

Abstract
Research on disease causation often attempts to isolate the effects of individual factors, including individual genes or environmental factors. This reductionist approach has generated many discoveries, but misses important interactive and cumulative effects that may help explain the broad range of variability in disease occurrence observed across studies and individuals. A disease rarely results from a single factor, and instead results from a broader combination of factors, characterized here as intrinsic (I) and extrinsic (E) factors. Intrinsic vulnerability or resilience emanates from a variety of both fixed and shifting biological factors including genetic traits, while extrinsic factors comprise all biologically-relevant external stressors encountered across the lifespan. The I×E concept incorporates the multi-factorial and dynamic nature of health and disease and provides a unified, conceptual basis for integrating results from multiple areas of research, including genomics, G×E, developmental origins of health and disease, and the exposome. We describe the utility of the I×E concept to better understand and characterize the cumulative impact of multiple extrinsic and intrinsic factors on individual and population health. New research methods increasingly facilitate the measurement of multifactorial and interactive effects in epidemiological and toxicological studies. Tiered or indicator-based approaches can guide the selection of potentially relevant I and E factors for study and quantification, and exposomics methods may eventually produce results that can be used to generate a response function over the life course. Quantitative data on I×E interactive effects should generate a better understanding of the variability in human response to environmental factors. The proposed I×E concept highlights the role for broader study design in order to identify extrinsic and intrinsic factors amenable to interventions at the individual and population levels in order to enhance resilience, reduce vulnerability and improve health.
Keywords: Social determinants of health, gene-environment interactions, exposome, risk, epidemiology, toxic chemical exposure
1. Introduction
In 1981, Doll and Peto [1] proposed sharply segmented environmental contributions to cancer (30% tobacco, 3% alcohol, 35% diet, 4% occupational, 10% infection, 7% reproductive/sexual behavior). This formulation of disease risk is now widely regarded to be incomplete because it disregards the interactive effects of multiple stressors, as well as the background of susceptibility that modulates risk.
While much research continues to examine contributions of individual genetic and environmental factors to disease, there has been a growing emphasis on studying the interplay and accumulation of multiple factors in disease causation. As researchers have historically viewed disease susceptibility as being mainly genetic, the majority of interaction studies in chronic disease risk have been gene-environment (G×E) interaction studies. Simonds et al. recently reviewed G×E studies related to cancer [2]. The authors identified 272 studies, focusing largely on breast, lung and colorectal cancers, and 4896 interactions, mostly in the areas of energy balance (dietary factors and anthropometrics), lifestyle (smoking and alcohol), and exogenous hormones (hormone replacement therapy and oral contraceptives). The authors reported a median interaction odds ratio (OR) of 1.3 for the 29 analyses for which p-values of interaction were reported, many of which did not remain significant after multiple test correction. Thus, the majority of these G×E studies, which examined interactions between single polymorphisms and environmental factors, revealed only low or modest effect sizes.
Concurrent consideration of multiple genetic variants and environmental factors may reveal a greater contribution of gene-environment interactions to disease outcome [3, 4]. Compared to the volume of research focused on individual factors and traits, relatively little research has been done in this area to date. Emerging scientific concepts and tools are making the simultaneous evaluation of multiple stressors over time more feasible and desirable. Examples of existing research paradigms that seek to address multiple additive or interacting stressors and susceptibility factors include the exposome, Developmental Origins of Health and Disease (DOHaD), and environmental health disparities research; cumulative risk assessment seeks to apply the outcomes of research to describe the impacts of multiple stressors and susceptibility factors on health risk. Each of these approaches is defined and described below.
The goal of this review is to expand upon current concepts to conceptualize the universe of potential susceptibility factors and environment stressors that impact on health outcomes and to explore the implications of this broader formulation for epidemiological and toxicological research and for the use of the resulting data in describing risk. A single unifying concept may help to guide the acquisition and integration of data from multiple research areas and the development of more complete models of disease causation.
2. Concepts and strategies related to cumulative impacts of multiple environmental stressors in the face of vulnerability
The exposome, a term first coined by Wild (2015), was redefined by Rappaport and Smith to be the “totality of environmental exposures from conception onwards” [5, 6]. They further proposed a methodology by which this concept could be operationalized and “exposures” could be identified from measurable amounts of biologically active chemicals (small molecules) in the internal environment of the body arising from both exogenous and endogenous sources [6]. Rappaport and Smith also described how the lifelong exposome may be characterized in a series of “snapshots” at critical time points throughout the lifecourse [6]. Mass spectrometry can identify and measure in blood samples thousands of chemicals from foods, drugs, pollutants, and endogenous processes [7, 8]. Others have considered how the exposome could be quantified in part using external measurements from monitors over extended periods, geospatial information and questionnaires [9]. Miller and Jones expanded the exposome concept to include the body’s responses to environmental influences – DNA mutations and adducts, epigenetic alterations and protein modifications – observable molecular manifestations of cumulative exposures [10], and this concept is being advanced in research [8, 9, 11–14].
The DOHaD concept, which emerged over 30 years ago, links conditions and diseases of late childhood and adulthood—including obesity, diabetes mellitus, cardiovascular disease, and cancer— with early life environmental conditions [15–17]. Persistent epigenetic modifications have been shown to mediate lifelong health effects resulting from adverse nutritional and diabetic intrauterine environments [18], and neurodevelopmental disorders resulting from exposure to environmental toxicants [19].
The environmental health disparities and justice concept links the effects of social constructs such as race and class with psychosocial stress and vulnerability to environmental exposures [20–23]. For example, variation in stressors and counter-balancing resources at the individual and community level, as a result of residential segregation, can modulate vulnerability to toxicant exposures. Morello-Frosch and colleagues described how extrinsic social constructs such as race and class and intrinsic biological susceptibility factors can interact with environmental hazard inequalities to exacerbate health disparities [22], and may be used to explain health disparities observed in disadvantaged communities [22, 24].
Cumulative risk assessment has been defined as the assessment of “combined risks from aggregate exposures to multiple agents or stressors, where agents or stressors may include chemical and nonchemical stressors” [25, 26], whereas cumulative impacts assessment skirts the risk assessment imperative to obtain probabilistic estimates of risk and instead obtains a more general characterization of harms from multiple stressors. Both applications can build on research findings described above along with environmental epidemiologic studies that ideally [26] combine measurements of multiple biomarkers to evaluate the effects of exposures and vulnerability factors on biological pathways relevant to disease. Researchers have examined cumulative impacts of extrinsic factors such as chemical and non-chemical stressors, and background intrinsic population vulnerability arising from differential susceptibility and exposure [22, 27–31]. Smith et al. (2015) proposed using exposomics tools to quantify cumulative risks and impacts and suggested engaging impacted communities in participatory exposome research [8].
Thus emanating from various areas of research are conceptual framings that recognize that chemical exposures, be they occupational or environmental, do not occur in isolation and can be modified by other lifestyle and biological factors, such as stress, obesity, concurrent tobacco smoking and chronic infections (e.g. hepatitis B virus). Collectively, these factors play a role in determining a person’s susceptibility to any given set of environmental chemical exposures, as illustrated in Figure 1. As such, cumulative risk can vary and depends on the susceptibility an individual has come into the world with and accrues through daily life. Thus, to understand human susceptibility to disease we must look beyond genetics and account for all forms of exposure as well as intrinsic factors that confer additional vulnerability, including sex, lifestage and health status.
Fig. 1.
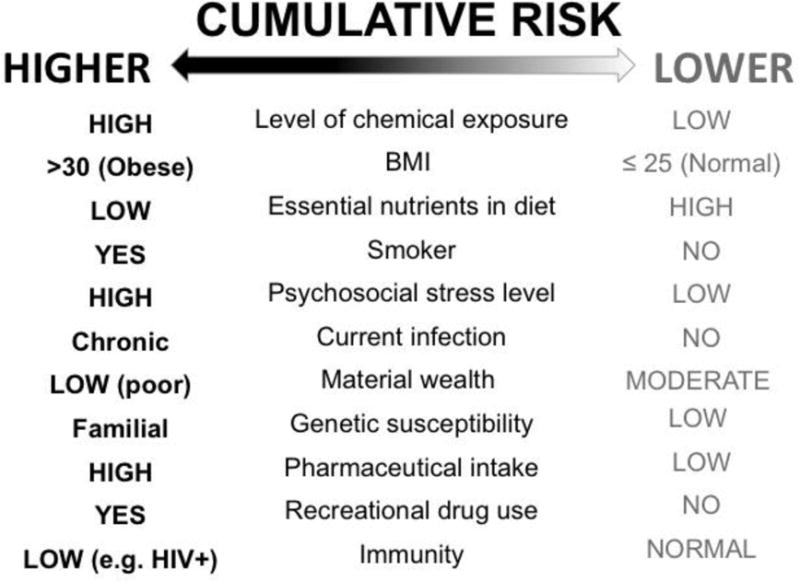
Higher vs. lower cumulative disease risk of hypothetical individuals at a single point in time based on combinations of multiple risk factors.
3. I×E – a unifying concept?
Given the understanding that multiple environmental factors combine with intrinsic susceptibility factors to increase risk of disease and death, a broad approach is needed to fully depict disease causation. Here, we describe a unifying concept, that we call I×E, illustrated in Fig. 2. The “environment” in gene-environment (G×E) studies typically encompasses a narrow range of factors such as occupational chemical exposures, infections, and lifestyle factors. Here we expand “environment” to include all extrinsic (E) factors, as illustrated in the upper panel in Fig. 2 where E factors are loosely grouped by type, e.g. diet, food contaminants, food security; different kinds of pollution; and behavioral factors. Similarly, expansion beyond the “gene” in gene-environment is necessary to account for the many inter-related intrinsic biological factors that contribute to disease susceptibility [2, 32, 33], e.g. obesity, allostatic load—a composite physiological factor signifying chronic stress [34], nutritional status, immune status, and even age. Thus “gene” or G becomes all intrinsic factors (I), as shown in the middle panel. An individual is exposed to multiple I and E factors which combine to influence health over time.
