterial cultures were supplemented with 1 mL solution of 5-fluoroindole in DMSO for a final concentration of 60 mg L–1. IPTG was added to 1 mM in all 6 × 330 mL bacterial cultures and cultures were incubated at 18 °C overnight, 250 rpm. For 15N-labeled protein, 15NH4Cl was substituted in the minimal media for NH4Cl. The protein was purified by the same method used for βPGM-His, providing 27.8 mg L–1 of 15N-βPGM, 13.0 mg L–1 of 5FWβPGM (method A) and 23.5 mg L–1 of 5FWβPGM (method B) (Fig. S6†).
Preparation of βPGM-W24F mutant
W24F mutant was prepared from the pET22b(+)_pgmB-His template and commercial oligonucleotide primers converting Trp24 to Phe24 (bold) and incorporating a silent mutation for BglI (underlined):  and
and 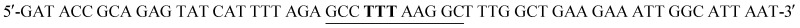 . Mutant preparation was performed according to the quikchange lightning site-directed mutagenesis procedure and confirmation of the mutation was accomplished by digesting plasmids with BglI and BstEII (Fig. S4†). The βPGM-W24F plasmid was transformed into E. coli BL21(DE3) and the protein was overexpressed and purified according to the same procedure used for 5FWβPGM, providing 18.5 mg L–1 of W24F 5FWβPGM (Fig. S6†).
. Mutant preparation was performed according to the quikchange lightning site-directed mutagenesis procedure and confirmation of the mutation was accomplished by digesting plasmids with BglI and BstEII (Fig. S4†). The βPGM-W24F plasmid was transformed into E. coli BL21(DE3) and the protein was overexpressed and purified according to the same procedure used for 5FWβPGM, providing 18.5 mg L–1 of W24F 5FWβPGM (Fig. S6†).
Preparation of βPGM-W216F mutant
W216F mutant was prepared from the pET22b(+)_pgmB-His template and commercial oligonucleotide primers converting Trp216 to Phe216 (bold) and incorporating a silent mutation for PstI (underlined): 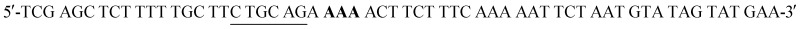 and
and 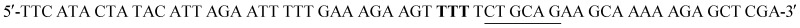 . Mutant preparation was performed according to the quikchange lightning site-directed mutagenesis procedure and confirmation of the mutation was accomplished by digesting plasmids with PstI (Fig. S5†). The βPGM-W216F plasmid was transformed into E. coli BL21(DE3) and the protein was overexpressed and purified according to the same procedure used for 5FWβPGM, using only method B for 19F-labelling, providing 45.8 mg L–1 of W216F 5FWβPGM (Fig. S7†).
. Mutant preparation was performed according to the quikchange lightning site-directed mutagenesis procedure and confirmation of the mutation was accomplished by digesting plasmids with PstI (Fig. S5†). The βPGM-W216F plasmid was transformed into E. coli BL21(DE3) and the protein was overexpressed and purified according to the same procedure used for 5FWβPGM, using only method B for 19F-labelling, providing 45.8 mg L–1 of W216F 5FWβPGM (Fig. S7†).
NMR methods
All NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer equipped with a 5 mm Smart Probe, operating at 470 MHz for fluorine. 1D 19F NMR were recorded at 5 °C with a zgbsfhigqn (for G6P complexes) or zgfhigqn (for G1CP and G1CFsP complexes) pulse program and acquired over 3000–31 150 transients with a recycle delay of 2.5 s. MgF3–TSA samples contained 1 mM (5FWβPGM and 5FWβPGM W24F) or 1.6 mM (5FWβPGM W216F) protein, 1 mM DTT, 5 mM MgCl2, 10 mM NH4F, 5 mM substrate and 10% D2O in 50 mM HEPES pH 7.2. AlF4–TSA complexes were prepared similarly with the addition of 1 equivalent AlCl3. All 19F NMR were referenced with NH4F (–119.5 ppm). 1H-15N HSQC were recorded at 27 °C with a hsqcetgpsi pulse program and acquired over 128 scans. 1 mM DSS was used to reference 1H NMR shifts. Samples contained 500 μM 5FW or unlabeled 15N βPGM, 5 mM MgCl2 and 10% D2O in 50 mM HEPES pH 7.2.