Fig. 2.
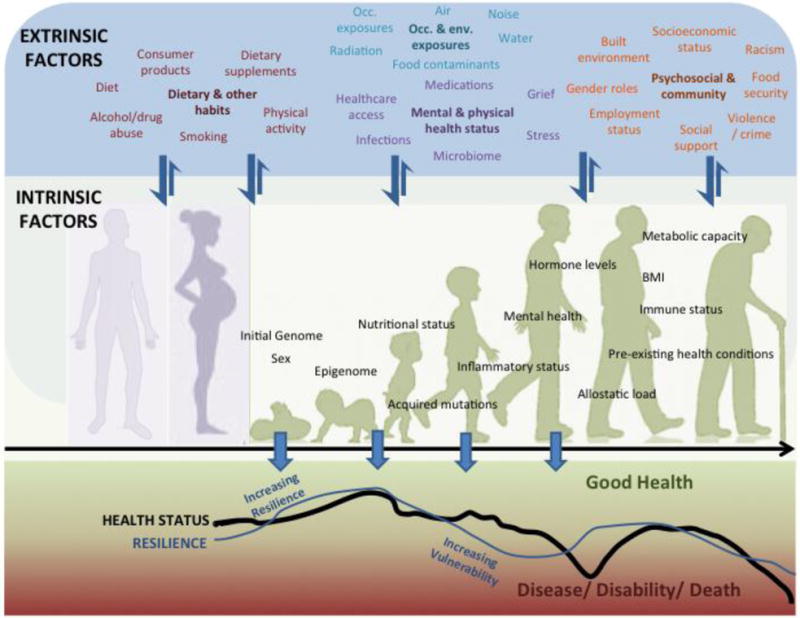
Extrinsic (E, upper panel) and intrinsic (I, middle panel) factors interact throughout the lifespan, enhancing vulnerability or resilience in a cumulative manner. Intrinsic genome and sex are fixed I factors whereas the other I factors are modifiable. Many E factors act on and influence the I factors, whereas E factors are modulated to a lesser degree by I factors (indicated by thick and thin arrows between the upper and middle panels). I×E interactions can vary over the lifespan beginning before conception via maternal and paternal effects (background schematic in middle panel). In a given individual, I×E interactions influence vulnerability and resilience, and consequentially health status, throughout life, as indicated by the fluctuating curves in the lower panel. The curves shown are not based on actual data and are hypothetical trajectories indicative of negative and positive effects on resilience and health status over the lifespan. Scenarios that could contribute to these fluctuations are described in the text in Section 3. Occ., occupational; Env., environmental.
Some I factors are fixed (sex and initial genome) and some are modifiable during the life course due to the actions of extrinsic factors, as when infectious agents or drugs modify immune status. Although the E factors act on and influence the I factors, there is some reciprocal relationship. For example, mental health status (I) influenced by chronic stress (E) can in turn influence dietary choices (E). Using arrows, a simplified relationship in which the effect of a large number of E factors are modulated by (to a lesser degree; thin arrows), and act upon (to a greater degree; thick arrows), a large number of I factors, is depicted in Fig. 2.
I×E interactions are complex and can vary over the lifespan (background schematic in middle panel), as I is modified by extrinsic factors, and age-dependent changes in function. Intrinsic factors in an individual are also accrued via parental transmission, such as via inherited epigenetic change through the female or male lineage [35]. The I×E concept includes exposures occurring before conception, as well as those occurring in utero or within the first years of life that can induce epigenetic or other changes which may result in health effects expressed later in life, thus incorporating the DOHaD concept.
The lower panel of Figure 2 illustrates that I×E interactions influence vulnerability and resilience, and consequentially health status. Factors causing increased vulnerability can lead to disability and disease. People exposed to a lesser degree and those with greater biological or social resilience are in general able to maintain a longer, healthier life without disability. The Healthy Aging Phenotype is an example of a multi-dimensional, age and gender-dependent biological resilience that is governed by interactions among genes (I), epigenetic status (I) and environmental (E) factors [36]. Resilience, vulnerability and health status may be positively and negatively impacted throughout the lifespan. The fluctuating curves represent possible trajectories for a hypothetical individual that could arise through various scenarios. For example, a healthy individual with low resilience in early life due to low parental socioeconomic status (SES) and lead exposure in the home may gain resilience through an enriched home environment and strong social support. As a young adult, decreased resilience and increased vulnerability, due to factors such as loss of employment, exposure to violence, and community environmental exposures may lead to a decline in health status in response to a challenge (e.g., infection) or the cumulative effects of different factors (e.g., poorer diet, chronic stress, less physical activity, smoking). Improved social, economic and medical support together with factors such as improved nutrition and physical activity, cessation of smoking, reduced environmental exposures, and stress reduction can increase resilience and improve health status. In this example resilience declines with age, perhaps exacerbated by grief or reduced social support, and is followed by a decline in health status, perhaps due to infection, or on-going chronic disease processes.
Thus, our unifying I×E concept illustrates how health and disease throughout the life-span can arise from a set of intrinsic factors, responding and modulating susceptibility to multiple extrinsic factors including traditional environmental stressors such as occupational and non-occupational chemical exposures, as well as nutritional, lifestyle, and socioeconomic factors (Fig. 2). We propose that this conceptualization will encourage researchers to move beyond the G×E approach and to study a greater number of potential I×E interactions using emerging research methodologies.
This conceptual framing can guide evaluation of data on factors and interactions relevant to a particular population/health outcome and identification of data gaps to be addressed in future studies. A systematic checklist or algorithm to assess potentially relevant extrinsic and intrinsic factors, and potential interaction among them at various stages across the lifespan could be developed (see Section 8). Examples of such framework approaches exist in the literature e.g. the U.S. Environmental Protection Agency’s Causal Analysis/Diagnosis Decision Information System for assessing aquatic systems [37] and a multilevel model of postmenopausal breast cancer incidence [38].
4. Illustration of the Concept: Examples of I×E studies
Here we illustrate the I×E concept through examples of studies examining the interaction of I factors other than genetics with E factors but do not provide systematic compilation of such studies, nor an evaluation of the strengths and weaknesses of individual studies, which is beyond the scope of this review.
4.1. Obesity (I) and environmental exposures (E) interact to affect a variety of diseases
In several studies, obesity (I) has been reported to exacerbate the effects of various environmental exposures (E). Multiple studies have reported that obesity and air pollution together increased the risk of several different diseases, including hypertension in children [39], asthma in children [40], and stroke and cardiovascular disease [41]. Obesity was found to modify the effects of indoor fine and coarse particulate matter exposure on chronic obstructive pulmonary disorder, producing more severe symptoms, greater rescue medication use, and greater proinflammatory effect than in non-obese individuals [42]. A substantive review of obesity, ozone exposure, and cardiopulmonary health reported that obesity was associated with decreased lung function and increased inflammatory mediators in the seven studies which met the criteria of examining the interaction of excess weight and ozone exposure on cardiopulmonary outcomes in adults [43]. In one study, obesity was observed to increase the risk of arsenic-associated lung and bladder cancer by over 10-fold in individuals with elevated arsenic exposure compared to non-obese individuals. This study found a 4-fold increase in the Rothman synergy index [44], a classic epidemiological measure of interaction based on departure from additive risks [45]. Finally, arsenic’s carcinogenicity and adverse pulmonary effects are synergistically higher in obese individuals, smokers, and those with concurrent occupational exposures [44, 46]. These findings suggest that obesity is a susceptibility factor in a number of diseases linked to environmental exposures, and is an area worthy of further exploration.
Obesity is an intrinsic factor modifiable by multiple extrinsic factors including diet, exercise, and certain chemical obesogens [47], and may be “programmed” by conditions in utero and early life. Obesity was found to be more prevalent among children with high exposure to both second-hand smoke and low levels of fiber (OR=2.6) or eicosapentaenoic acid (EPA, a polyunsaturated fatty acid precursor for physiologically active lipid compounds) (OR=2.6) compared to those with high levels of fiber or EPA [48]. The interactions were significant by both additive and multiplicative analyses.
4.2. Preexisting disease (I) and environmental exposure (E) interactions
I×E interactions can be complex. Preexisting disease status (I) may affect capacity for physical activity (E), which in turn can affect a variety of intrinsic factors. Further, extrinsic factors can become intrinsic factors. For example, exposure to hepatitis B virus (HBV) (E) can result in infection, and, depending upon intrinsic factors (e.g., immune status, life stage), an individual can develop chronic hepatitis. Chronic hepatic inflammation becomes an intrinsic factor, which can increase vulnerability to other hepatotoxicants, and can lead to cirrhosis of the liver and cancer. A synergistic interaction has been reported between chronic HBV infection (I) and aflatoxin B1 exposure (E) resulting in an increased risk of hepatocellular carcinoma [49]. This association was found across 5 large cohort studies in which odds ratios ranged from 40.7 to 70.0 with a mean of 59.6 compared with HBV alone (mean OR=10) or aflatoxin B1 alone (mean OR=13.7) [49]. Off-target effects of medication (E) taken to treat HBV can also impact intrinsic factors, which in turn can affect health. The balance of susceptibility versus resilience operates both at an individual and a population level. For example, in countries where HBV is prevalent and dietary aflatoxin B1 exposure is poorly controlled, “background” rates of hepatic carcinoma in the population are high [49]. In other populations, different I and E factors and their interactions result in higher “background” rates of breast cancer or cardiovascular disease. Thus, the rate of a disease in a population should be considered to be a marker of the balance of susceptibility versus resilience in that population, and the importance of I×E effects overall may be estimated based on population variation in “background” risk of disease.
4.3. Social resilience (I) and environmental exposures (E)
Social resilience is another key determinant of health outcome [50]. Although biological and social resilience are distinct, they are intertwined. Environmental exposures (E) may interact with social stressors (E), as well as with susceptibility factors (I) to worsen disease risk in highly vulnerable populations [24]. Low SES (E) increases allostatic load (I) [51], which can affect immune and metabolic status (I) [52], as well as amplifying the adverse effects of environmental chemical exposures (E) [31]. Thus low SES may sometimes be treated as a surrogate for allostatic load. In 2003, a review described how low socioeconomic position may worsen the adverse effects of air pollution [53]. This was further seen in later studies that included stressors related to SES, particularly in relation to childhood asthma. Among 413 children in an urban community-based pregnancy cohort in Massachusetts, higher rates of asthma diagnosis were reported for children chronically exposed to high levels of violence and high levels of traffic-related air pollution [54]. In another study of 73 children with asthma over a 6 month period, chronically increased family stress was found to interact with traffic-related pollution exposure to exacerbate asthma symptoms in children; this study reported no effect of stress or pollution alone [55]. In a 3-year study of 2,497 children, the risk of onset of childhood asthma attributable to in utero exposure to high levels of traffic-related pollution was found to be significantly higher for those born to parents with high levels of perceived stress compared with those with low levels of stress (HR=1.51 vs 1.05) [30].
Socioeconomic stress may also exacerbate the risk of endpoints besides asthma. Low neighborhood SES was reported to amplify the risk of air pollution-related preterm births (OR=1.3 in low vs high SES neighborhoods with high air pollution levels) [56] and adult mortality (RR=2.62 in low income-high pollutant vs high income-low pollutant) [57]. The working memory component of IQ in children was found to be inversely impacted by significant interactions between high prenatal exposure to PAHs (measured as cord blood-DNA adducts) and high levels of prenatal hardship (β=−8.07) and between high DNA adduct levels in cord blood and recurrent material hardship (β=−9.82) [58]. For neurobehavioral development in children, another study reported that maternal psychological distress during pregnancy interacted with PAH exposure, and found a βinteraction=4.37 (p-value=0.021) for somatic complaints and βinteraction=3.31 (p-value=0.04) for aggressive behavior [59].
5. A more precise representation of IxE by exposomics
Quantitative measurements of both extrinsic and intrinsic factors are needed so that their relative importance in affecting health outcomes can be better understood and characterized. The disparity in coverage of genetics and environmental factors in GxE studies was previously discussed [60]. Measurement of proportional effects of multiple factors is more precise if they are continuous measures rather than binary or categorical classifications such as those derived from a job exposure matrix or recall questionnaire [61–63]. Quantitative measurements of intrinsic factors such as metabolic health or disease status beyond yes/no diagnosis are also crucial. The latter requires the establishment of biomarkers of underlying disease processes or predictors of outcome. Zeise et al. previously discussed opportunities for incorporating evidence from in vitro, experimental in vivo, and GWAS studies on differences in intrinsic factors such as molecular transport systems, enzyme activity, and DNA repair capacity in modeling pharmacokinetic and/or pharmacodynamic variability [64]. Exposomics offers another means to quantify a range of factors.
In the exposome paradigm, the internal biochemical environment reflects all exposures from both exogenous and endogenous sources [6]. Going beyond that, the internal biochemical environment in the I×E concept provides a lens for representing interacting mixtures of I and E factors at any given time. Thus, in a manner analogous to the “snapshots” proposed for the exposome [6], representations of multiple contributing E and I factors and their interactions would be identified and measured over time. Use of exposomics tools such as targeted and untargeted metabolomics or other assays [7, 13, 14] could quantify both I and E factors, thereby allowing for much improved assessments of the importance of different environmental factors including non-chemical stressors in modulating risk. The expanded concept of exposomics by Miller and Jones, which includes the proteome and adductome, improves the capacity of exposomics to reflect cumulative exposures [10]. Once the relationships in the internal environment are understood, evaluations can describe the relationship of external exposure to the internal milieu to move toward characterizations of external exposures – sources and circumstances – that lead to modification of intrinsic factors, increased susceptibility and risk.
There have been many recent improvements in how samples are processed and analyzed, and new bioinformatics tools and databases to store exposome data, analyze it, and integrate it with data on other omics and pathways are being developed [12, 13, 65–68]. Recently, the National Institutes of Health (NIH) launched the “Big Data to Knowledge” (BD2K) program under which a consortium of research centers are devising methods to streamline the synthesis and analysis of large-scale data from a variety of biomedical sources [69–71]. For example, a big-data platform, BD2K Patient Centered Information Commons (http://pic-sure.org), was created to enable programmatic access to exposome and phenome data from the National Health and Nutrition Examination Survey (NHANES) via a web browser (https://nhanes.hms.harvard.edu) [72].
6. Increasing the focus on I×E in epidemiologic research
The IxE conceptualization can guide the design of epidemiological investigations in addressing the multiple stressors and susceptibilities leading to response. The tools and capacities in many cases exist, as evidenced by various published epidemiological studies of multiple exposures and their interactions with genomic and other intrinsic factors. Some specific challenges and trends are explored below.
6.1 Lessons learned from G×E studies
Some of the challenges of I×E-based research can be anticipated from those noted previously for G×E studies. For example, many of the G×E interaction ORs estimated by Simonds et al. were of small magnitude and were not significant after multiple test correction [2]. A review of studies on gene-obesity interactions showed that although interactions between specific genes and a variety of environmental, lifestyle and treatment exposures were reported, findings were limited by issues related to “statistical modelling, confounding, low replication rate, underpowered analyses, biological assumptions and measurement precision” [73]. A systematic review of gene-macronutrient interaction studies in type-2 diabetes (T2D) also revealed that many reported interactions could not be replicated and highlighted the importance of improving standards for examining and reporting interactions, multiple test correction and independent replication [74]. Ultimately, the requirement for large epidemiological studies to address study power limitations has fostered the development of multicenter consortia, tissue databanks and data-sharing procedures [75].
6.2. Explicit examination of the effects of “confounders” as possible mediators of effect
The classic epidemiological study design was based on the objective of measuring the impact of a single factor (such as a well-defined chemical exposure). Other factors affecting the observed outcome were regarded as potential “confounders” that were, if possible, eliminated by selection of a relatively homogeneous study population that minimized variability. Consider, for example, the number of studies that deliberately excluded women, asthmatics, or non-whites. To the extent that this was not possible, mathematical approaches controlled for, and sought to eliminate the impact of these confounding factors. Thus, an estimate of the impact of the single factor of interest was presented, unmodified by other influences or averaged over a relatively homogeneous study population. Even if quantitative estimates of the impacts of these other potential modifiers of risk were obtainable from the data, these were often not reported or were only listed in table footnotes. However, controlling for confounders that are in fact mediators and part of the exposure-response pathway results in essentially ‘controlling-away’ a significant finding. Studies that control for related factors may miss both multiplicative and additive effects (commonly identified by the study design as effect modification and confounding, respectively). Some epidemiology studies do look at effect modification, but the statistical bar is high for modeling interactive terms, and additive effects may go undetected (i.e., false negatives).
Standard techniques for achieving a multifactorial analysis exist (see for example Johnson and Wichern, 2007 [76]), but there are challenges to implementing such a study: more power, and therefore larger sample size, is often required. Recent developments in the range of available computational and analytical tools, such as the use of artificial intelligence (AI) techniques for identifying associations in large datasets, and the application of open-ended multifactorial approaches such as principal component analysis present an opportunity for method development in this area. Various efforts are underway to coordinate, standardize and develop processes for the use and sharing of “big data” from multi-consortia, large data set collaborations [75].
In evaluating I and E, factors of interest need to be explicitly examined both for their individual impacts on the disease endpoint(s) of interest, and for their role as modifiers of the impact of other contributing factors. Findings from such studies would support models that characterize responses to a number of different intrinsic and extrinsic contributing factors [75].
Finally, when studying multifactorial causes of ill-health, it is important to integrate consideration of IxE disease determinants both as individual-level attributes and group-level features to which individuals belong [77]. Diez-Roux and colleagues (2004) point to the concept of herd immunity, an important group-level factor that affects population differences in disease incidence, but also determines an individual’s risk of contracting a particular infectious disease. Similarly, there has been increasing scientific interest in the field of environmental epidemiology to integrate environmental health with social epidemiological methods to better elucidate the pathways by which group-level or macro-social factors, such as neighborhood characteristics, racial segregation, and income inequality, may mediate or interact with environmental hazard exposures in ways that affect health disparities [78]. Multilevel modeling enables the simultaneous examination of group-level and individual-level factors on individual-level outcomes, while accounting for the non-independence of observations within groups and allowing for inferences regarding intergroup variation [77].
6.3. Study design to account for temporality
Single exposure measurements are often taken at one point in time in epidemiologic studies, but measurements at different times may be needed, depending on the nature of the interaction. This is likely to be especially important in studies of developmental impacts, which may create health states or enhanced sensitivity reflecting permanently altered intrinsic factors after an initial, extrinsic, early-in-life exposure. The ideal study design is a prospective cohort, which would allow repeated sampling and monitoring during critical windows of susceptibility. As continuous monitoring of biomarker or exposure data is seldom feasible, cross-sectional “snapshot” analyses of the study population are important. Large prospective studies are difficult and expensive to implement, especially over the timescale needed to measure developmental impacts, limiting their feasibility in many situations. Other study types such as ecological and time-series studies of effect incidence, and case-control studies traditionally are designed to address single exposure factors and single health endpoints. However, these design approaches can be expanded to examine multiple factors and the interactions between them, subject to the necessary power for multifactorial analyses. Meta-analysis is also an important tool for achieving the desired power and breadth of coverage for analysis of the interaction between various intrinsic and extrinsic factors (see for example Munsell et al., 2014 [79] who review a number of studies of various designs and provide a meta-analysis relating body mass, estrogen use, breast cancer and hormone receptor status).