βPGM Km and kcat determination
All kinetic data was obtained using spectrophotometric analysis. The turnover of βG1P (commercially available) to G6P was monitored by the accumulation of NADPH (ε340 = 6.22 mM–1 cm–1) via a coupled reaction to glucose-6-phosphate dehydrogenase (G6PDH). Kinetic assays were performed in 100 μL total volume at 25 °C in 50 mM HEPES pH 7.2 and contained 2 mM MgCl2, 0.5 mM NADP+, 5 U mL–1 G6PDH, 150 μM α-fructose-1,6-bisphosphate, 50 nM 5FWβPGM, and 0–200 μM βG1P unless otherwise stated. Reactions were initiated with the addition of substrate. Steady state rates were obtained from the linear portions of time vs. A340 plots. Calculated errors are a result of standard error.
βPGM inhibition assays
Assays were performed in 100 μL total volume at 25 °C in 50 mM HEPES pH 7.2. IC50 assays included 2 mM MgCl2, 0.5 mM NADP+, 5 U mL–1 G6PDH, 150 μM α-fructose-1,6-bisphosphate, 10 μM βG1P, 50 nM 5FWβPGM and 0–5 mM inhibitor. Assays for Ki determination included 2 mM MgCl2, 0.5 mM NADP+, 5 U mL–1 G6PDH, 150 μM fructose-1,6-bisphosphate, 50 nM βPGM, 0–200 μM βG1P and were evaluated for different inhibitor concentrations (0–20 μM). All reactions were initiated with the addition of substrate. Calculated errors are a result of standard error.
Crystallization, data collection and refinement
Purified 5FWβPGM was concentrated to 10 mg mL–1 and used for sitting drop vapour diffusion crystallization experiments. Orthorhombic crystals of the 5FWβPGM-MgF3––G6P complex were obtained from already established conditions29 and cryo-cooled directly.31 New crystal conditions for the apo-protein and 5FWβPGM–MgF3––G6P complex were established at the EMBL High Throughput Crystallization (HTX) Laboratory (Grenoble, France). The protein was set up in sitting-drop vapour-diffusion plates, with drops consisting of 100 nL sample plus 100 nL of reservoir solution. A number of crystals in different crystallization conditions were obtained. Crystals of the apo-protein grew at 20 °C in 1–7 days from a solution of 0.2 M calcium chloride, 20% (w/v) PEG 6000 and 0.1 M tris (pH 8). Crystals of a new monoclinic crystal form of the 5FWβPGM–MgF3––G6P complex grew in 20% (w/v) PEG 5000MME and 0.1 M sodium acetate. Crystals were harvested and cryocooled automatically using the CrystalDirect robot. X-ray diffraction data were collected by the autonomous ESRF beamline MASSIF-1 32 using automatic protocols for the location and optimal centring of crystals.33 The beam diameter was selected automatically to match the crystal volume of highest homogeneous quality. Strategy calculations accounted for flux and crystal volume in the parameter prediction for complete data sets. Data reduction was performed with XDS34 followed by AIMLESS.35 The crystals of the apo-protein belonged to the orthorhombic space group P21212, with two molecules in the asymmetric unit coordinating a Ca2+. The new crystal form of the 5FWβPGM–MgF3––G6P complex was in the monoclinic space group P21 with two molecules in the asymmetric unit (Table S1†). The structures were solved using PHASER_MR36 implemented in CCP4 using the coordinates of the open conformation of β-PGM (PDB code ; 2WHE;29 apo-protein) and the native βPGM–MgF3––G6P structure (pdb code ; 2WF5;29 5FWβPGM–MgF3––G6P complex) as initial search models. Refinement was carried out using REFMAC5 37 and PHENIX38 and by manual rebuilding with COOT.39
Data deposition
The atomic coordinates and structure factors have been deposited in the protein data bank with codes 5FWβPGM apo-protein: 5OLW, 5FWβPGM–MgF3––G6P complex: ; 5OLX and 5FWβPGM–MgF3––G6P complex monoclinic crystal form: ; 5OLY.
Conflicts of interest
There are no conflicts of interest to declare.
Supplementary Material
Acknowledgments
We thank NSERC, CIHR, and NSHRF for financial support of this research. We thank Nicole E. McCormick for her help with mutant preparation and protein expression.