6.4. Selection of vulnerable populations and relevant I×E factors for study
Recent migrants, disadvantaged socioeconomic groups with high environmental chemical exposures, and pregnant women are potential high priority populations for quantitative study of I×E interactions using exposomics. Other vulnerable populations can be selected using indicator and map-based approaches. These methods use geographic information systems (GIS) mapping to integrate chemical and nonchemical stressors, vulnerability, and background risk factors and generate semi-quantitative indicators that highlight communities with enhanced vulnerability [24, 80, 81]. An example is the California Communities Environmental Health Screening Tool (CalEnviroScreen), developed by the California Office of Environmental Health Hazard Assessment, which maps measures of cumulative impact at the census tract level based on 20 indicators of “pollution burden” (exposures and environmental threats) and “population characteristics” (sensitive populations and socioeconomic factors) [81]. A screening tool such as this one could help identify communities for in-depth epidemiological study of the interactions between the various stressors, and to select factors for study. Tiered or phased approaches have been used to identify relevant factors in cumulative risk assessments, with the different tiers applying increasingly more conservative filters based on availability of data supporting an effect [82–85].
We suggested in section 3 that a systematic approach based on the I×E concept could be developed to assess extrinsic and intrinsic factors and potential interaction among them at various stages across the lifespan for further consideration. In selecting I and E factors for study, consideration can be given to their contribution to a hypothesized common general mechanism of toxicity. For example, the interaction of arsenic/obesity in lung and bladder cancer risk identified in a human population study was based on a hypothesized common mechanism of inflammation [44]. Another approach is to consider interaction among factors that affect common adverse outcomes such as lead/methylmercury/polychlorinated biphenyls, nutritional factors, social deprivation and IQ [86] or multiple endocrine disruptors and reproductive tract development [87]. In recognition of the fact that stressors may act at different points in carcinogenesis, synergies of chemicals acting via linked but dissimilar sequences/processes, in different targets/tissues could also be considered [88]. For example, chemicals that alter cell signaling and those that increase oxidative stress may interact to increase cancer risk.
Other approaches to identifying relevant I and E interactions for further study include Environment-Wide Association Studies (EWAS) and the exposome globe approach. In the EWAS approach associations between exposures and phenotypes can be examined, e.g. type 2 diabetes and multiple environmental factors [89, 90], in a non-hypothesis based manner. For example, an EWAS of 76 environmental contaminants or lifestyle factors in 70 Caucasian adults living in Sweden reported significant additive interactions between dietary saturated fat and levels of p,p′-dichlorodiphenyldichloroethene, polychlorinated biphenyls and exercise, that lead to high prevalence of metabolic syndrome in an elderly population [91]. Using the “exposome globe” approach, a visual depiction of the network of replicated correlations between individual exposures in the exposome, clusters of exposure can be visualized [92]. In this area, the use of statistical machine learning methods, such as regularized regression and tree-based methods, to identify exposures and mixtures of exposures associated with phenotypes and outcomes recently has been discussed [93].
7. Usefulness of I×E studies in animals
The study of multifactorial impacts in humans is important, but obviously has limitations in terms of the number of intrinsic and extrinsic factors that can be evaluated. An advantage of animal studies is that they can overcome some of the limitations of human studies and allow exploration of exposure scenarios and phenotypic effects that are challenging to explore in human studies, such as early-life/late-life effects in the same individuals exposed to stressor combinations, or pathology of organs and tissues not generally accessible in humans, or the interaction of multiple toxicological mechanisms [73]. For example, early-life environmental tobacco smoke exposure plus later exposure to asbestos was shown to increase the risk of lung disease via immune effects in mice [94]. Intrinsic factors resulting from a high fat diet (HFD) also have a profound effect on the toxicity of numerous chemicals in different organ systems, resulting for example in an elevated risk of mammary cancer in mice following maternal dioxin exposure [95] and of steatogenesis in mice following valproic acid exposure in adulthood [96]. Recognition of I×E interactions observed in animals such as these has important implications for the design and interpretation of toxicology studies, which are usually performed in healthy, young adult animals fed low fat diets. Observations in humans of profound toxicity associated with environmental chemical exposures in individuals with pre-existing conditions, such as obesity and diabetes, who are eating a HFD, also highlight limitations of the current toxicity testing paradigm, where healthy animals are typically exposed to a single chemical, in predicting human health effects resulting from I×E interactions [97].
One challenge of animal studies has been the limited ability to approximate human genetic variability, and the interaction of genetic variability with other intrinsic as well as extrinsic factors. Genetically diverse mouse models such as Collaborative Cross strains [98] and Diversity Outbred mice [99] were developed to address this. They have enabled the examination of effects of environmental exposures against variable genetic backgrounds [75], e.g., susceptibility to micronuclei formation for benzene [100], liver toxicity for trichloroethylene [101], and the effect of diet on intestinal cardiometabolic-related microbiota [102]. Using the Collaborative Cross mouse population model, a complex and highly variable relationship was identified between perchloroethylene and the toxicokinetics and toxicodynamics of its primary oxidative metabolite trichloroacetate at the population level [103]. Similarly studies with the Collaborative Cross can be designed to study the interaction of genetic susceptibility with other intrinsic factors and environmental stressors. The Mouse Phenome Database hosts data on molecular and clinical phenotypes collected across genotypes, tissues, ages, environmental exposures, interventions, and treatments in Collaborative Cross strains and Diversity Outbred mice [104].
Another challenging but important I×E scenario to study in animals is the interaction of embodied stress with other environmental factors. Multiple mouse studies have examined the effects of exposures in combination with stress and elucidated underlying mechanisms. Studies in male mice have shown that a combination of chronic social defeat stress and a high fat diet resulted in altered expression of hepatic genes involved in lipid metabolism, dysregulation of lipid profile, and modulation of effects on body weight [105, 106]. In rats, enhanced effects in behavioral, neurochemical and glucocorticoid outcomes occurred in response to combined maternal lead (Pb) and prenatal restraint stress [107–111]. Notable differences in neurotransmitter changes induced by developmental Pb exposure with and without prenatal stress were observed to depend on the nature of behavioral experiences [108, 112]. On the positive side, repeated learning behavioral experience was observed to attenuate the effects of combined Pb exposure and prenatal stress [113]. In another series of studies, combined low level maternal Pb exposure and prenatal stress followed by offspring stress was found to significantly increase impulsive behavior in males [114]. Brain regions and neurotransmitter systems that mediate learning/behavioral flexibility were found to be more greatly impacted in males, illustrating the potential modifying effect of sex as an intrinsic factor. Studies such as these can generate potentially useful data and hypotheses relevant to IxE interactions that can then be tested in human epidemiological research.
8. Utility of I×E-based research in cumulative risk and impact assessment
I×E-guided research can help to address some of the challenges identified in assessing cumulative risk, such as a need for the incorporation of data on background exposures, vulnerability factors, and non-chemical stressors such as psychosocial stress; a need for improved biomarkers of exposure, susceptibility, and effect; and difficulty incorporating some epidemiologic research findings into risk assessment [85]. Intrinsic and extrinsic factors have traditionally been addressed in risk assessment as two separate problems: one involving cumulative risks from extrinsic factors and the other involving inter-individual variability from intrinsic factors. For non-cancer outcomes, the latter is estimated by generic variability factors for toxicokinetics and toxicodynamics [115, 116], which were found to not adequately address children [117–119]. Many of the potential interactions in Figure 2 and the examples provided in the paper expand beyond the traditional notion of toxicokinetics and toxicodynamics, and the I×E concept introduces multiple intrinsic and extrinsic stressors as potential contributors to the range of population variability, vulnerability and risk. The implication of the I×E concept is that the total range of variability and vulnerability in the population may be greater than that captured by the current default uncertainty factors, in part due to the sheer numbers of potential interactions that are not currently assessed. Few studies have been done to date to test this hypothesis, but some suggest that even fairly simple combinations can result in effects that differ across the population by more than 10-fold [44, 54]. Ultimately, a risk assessment approach that attempts to explicitly mathematically model I×E interactions across population distributions might be more useful, but would require considerable data and analytical effort, and is not currently feasible. More feasible options, while the research in this area is developing, would include the following practices:
-
Adopt a mathematical description for human variability to model dose-response for non-carcinogens
The fact that any single chemical, industrial site, or other factor evaluated in a risk assessment exists against a background of a large volume of extrinsic exposure, homeostatic impairment and pre-existing disease in the population calls into question the use of a threshold assumption for many non-carcinogens. The National Research Council (2009) recommended a unified probabilistic approach that explicitly and formally models human variability to estimate risk, for carcinogens and non-carcinogens [85]. Chiu and Slob (2015) [120] developed a related probabilistic approach for a general class of chemicals into a unified three-step framework for dose-response extrapolation. Their framework is relatively straightforward to compute, and also explicitly addresses variability across a population; however it does not consider the potential addition of the effect of the chemical exposure to that of other similarly acting chemicals in the “background” of exposures experienced by individuals within the population and so it is most applicable for rare health outcomes.
-
Adoption of a structured algorithm to assess I×E factors and interactions
A systematic inquiry based on the I×E concept could be developed to assess extrinsic and intrinsic factors, and potential interactions among them at various stages across the lifespan. The risk assessors could examine:- Are there studies of the chemical’s effects in vulnerable human populations? E.g., pregnant mothers, newborns, children, the elderly, obese individuals, those with preexisting conditions, genetically susceptible subjects, those with relevant co-exposures. What is the effect of this vulnerability on dose-dependent effects? Are effects seen at lower doses? Can the vulnerability be quantified?
- Are there studies of the chemical’s effects in susceptible animals? E.g., early life and in utero exposure, studies in the Collaborative Cross, studies in animals given a high-fat diet or one low in essential nutrients, studies in stressed animals, studies in animals with other co-exposures. What is the effect of this susceptibility on dose-dependent effects? Are effects seen at lower doses? Can the difference be quantified?
- What are the general toxicity mechanisms that are affected by the chemical, and what intrinsic (I) and extrinsic factors (E) can be expected to contribute to vulnerability, based on those mechanisms? E.g., if genotoxicity is one mechanism by which a chemical is thought to act, are there deficiencies in DNA repair that would likely amplify the effects of the chemical? If a chemical acts on a hormonal pathway, are there other chemical stressors that can contribute to the same general mechanism?
- How is the chemical activated, distributed, metabolized, and excreted? E.g., if a chemical is activated via a certain CYP isoform, are there medications or other agents that are known to induce that isoform?
- Is there evidence that the chemical possesses any of the ten key characteristics of carcinogens [28]? How about key characteristics of other major classes of toxicants such as endocrine disruptors? If so, what other intrinsic and extrinsic factors could amplify these effects.
- Are there biomarkers of susceptibility that could be used to quantify the effect? E.g., polymorphisms in a gene or set of genes that amplify or reduce the effects of the chemical; a biomarker of inflammation that is associated with an increased risk in combination with chemical exposure etc.
- Are there potential interactions between the chemical agent and infectious agents in producing disease. E.g., hepatitis B infection is already known to interact with hepatocellular carcinogens such as aflatoxin B1 to cause liver cancer, so the infection may also confer vulnerability to other hepatocellular carcinogens.
- Are there members of the population with background exposure to other chemical or non-chemical stressors that affect the same target tissue/organ/system, or act via the same toxicity mechanisms? E.g., persons on a specific diet or with concurrent occupational exposures.
-
Are the health outcomes caused by the chemical relatively common?A structured algorithm that includes the considerations above could help the risk assessors consider which I×E interactions may be most relevant to a given assessment. Where adequate data exist, quantitative assessments should be attempted. In the absence of data, these issues would be addressed in the assessment using adequate factors to account for variability, vulnerability, and uncertainty. The responses to these questions would guide discussion in the risk assessments of the potential areas of interactive and cumulative effects. If data do not exist to quantify I×E interactions, the need for additional data would be articulated, along with a qualitative discussion of the impact of the resulting uncertainty on the assessment.
-
Employ indicator-based approaches to describe impact
Index/indicator-based approaches have the advantage of being able to incorporate an almost unlimited number of stressors for which data are available, and to combine very different types of stressors (chemical, psychosocial, health) into one analysis. Such approaches are not intended to produce estimates of health risk for any given population. The CalEnviroScreen tool described above facilitates comparisons of communities differentially impacted by intrinsic and environmental factors, and directs attention to communities substantially impacted by cumulative stressors.
9. Concluding Remarks
While recognizing that various I and E factors can have significant contributions, understanding the role (i.e., if a cause and effect relationship exists) and magnitude of effect of individual factors remains an important aspect of epidemiologic research and risk and impact assessments. In some cases, even where there are multiple component causes, removal of one cause can have an impact, e.g., smoking cessation and reducing exposure to environmental tobacco smoke remain important public health actions even when other lung cancer risk factors such as air pollution are present.
Similarly, the demonstration of a synergistic relationship between cigarette smoking and asbestos exposure highlights the need for smoking prevention and cessation for workers in industries with high asbestos exposure [121]. Neonatal vaccination against HBV is another example, as prevention of chronic HBV infection and chronic liver inflammation substantially reduces the risk of liver cancer resulting from the interaction of inflammation and environmental chemical exposures such as aflatoxin B1. Thus, I×E complements other research efforts seeking to understand what are the important risk factors, what risk factors are present, how do the risk factors add or otherwise interact, and which risk factors can be targeted through public health action.
We propose the I×E concept to promote a more robust and nuanced understanding of the combined impacts of intrinsic and extrinsic factors, including their potentially additive and multiplicative effects, on the health of diverse populations. Epidemiologic and toxicological studies should be designed and sufficiently powered to assess multifactorial effects, and enhanced technologies and novel methods in exposure science should be leveraged in their implementation. As methods improve for integrating I×E more systematically into research and risk assessment, use of indicator/index approaches can facilitate valuable spatial screening to inform decisions in ways that advance environmental equity goals by improving existing conditions and reducing future harm. Ultimately, improved understanding of I×E interactions throughout the life span may facilitate timely public health interventions that can effectively tip the scales toward resilience and health and away from vulnerability and disease.
Acknowledgments
This work was supported by the Research Translation Core of the Berkeley Superfund Research Center through National Institute of Environmental Health Sciences [Grant P42 ES004705 to M.T.S]; UCSF PEEC Children’s Center through U.S. EPA [Grant RD83543301 to R. M-F]; and National Institute of Environmental Health Sciences [Grant P01 ES022841 to R. M-F].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66:1191–1308. [PubMed] [Google Scholar]
- 2.Simonds NI, Ghazarian AA, Pimentel CB, Schully SD, Ellison GL, Gillanders EM, Mechanic LE. Review of the Gene-Environment Interaction Literature in Cancer: What Do We Know? Genet Epidemiol. 2016;40:356–365. doi: 10.1002/gepi.21967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu M, Shi X, Yang F, Wang J, Xu Y, Wei D, Yang K, Zhang Y, Wang X, Liang S, Chen X, Sun L, Zhu X, Zhao C, Zhu L, Tang L, Zheng C, Yang Z. The Cumulative Effect of Gene-Gene and Gene-Environment Interactions on the Risk of Prostate Cancer in Chinese Men. Int J Environ Res Public Health. 2016;13:162. doi: 10.3390/ijerph13020162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Narayan S, Sinsheimer JS, Paul KC, Liew Z, Cockburn M, Bronstein JM, Ritz B. Genetic variability in ABCB1, occupational pesticide exposure, and Parkinson’s disease. Environ Res. 2015;143:98–106. doi: 10.1016/j.envres.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wild CP. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomarkers Prev. 2005;14:1847–1850. doi: 10.1158/1055-9965.EPI-05-0456. [DOI] [PubMed] [Google Scholar]
- 6.Rappaport SM, Smith MT. Environment and disease risks. Science. 2010;330:460–461. doi: 10.1126/science.1192603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rappaport SM, Barupal DK, Wishart D, Vineis P, Scalbert A. The blood exposome and its role in discovering causes of disease. Environ Health Perspect. 2014;122:769–774. doi: 10.1289/ehp.1308015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith MT, de la Rosa R, Daniels SI. Using exposomics to assess cumulative risks and promote health. Environ Mol Mutagen. 2015;56:715–723. doi: 10.1002/em.21985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner MC, Nieuwenhuijsen M, Anderson K, Balshaw D, Cui Y, Dunton G, Hoppin JA, Koutrakis P, Jerrett M. Assessing the Exposome with External Measures: Commentary on the State of the Science and Research Recommendations. Annu Rev Public Health. 2017;38:215–239. doi: 10.1146/annurev-publhealth-082516-012802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller GW, Jones DP. The nature of nurture: refining the definition of the exposome. Toxicol Sci. 2014;137:1–2. doi: 10.1093/toxsci/kft251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Go YM, Chandler JD, Jones DP. The cysteine proteome. Free Radic Biol Med. 2015;84:227–245. doi: 10.1016/j.freeradbiomed.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Go YM, Jones DP. Redox biology: interface of the exposome with the proteome, epigenome and genome. Redox Biol. 2014;2:358–360. doi: 10.1016/j.redox.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones DP. Sequencing the exposome: A call to action. Toxicol Rep. 2016;3:29–45. doi: 10.1016/j.toxrep.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uppal K, Walker DI, Liu K, Li S, Go YM, Jones DP. Computational Metabolomics: A Framework for the Million Metabolome. Chem Res Toxicol. 2016;29:1956–1975. doi: 10.1021/acs.chemrestox.6b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grandjean P, Barouki R, Bellinger DC, Casteleyn L, Chadwick LH, Cordier S, Etzel RA, Gray KA, Ha EH, Junien C, Karagas M, Kawamoto T, Paige Lawrence B, Perera FP, Prins GS, Puga A, Rosenfeld CS, Sherr DH, Sly PD, Suk W, Sun Q, Toppari J, van den Hazel P, Walker CL, Heindel JJ. Life-Long Implications of Developmental Exposure to Environmental Stressors: New Perspectives. Endocrinology. 