Footnotes
†Electronic supplementary information (ESI) available: Supporting Figures, HQSC NMR spectra, LC-MS/MS data and X-ray diffraction refinement data are described. See DOI: 10.1039/c7sc04204c
References
- Bowler M. W., Cliff M. J., Waltho J. P., Blackburn G. M. New J. Chem. 2010;34:784. [Google Scholar]
- Jin Y., Richards N. G., Waltho J. P., Blackburn G. M. Angew. Chem., Int. Ed. 2017;56:4110. doi: 10.1002/anie.201606474. [DOI] [PubMed] [Google Scholar]
- Glassier M. E., Gerlt J. A., Babbitt P. C. Curt. Opin. Chem. Biol. 2006;10:492. doi: 10.1016/j.cbpa.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Lahiri S., Zhang G., Dunaway-Mariano D., Allen K. Biochemistry. 2002;41:8351. doi: 10.1021/bi0202373. [DOI] [PubMed] [Google Scholar]
- Lahiri S. D., Zhang G. F., Dunaway-Mariano D., Allen K. N. Science. 2003;299:2067. doi: 10.1126/science.1082710. [DOI] [PubMed] [Google Scholar]
- Dai J., Finci L., Zhang C., Lahiri S., Zhang G., Peisach E., Allen K. N., Dunaway-Mariano D. Biochemistry. 2009;48:1984. doi: 10.1021/bi801653r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y., Bhattasali D., Pellegrini E., Forget S. M., Baxter N. J., Cliff M. J., Bowler M. W., Jakeman D. L., Blackburn G. M., Waltho J. P. Proc. Natl. Acad. Sci. U. S. A. 2014;111:12384. doi: 10.1073/pnas.1402850111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter N. J., Olguin L. F., Golicnik M., Feng G., Hounslow A. M., Bermel W., Blackburn G. M., Hollfelder F., Waltho J. P., Williams N. H. Proc. Natl. Acad. Sci. U. S. A. 2006;103:14732. doi: 10.1073/pnas.0604448103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter N. J., Blackburn G. M., Marston J. P., Hounslow A. M., Cliff M. J., Bermel W., Williams N. H., Hollfelder F., Wemmer D. E., Waltho J. P. J. Am. Chem. Soc. 2008;130:3952. doi: 10.1021/ja078000n. [DOI] [PubMed] [Google Scholar]
- Griffin J. L., Bowler M. W., Baxter N. J., Leigh K. N., Dannatt H. R. W., Hounslow A. M., Blackburn G. M., Webster C. E., Cliff M. J., Waltho J. P. Proc. Natl. Acad. Sci. U. S. A. 2012;109:6910. doi: 10.1073/pnas.1116855109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matei E., Gronenborn A. M. Angew. Chem., Int. Ed. 2016;55:150. doi: 10.1002/anie.201508464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee C. T., Koleski E. J., Pomerantz W. C. Angew. Chem., Int. Ed. 2015;54:3735. doi: 10.1002/anie.201411658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F., Yu X., Liu C., Qu C. X., Gong Z., Liu H. D., Li F. H., Wang H. M., He D. F., Yi F. Nat. Commun. 2015;6:1. doi: 10.1038/ncomms9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung E. W., Yagi H., Harjani J. R., Mulcair M. D., Scanlon M. J., Baell J. B., Norton R. S. Chem. Biol. Drug Des. 2014;84:616. doi: 10.1111/cbdd.12355. [DOI] [PubMed] [Google Scholar]
- Curtis-Maroma R., Dokoa D., Rowea M. L., Richardsa K. L., Williamson R. A., Howard M. J. Org. Biomol. Chem. 2014;12:3808. doi: 10.1039/c4ob00699b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra N. K., Urick A. K., Ember S. W. J., Schonbrunn E., Pomerantz W. C. ACS Chem. Biol. 2014;9:2755. doi: 10.1021/cb5007344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez J., Haapalainen A., Cahill S. M., Ho M. C., Yan F. N., Almo S. C., Schramm V. L. Chem. Biol. 2013;20:212. doi: 10.1016/j.chembiol.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. J., Horst R., Katritch V. Science. 2012;335:1106. doi: 10.1126/science.1215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitevski-LeBlanc J. L., Prosser R. S. Prog. Nucl. Magn. Reson. Spectrosc. 2012;62:1. doi: 10.1016/j.pnmrs.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Visser N. V., Westphal A. H., Nabuurs S. M., van Hoek A., van Mierlo C. P. M., Visser A. J. W. G., Broos J., van Amerongen H. FEBS Lett. 2009;583:2785. doi: 10.1016/j.febslet.2009.07.022. [DOI] [PubMed] [Google Scholar]