2015;156:3408–3415. doi: 10.1210/EN.2015-1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore SE. Early life nutritional programming of health and disease in The Gambia. J Dev Orig Health Dis. 2016;7:123–131. doi: 10.1017/S2040174415007199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walker CL, Ho SM. Developmental reprogramming of cancer susceptibility. Nat Rev Cancer. 2012;12:479–486. doi: 10.1038/nrc3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.El Hajj N, Schneider E, Lehnen H, Haaf T. Epigenetics and life-long consequences of an adverse nutritional and diabetic intrauterine environment. Reproduction. 2014;148:R111–120. doi: 10.1530/REP-14-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran NQV, Miyake K. Neurodevelopmental Disorders and Environmental Toxicants: Epigenetics as an Underlying Mechanism. Int J Genomics. 2017;2017:7526592. doi: 10.1155/2017/7526592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gee GC, Payne-Sturges DC. Environmental health disparities: a framework integrating psychosocial and environmental concepts. Environ Health Perspect. 2004;112:1645–1653. doi: 10.1289/ehp.7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gravlee CC. How race becomes biology: embodiment of social inequality. Am J Phys Anthropol. 2009;139:47–57. doi: 10.1002/ajpa.20983. [DOI] [PubMed] [Google Scholar]
- 22.Morello-Frosch R, Zuk M, Jerrett M, Shamasunder B, Kyle AD. Understanding the cumulative impacts of inequalities in environmental health: implications for policy. Health Aff (Millwood) 2011;30:879–887. doi: 10.1377/hlthaff.2011.0153. [DOI] [PubMed] [Google Scholar]
- 23.Sexton K, Gong H, Jr, Bailar JC, 3rd, Ford JG, Gold DR, Lambert WE, Utell MJ. Air pollution health risks: do class and race matter? Toxicol Ind Health. 1993;9:843–878. doi: 10.1177/074823379300900509. [DOI] [PubMed] [Google Scholar]
- 24.Solomon GM, Morello-Frosch R, Zeise L, Faust JB. Cumulative Environmental Impacts: Science and Policy to Protect Communities. Annu Rev Public Health. 2016;37:83–96. doi: 10.1146/annurev-publhealth-032315-021807. [DOI] [PubMed] [Google Scholar]
- 25.U.S. EPA. Framework for cumulative risk assessment. U.S Environmental Protection Agency, Office of Research and Development, National Center for Environmental Assessment; Washington, DC: 2003. [Google Scholar]
- 26.National Research Council. Toxicity testing in the 21st century: A vision and a strategy. Washington D.C.: 2007. pp. 75–80. [Google Scholar]
- 27.US Environmental Protection Agency. Executive Order 12898. 1994 [Google Scholar]
- 28.Cote I, Andersen ME, Ankley GT, Barone S, Birnbaum LS, Boekelheide K, Bois FY, Burgoon LD, Chiu WA, Crawford-Brown D, Crofton KM, DeVito M, Devlin RB, Edwards SW, Guyton KZ, Hattis D, Judson RS, Knight D, Krewski D, Lambert J, Maull EA, Mendrick D, Paoli GM, Patel CJ, Perkins EJ, Poje G, Portier CJ, Rusyn I, Schulte PA, Simeonov A, Smith MT, Thayer KA, Thomas RS, Thomas R, Tice RR, Vandenberg JJ, Villeneuve DL, Wesselkamper S, Whelan M, Whittaker C, White R, Xia M, Yauk C, Zeise L, Zhao J, DeWoskin RS. The Next Generation of Risk Assessment Multi-Year Study-Highlights of Findings, Applications to Risk Assessment, and Future Directions. Environ Health Perspect. 2016;124:1671–1682. doi: 10.1289/EHP233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sadd JL, Pastor M, Morello-Frosch R, Scoggins J, Jesdale B. Playing it safe: assessing cumulative impact and social vulnerability through an environmental justice screening method in the South Coast Air Basin, California. Int J Environ Res Public Health. 2011;8:1441–1459. doi: 10.3390/ijerph8051441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shankardass K, McConnell R, Jerrett M, Milam J, Richardson J, Berhane K. Parental stress increases the effect of traffic-related air pollution on childhood asthma incidence. Proc Natl Acad Sci U S A. 2009;106:12406–12411. doi: 10.1073/pnas.0812910106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zota AR, Shenassa ED, Morello-Frosch R. Allostatic load amplifies the effect of blood lead levels on elevated blood pressure among middle-aged U.S. adults: a cross-sectional study. Environ Health. 2013;12:64. doi: 10.1186/1476-069X-12-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez-Bulnes A, Astiz S, Ovilo C, Garcia-Contreras C, Vazquez-Gomez M. Nature and Nurture in the Early-Life Origins of Metabolic Syndrome. Curr Pharm Biotechnol. 2016;17:573–586. doi: 10.2174/1389201017666160301103835. [DOI] [PubMed] [Google Scholar]
- 33.Lill CM. Genetics of Parkinson’s disease. Mol Cell Probes. 2016;30:386–396. doi: 10.1016/j.mcp.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 34.McEwen BS, Stellar E. Stress and the individual. Mechanisms leading to disease. Arch Intern Med. 1993;153:2093–2101. [PubMed] [Google Scholar]
- 35.Braun K, Champagne FA. Paternal influences on offspring development: behavioural and epigenetic pathways. J Neuroendocrinol. 2014;26:697–706. doi: 10.1111/jne.12174. [DOI] [PubMed] [Google Scholar]
- 36.Franco OH, Karnik K, Osborne G, Ordovas JM, Catt M, van der Ouderaa F. Changing course in ageing research: The healthy ageing phenotype. Maturitas. 2009;63:13–19. doi: 10.1016/j.maturitas.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 37.U.S.E.E.P. agency. Causal Analysis/Diagnosis Decision Information System (CADDIS) Office of research and Development; Washington, DC: 2017. [Google Scholar]
- 38.Hiatt RA, Porco TC, Liu F, Balke K, Balmain A, Barlow J, Braithwaite D, Diez-Roux AV, Kushi LH, Moasser MM, Werb Z, Windham GC, Rehkopf DH. A multilevel model of postmenopausal breast cancer incidence. Cancer Epidemiol Biomarkers Prev. 2014;23:2078–2092. doi: 10.1158/1055-9965.EPI-14-0403. [DOI] [PubMed] [Google Scholar]
- 39.Dong GH, Wang J, Zeng XW, Chen L, Qin XD, Zhou Y, Li M, Yang M, Zhao Y, Ren WH, Hu QS. Interactions Between Air Pollution and Obesity on Blood Pressure and Hypertension in Chinese Children. Epidemiology. 2015;26:740–747. doi: 10.1097/EDE.0000000000000336. [DOI] [PubMed] [Google Scholar]
- 40.Jung KH, Perzanowski M, Rundle A, Moors K, Yan B, Chillrud SN, Whyatt R, Camann D, Perera FP, Miller RL. Polycyclic aromatic hydrocarbon exposure, obesity and childhood asthma in an urban cohort. Environ Res. 2014;128:35–41. doi: 10.1016/j.envres.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin XD, Qian Z, Vaughn MG, Trevathan E, Emo B, Paul G, Ren WH, Hao YT, Dong GH. Gender-specific differences of interaction between obesity and air pollution on stroke and cardiovascular diseases in Chinese adults from a high pollution range area: A large population based cross sectional study. Sci Total Environ. 2015;529:243–248. doi: 10.1016/j.scitotenv.2015.05.041. [DOI] [PubMed] [Google Scholar]
- 42.McCormack MC, Belli AJ, Kaji DA, Matsui EC, Brigham EP, Peng RD, Sellers C, Williams DL, Diette GB, Breysse PN, Hansel NN. Obesity as a susceptibility factor to indoor particulate matter health effects in COPD. Eur Respir J. 2015;45:1248–1257. doi: 10.1183/09031936.00081414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koman PD, Mancuso P. Ozone Exposure, Cardiopulmonary Health, and Obesity: A Substantive Review. Chem Res Toxicol. 2017;30:1384–1395. doi: 10.1021/acs.chemrestox.7b00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Steinmaus C, Castriota F, Ferreccio C, Smith AH, Yuan Y, Liaw J, Acevedo J, Perez L, Meza R, Calcagno S, Uauy R, Smith MT. Obesity and excess weight in early adulthood and high risks of arsenic-related cancer in later life. Environ Res. 2015;142:594–601. doi: 10.1016/j.envres.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rothman KJ. The estimation of synergy or antagonism. Am J Epidemiol. 1976;103:506–511. doi: 10.1093/oxfordjournals.aje.a112252. [DOI] [PubMed] [Google Scholar]
- 46.Ferreccio C, Yuan Y, Calle J, Benitez H, Parra RL, Acevedo J, Smith AH, Liaw J, Steinmaus C. Arsenic, tobacco smoke, and occupation: associations of multiple agents with lung and bladder cancer. Epidemiology. 2013;24:898–905. doi: 10.1097/EDE.0b013e31829e3e03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cano-Sancho G, Salmon AG, La Merrill MA. Association between Exposure to p,p′-DDT and Its Metabolite p,p′-DDE with Obesity: Integrated Systematic Review and Meta-Analysis. Environ Health Perspect. 2017;125:096002. doi: 10.1289/EHP527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore BF, Clark ML, Bachand A, Reynolds SJ, Nelson TL, Peel JL. Interactions between Diet and Exposure to Secondhand Smoke on the Prevalence of Childhood Obesity: Results from NHANES, 2007–2010. Environ Health Perspect. 2016;124:1316–1322. doi: 10.1289/ehp.1510138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kew MC. Synergistic Interaction Between Aflatoxin and Hepatitis B Virus in Hepatocarcinogenesis. In: Razzaghi-Abyaneh M, editor. Aflatoxins - Recent Advances and Future Prospects. InTech; Rijeka, Croatia: 2013. [Google Scholar]
- 50.Keck M, Sakdapolrak P. What is social resilience? Lessons learned and ways forward. Erdkunde. 2013;67:5–19. [Google Scholar]
- 51.Lynch JW, Kaplan GA, Shema SJ. Cumulative impact of sustained economic hardship on physical, cognitive, psychological, and social functioning. N Engl J Med. 1997;337:1889–1895. doi: 10.1056/NEJM199712253372606. [DOI] [PubMed] [Google Scholar]
- 52.McEwen BS. Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci. 1998;840:33–44. doi: 10.1111/j.1749-6632.1998.tb09546.x. [DOI] [PubMed] [Google Scholar]
- 53.O’Neill MS, Jerrett M, Kawachi I, Levy JI, Cohen AJ, Gouveia N, Wilkinson P, Fletcher T, Cifuentes L, Schwartz J, P. Workshop on Air, C. Socioeconomic, Health, wealth, and air pollution: advancing theory and methods. Environ Health Perspect. 2003;111:1861–1870. doi: 10.1289/ehp.6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clougherty JE, Levy JI, Kubzansky LD, Ryan PB, Suglia SF, Canner MJ, Wright RJ. Synergistic effects of traffic-related air pollution and exposure to violence on urban asthma etiology. Environ Health Perspect. 2007;115:1140–1146. doi: 10.1289/ehp.9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen E, Schreier HM, Strunk RC, Brauer M. Chronic traffic-related air pollution and stress interact to predict biologic and clinical outcomes in asthma. Environ Health Perspect. 2008;116:970–975. doi: 10.1289/ehp.11076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ponce NA, Hoggatt KJ, Wilhelm M, Ritz B. Preterm birth: the interaction of traffic-related air pollution with economic hardship in Los Angeles neighborhoods. Am J Epidemiol. 2005;162:140–148. doi: 10.1093/aje/kwi173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Finkelstein MM, Jerrett M, DeLuca P, Finkelstein N, Verma DK, Chapman K, Sears MR. Relation between income, air pollution and mortality: a cohort study. CMAJ. 2003;169:397–402. [PMC free article] [PubMed] [Google Scholar]
- 58.Vishnevetsky J, Tang D, Chang HW, Roen EL, Wang Y, Rauh V, Wang S, Miller RL, Herbstman J, Perera FP. Combined effects of prenatal polycyclic aromatic hydrocarbons and material hardship on child IQ. Neurotoxicol Teratol. 2015;49:74–80. doi: 10.1016/j.ntt.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perera FP, Wang S, Rauh V, Zhou H, Stigter L, Camann D, Jedrychowski W, Mroz E, Majewska R. Prenatal exposure to air pollution, maternal psychological distress, and child behavior. Pediatrics. 2013;132:e1284–1294. doi: 10.1542/peds.2012-3844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rappaport SM. Genetic Factors Are Not the Major Causes of Chronic Diseases. PLoS One. 2016;11:e0154387. doi: 10.1371/journal.pone.0154387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith RB, Edwards SC, Best N, Wright J, Nieuwenhuijsen MJ, Toledano MB. Birth Weight, Ethnicity, and Exposure to Trihalomethanes and Haloacetic Acids in Drinking Water during Pregnancy in the Born in Bradford Cohort. Environ Health Perspect. 2016;124:681–689. doi: 10.1289/ehp.1409480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vermeulen R, Li G, Lan Q, Dosemeci M, Rappaport SM, Bohong X, Smith MT, Zhang L, Hayes RB, Linet M. Detailed exposure assessment for a molecular epidemiology study of benzene in two shoe factories in China. Annals of Occupational Hygiene. 2004;48:105. doi: 10.1093/annhyg/meh005. [DOI] [PubMed] [Google Scholar]
- 63.Cantor KP, Villanueva CM, Silverman DT, Figueroa JD, Real FX, Garcia-Closas M, Malats N, Chanock S, Yeager M, Tardon A, Garcia-Closas R, Serra C, Carrato A, Castano-Vinyals G, Samanic C, Rothman N, Kogevinas M. Polymorphisms in GSTT1, GSTZ1, and CYP2E1, disinfection by-products, and risk of bladder cancer in Spain. Environ Health Perspect. 2010;118:1545–1550. doi: 10.1289/ehp.1002206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zeise L, Bois FY, Chiu WA, Hattis D, Rusyn I, Guyton KZ. Addressing human variability in next-generation human health risk assessments of environmental chemicals. Environ Health Perspect. 2013;121:23–31. doi: 10.1289/ehp.1205687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huan T, Forsberg EM, Rinehart D, Johnson CH, Ivanisevic J, Benton HP, Fang M, Aisporna A, Hilmers B, Poole FL, Thorgersen MP, Adams MWW, Krantz G, Fields MW, Robbins PD, Niedernhofer LJ, Ideker T, Majumder EL, Wall JD, Rattray NJW, Goodacre R, Lairson LL, Siuzdak G. Systems biology guided by XCMS Online metabolomics. Nat Methods. 2017;14:461–462. doi: 10.1038/nmeth.4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Warth B, Levin N, Rinehart D, Teijaro J, Benton HP, Siuzdak G. Metabolizing Data in the Cloud. Trends Biotechnol. 2017;35:481–483. doi: 10.1016/j.tibtech.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Warth B, Spangler S, Fang M, Johnson CH, Forsberg EM, Granados A, Martin RL, Domingo-Almenara X, Huan T, Rinehart D, Montenegro-Burke JR, Hilmers B, Aisporna AE, Hoang LT, Uritboonthai W, Benton HP, Richardson SD, Williams AJ, Siuzdak G. Exposome-Scale Investigations Guided by Global Metabolomics, Pathway Analysis, and Cognitive Computing. Anal Chem. 2017 doi: 10.1021/acs.analchem.7b02759. [DOI] [PubMed] [Google Scholar]
- 68.Wishart D, Arndt D, Pon A, Sajed T, Guo AC, Djoumbou Y, Knox C, Wilson M, Liang Y, Grant J, Liu Y, Goldansaz SA, Rappaport SM. T3DB: the toxic exposome database. Nucleic Acids Res. 2015;43:D928–934. doi: 10.1093/nar/gku1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bourne PE, Bonazzi V, Dunn M, Green ED, Guyer M, Komatsoulis G, Larkin J, Russell B. The NIH Big Data to Knowledge (BD2K) initiative. J Am Med Inform Assoc. 2015;22:1114. doi: 10.1093/jamia/ocv136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bui AAT, Van Horn JD, N.B.K.C. Consortium Envisioning the future of ‘big data’ biomedicine. J Biomed Inform. 2017;69:115–117. doi: 10.1016/j.jbi.2017.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Margolis R, Derr L, Dunn M, Huerta M, Larkin J, Sheehan J, Guyer M, Green ED. The National Institutes of Health’s Big Data to Knowledge (BD2K) initiative: capitalizing on biomedical big data. J Am Med Inform Assoc. 2014;21:957–958. doi: 10.1136/amiajnl-2014-002974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patel CJ, Pho N, McDuffie M, Easton-Marks J, Kothari C, Kohane IS, Avillach P. A database of human exposomes and phenomes from the US National Health and Nutrition Examination Survey. Sci Data. 2016;3:160096. doi: 10.1038/sdata.2016.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reddon H, Gueant JL, Meyre D. The importance of gene-environment interactions in human obesity. Clin Sci (Lond) 2016;130:1571–1597. doi: 10.1042/CS20160221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li SX, Imamura F, Ye Z, Schulze MB, Zheng J, Ardanaz E, Arriola L, Boeing H, Dow C, Fagherazzi G, Franks PW, Agudo A, Grioni S, Kaaks R, Katzke VA, Key TJ, Khaw KT, Mancini FR, Navarro C, Nilsson PM, Onland-Moret NC, Overvad K, Palli D, Panico S, Quiros JR, Rolandsson O, Sacerdote C, Sanchez MJ, Slimani N, Sluijs I, Spijkerman AM, Tjonneland A, Tumino R, Sharp SJ, Riboli E, Langenberg C, Scott RA, Forouhi NG, Wareham NJ. Interaction between genes and macronutrient intake on the risk of developing type 2 diabetes: systematic review and findings from European Prospective Investigation into Cancer (EPIC)-InterAct. Am J Clin Nutr. 2017;106:263–275. doi: 10.3945/ajcn.116.150094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Committee on Incorporating 21st Century Science into Risk-Based Evaluations, editor. National Research Council. Using 21st Century Science to Improve Risk-Related Evaluations. National Academy Press; Washington, D.C.: 2017. [Google Scholar]
- 76.Jonsonn RA, Wichern DW. Applied multivariate statistical analysis. Pearson Prentice Hall; New Jersey: 2007. [Google Scholar]
- 77.Diez Roux AV. The study of group-level factors in epidemiology: rethinking variables, study designs, and analytical approaches. Epidemiol Rev. 2004;26:104–111. doi: 10.1093/epirev/mxh006. [DOI] [PubMed] [Google Scholar]
- 78.Cushing L, Morello-Frosch R, Wander M, Pastor M. The haves, the have-nots, and the health of everyone: the relationship between social inequality and environmental quality. Annu Rev Public Health. 2015;36:193–209. doi: 10.1146/annurev-publhealth-031914-122646. [DOI] [PubMed] [Google Scholar]
- 79.Munsell MF, Sprague BL, Berry DA, Chisholm G, Trentham-Dietz A. Body mass index and breast cancer risk according to postmenopausal estrogen-progestin use and hormone receptor status. Epidemiol Rev. 2014;36:114–136. doi: 10.1093/epirev/mxt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sadd J, Morello-Frosch R, Pastor M, Matsuoka M, Prichard M, Carter V. The Truth, the Whole Truth, and Nothing but the Ground-Truth: Methods to Advance Environmental Justice and Researcher-Community Partnerships. Health Educ Behav. 2014;41:281–290. doi: 10.1177/1090198113511816. [DOI] [PubMed] [Google Scholar]
- 81.OEHHA and California EPA. Update to the California Communities Environmental Health Screening Tool. CalEnviroScreen 3.0. 2017 available at: https://oehha.ca.gov/media/downloads/calenviroscreen/report/ces3report.pdf.