- Frieden C., Hoeltzli S. D., Ban J. G. Methods Enzymol. 2004;380:400. doi: 10.1016/S0076-6879(04)80018-1. [DOI] [PubMed] [Google Scholar]
- Sarker M., Orrell K. E., Xu L., Tremblay M., Bak J. J., Liu X., Rainey J. K. Biochemistry. 2016;55:3048. doi: 10.1021/acs.biochem.6b00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arntson K. E., Pomerantz W. C. K. J. Med. Chem. 2016;59:5158. doi: 10.1021/acs.jmedchem.5b01447. [DOI] [PubMed] [Google Scholar]
- Crowley P. B., Kyne C., Monteith W. B. Chem. Commun. 2012;48:10681. doi: 10.1039/c2cc35347d. [DOI] [PubMed] [Google Scholar]
- Sorensen M. D., Kristensen S. M., Bjorn S., Noris K., Olsen O., Led J. J. J. Biol. NMR. 1996;8:391. doi: 10.1007/BF00228142. [DOI] [PubMed] [Google Scholar]
- Hansson H., Mattson P. T., Allard P., Haapaniemi P., Vihinen M., Smith C. I. E., Hard T. Biochemistry. 1998;37:2912. doi: 10.1021/bi972409f. [DOI] [PubMed] [Google Scholar]
- Zhang G. F., Dai J., Wang L. B., Dunaway-Mariano D., Tremblay L. W., Allen K. N. Biochemistry. 2005;44:9404. doi: 10.1021/bi050558p. [DOI] [PubMed] [Google Scholar]
- Golicnik M., Olguin L. F., Feng G., Baxter N. J., Waltho J. P., Williams N. H., Hollfelder F. J. Am. Chem. Soc. 2009;131:1575. doi: 10.1021/ja806421f. [DOI] [PubMed] [Google Scholar]
- Baxter N. J., Bowler M. W., Alizadeh T., Cliff M. J., Hounslow A. M., Wu B., Berkowitz D. B., Williams N. H., Blackburn G. M., Waltho J. P. Proc. Natl. Acad. Sci. U. S. A. 2010;107:4555. doi: 10.1073/pnas.0910333106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S. D., Zhang G., Radstrom P., Dunaway-Mariano D., Allen K. N. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002;58:324. doi: 10.1107/s0907444901019989. [DOI] [PubMed] [Google Scholar]
- Pellegrini E., Piano D., Bowler M. W. Acta Crystallogr. 2011;67:902. doi: 10.1107/S0907444911031210. [DOI] [PubMed] [Google Scholar]
- Bowler M. W., Nurizzo D., Barrett R., Beteva A., Bodin M., Caserotto H., Delageniere S., Dobias F., Flot D., Giraud T., Guichard N., Guijarro M., Lentini M., Leonard G. A., McSweeney S., Oskarsson M., Schmidt W., Snigirev A., von Stetten D., Surr J., Svensson O., Theveneau P., Mueller-Dieckmann C. J. Synchrotron Radiat. 2015;22:1540. doi: 10.1107/S1600577515016604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson O., Malbet-Monaco S., Popov A., Nurizzo D., Bowler M. W. Acta Crystallogr., Sect. D: Struct. Biol. 2015;71:1757. doi: 10.1107/S1399004715011918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Acta Crystallogr., Sect. D: Struct. Biol. 2010;66:125. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. R., Murshudov G. N. Acta Crystallogr., Sect. D: Struct. Biol. 2013;69:1204. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. J. Appl. Crystallogr. 2007;40:658. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G., Vagin A., Dodson E. Acta Crystallogr., Sect. D: Struct. Biol. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. Acta Crystallogr., Sect. D: Struct. Biol. 2012;68:352. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Acta Crystallogr., Sect. D: Struct. Biol. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.