- 82.Meek ME, Boobis AR, Crofton KM, Heinemeyer G, Raaij MV, Vickers C. Risk assessment of combined exposure to multiple chemicals: A WHO/IPCS framework. Regul Toxicol Pharmacol. 2011 doi: 10.1016/j.yrtph.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 83.Menzie CA, MacDonell MM, Mumtaz M. A phased approach for assessing combined effects from multiple stressors. Environ Health Perspect. 2007;115:807–816. doi: 10.1289/ehp.9331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Moretto A, Bachman A, Boobis A, Solomon KR, Pastoor TP, Wilks MF, Embry MR. A framework for cumulative risk assessment in the 21st century. Crit Rev Toxicol. 2017;47:85–97. doi: 10.1080/10408444.2016.1211618. [DOI] [PubMed] [Google Scholar]
- 85.National Research Council, Science and Decisions: Advancing Risk Assessment, Committee on Improving Risk Analysis Approaches used by the U.S. EPA. Board on Environmental Studies and Toxicology. Division on Earth and Life Studies; Washington D.C.: 2009. [Google Scholar]
- 86.B.o.E.S.a. Toxicology, editor. National Research Council. Phthalates and cumulative risk assessment. National Academy Press; Washington, D.C.: 2008. [Google Scholar]
- 87.Rider CV, Furr JR, Wilson VS, Gray LE., Jr Cumulative effects of in utero administration of mixtures of reproductive toxicants that disrupt common target tissues via diverse mechanisms of toxicity. Int J Androl. 2010;33:443–462. doi: 10.1111/j.1365-2605.2009.01049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goodson WH, 3rd, Lowe L, Carpenter DO, Gilbertson M, Manaf Ali A, Lopez de Cerain Salsamendi A, Lasfar A, Carnero A, Azqueta A, Amedei A, Charles AK, Collins AR, Ward A, Salzberg AC, Colacci A, Olsen AK, Berg A, Barclay BJ, Zhou BP, Blanco-Aparicio C, Baglole CJ, Dong C, Mondello C, Hsu CW, Naus CC, Yedjou C, Curran CS, Laird DW, Koch DC, Carlin DJ, Felsher DW, Roy D, Brown DG, Ratovitski E, Ryan EP, Corsini E, Rojas E, Moon EY, Laconi E, Marongiu F, Al-Mulla F, Chiaradonna F, Darroudi F, Martin FL, Van Schooten FJ, Goldberg GS, Wagemaker G, Nangami GN, Calaf GM, Williams G, Wolf GT, Koppen G, Brunborg G, Lyerly HK, Krishnan H, Ab Hamid H, Yasaei H, Sone H, Kondoh H, Salem HK, Hsu HY, Park HH, Koturbash I, Miousse IR, Scovassi AI, Klaunig JE, Vondracek J, Raju J, Roman J, Wise JP, Sr, Whitfield JR, Woodrick J, Christopher JA, Ochieng J, Martinez-Leal JF, Weisz J, Kravchenko J, Sun J, Prudhomme KR, Narayanan KB, Cohen-Solal KA, Moorwood K, Gonzalez L, Soucek L, Jian L, D’Abronzo LS, Lin LT, Li L, Gulliver L, McCawley LJ, Memeo L, Vermeulen L, Leyns L, Zhang L, Valverde M, Khatami M, Romano MF, Chapellier M, Williams MA, Wade M, Manjili MH, Lleonart ME, Xia M, Gonzalez MJ, Karamouzis MV, Kirsch-Volders M, Vaccari M, Kuemmerle NB, Singh N, Cruickshanks N, Kleinstreuer N, van Larebeke N, Ahmed N, Ogunkua O, Krishnakumar PK, Vadgama P, Marignani PA, Ghosh PM, Ostrosky-Wegman P, Thompson PA, Dent P, Heneberg P, Darbre P, Sing Leung P, Nangia-Makker P, Cheng QS, Robey RB, Al-Temaimi R, Roy R, Andrade-Vieira R, Sinha RK, Mehta R, Vento R, Di Fiore R, Ponce-Cusi R, Dornetshuber-Fleiss R, Nahta R, Castellino RC, Palorini R, Abd Hamid R, Langie SA, Eltom SE, Brooks SA, Ryeom S, Wise SS, Bay SN, Harris SA, Papagerakis S, Romano S, Pavanello S, Eriksson S, Forte S, Casey SC, Luanpitpong S, Lee TJ, Otsuki T, Chen T, Massfelder T, Sanderson T, Guarnieri T, Hultman T, Dormoy V, Odero-Marah V, Sabbisetti V, Maguer-Satta V, Rathmell WK, Engstrom W, Decker WK, Bisson WH, Rojanasakul Y, Luqmani Y, Chen Z, Hu Z. Assessing the carcinogenic potential of low-dose exposures to chemical mixtures in the environment: the challenge ahead. Carcinogenesis. 2015;36(Suppl 1):S254–296. doi: 10.1093/carcin/bgv039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patel CJ, Bhattacharya J, Butte AJ. An Environment-Wide Association Study (EWAS) on type 2 diabetes mellitus. PLoS One. 2010;5:e10746. doi: 10.1371/journal.pone.0010746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Patel CJ, Chen R, Kodama K, Ioannidis JP, Butte AJ. Systematic identification of interaction effects between genome- and environment-wide associations in type 2 diabetes mellitus. Hum Genet. 2013;132:495–508. doi: 10.1007/s00439-012-1258-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lind PM, Riserus U, Salihovic S, Bavel B, Lind L. An environmental wide association study (EWAS) approach to the metabolic syndrome, Environ Int. 2013;55:1–8. doi: 10.1016/j.envint.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 92.Patel CJ, Manrai AK. Development of exposome correlation globes to map out environment-wide associations. Pac Symp Biocomput. 2015:231–242. [PMC free article] [PubMed] [Google Scholar]
- 93.Patel CJ. Analytic Complexity and Challenges in Identifying Mixtures of Exposures Associated with Phenotypes in the Exposome Era. Curr Epidemiol Rep. 2017;4:22–30. doi: 10.1007/s40471-017-0100-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brown TA, Holian A, Pinkerton KE, Lee JW, Cho YH. Early life exposure to environmental tobacco smoke alters immune response to asbestos via a shift in inflammatory phenotype resulting in increased disease development. Inhal Toxicol. 2016;28:349–356. doi: 10.1080/08958378.2016.1175526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.La Merrill M, Harper R, Birnbaum LS, Cardiff RD, Threadgill DW. Maternal dioxin exposure combined with a diet high in fat increases mammary cancer incidence in mice. Environ Health Perspect. 2010;118:596–601. doi: 10.1289/ehp.0901047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sathiya Priya C, Bhavani K, Anuradha CV. High-calorie diet inflates steatogenic effects of valproic acid in mice. Toxicol Mech Methods. 2016;26:112–121. doi: 10.3109/15376516.2015.1128034. [DOI] [PubMed] [Google Scholar]
- 97.Smith MT. Mechanisms of troglitazone hepatotoxicity. Chem Res Toxicol. 2003;16:679–687. doi: 10.1021/tx034033e. [DOI] [PubMed] [Google Scholar]
- 98.Threadgill DW, Miller DR, Churchill GA, de Villena FP. The collaborative cross: a recombinant inbred mouse population for the systems genetic era. ILAR J. 2011;52:24–31. doi: 10.1093/ilar.52.1.24. [DOI] [PubMed] [Google Scholar]
- 99.Churchill GA, Gatti DM, Munger SC, Svenson KL. The Diversity Outbred mouse population. Mamm Genome. 2012;23:713–718. doi: 10.1007/s00335-012-9414-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.French JE, Gatti DM, Morgan DL, Kissling GE, Shockley KR, Knudsen GA, Shepard KG, Price HC, King D, Witt KL, Pedersen LC, Munger SC, Svenson KL, Churchill GA. Diversity Outbred Mice Identify Population-Based Exposure Thresholds and Genetic Factors that Influence Benzene-Induced Genotoxicity. Environ Health Perspect. 2015;123:237–245. doi: 10.1289/ehp.1408202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yoo HS, Bradford BU, Kosyk O, Shymonyak S, Uehara T, Collins LB, Bodnar WM, Ball LM, Gold A, Rusyn I. Comparative analysis of the relationship between trichloroethylene metabolism and tissue-specific toxicity among inbred mouse strains: liver effects. J Toxicol Environ Health A. 2015;78:15–31. doi: 10.1080/15287394.2015.958417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.O’Connor A, Quizon PM, Albright JE, Lin FT, Bennett BJ. Responsiveness of cardiometabolic-related microbiota to diet is influenced by host genetics. Mamm Genome. 2014;25:583–599. doi: 10.1007/s00335-014-9540-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cichocki JA, Furuya S, Venkatratnam A, McDonald TJ, Knap AH, Wade T, Sweet S, Chiu WA, Threadgill DW, Rusyn I. Characterization of Variability in Toxicokinetics and Toxicodynamics of Tetrachloroethylene Using the Collaborative Cross Mouse Population. Environ Health Perspect. 2017;125:057006. doi: 10.1289/EHP788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bogue MA, Churchill GA, Chesler EJ. Collaborative Cross and Diversity Outbred data resources in the Mouse Phenome Database. Mamm Genome. 2015;26:511–520. doi: 10.1007/s00335-015-9595-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chuang JC, Cui H, Mason BL, Mahgoub M, Bookout AL, Yu HG, Perello M, Elmquist JK, Repa JJ, Zigman JM, Lutter M. Chronic social defeat stress disrupts regulation of lipid synthesis. J Lipid Res. 2010;51:1344–1353. doi: 10.1194/jlr.M002196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Balsevich G, Uribe A, Wagner KV, Hartmann J, Santarelli S, Labermaier C, Schmidt MV. Interplay between diet-induced obesity and chronic stress in mice: potential role of FKBP51. J Endocrinol. 2014;222:15–26. doi: 10.1530/JOE-14-0129. [DOI] [PubMed] [Google Scholar]
- 107.Cory-Slechta DA, Virgolini MB, Rossi-George A, Thiruchelvam M, Lisek R, Weston D. Lifetime consequences of combined maternal lead and stress. Basic Clin Pharmacol Toxicol. 2008;102:218–227. doi: 10.1111/j.1742-7843.2007.00189.x. [DOI] [PubMed] [Google Scholar]
- 108.Cory-Slechta DA, Virgolini MB, Rossi-George A, Weston D, Thiruchelvam M. Experimental manipulations blunt time-induced changes in brain monoamine levels and completely reverse stress, but not Pb+/-stress-related modifications to these trajectories. Behav Brain Res. 2009;205:76–87. doi: 10.1016/j.bbr.2009.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rossi-George A, Virgolini MB, Weston D, Cory-Slechta DA. Alterations in glucocorticoid negative feedback following maternal Pb, prenatal stress and the combination: a potential biological unifying mechanism for their corresponding disease profiles. Toxicol Appl Pharmacol. 2009;234:117–127. doi: 10.1016/j.taap.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Virgolini MB, Rossi-George A, Lisek R, Weston DD, Thiruchelvam M, Cory-Slechta DA. CNS effects of developmental Pb exposure are enhanced by combined maternal and offspring stress. Neurotoxicology. 2008;29:812–827. doi: 10.1016/j.neuro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Virgolini MB, Rossi-George A, Weston D, Cory-Slechta DA. Influence of low level maternal Pb exposure and prenatal stress on offspring stress challenge responsivity. Neurotoxicology. 2008;29:928–939. doi: 10.1016/j.neuro.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Cory-Slechta DA, Merchant-Borna K, Allen JL, Liu S, Weston D, Conrad K. Variations in the nature of behavioral experience can differentially alter the consequences of developmental exposures to lead, prenatal stress, and the combination. Toxicol Sci. 2013;131:194–205. doi: 10.1093/toxsci/kfs260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cory-Slechta DA, Weston D, Liu S, Allen JL. Brain hemispheric differences in the neurochemical effects of lead, prenatal stress, and the combination and their amelioration by behavioral experience. Toxicol Sci. 2013;132:419–430. doi: 10.1093/toxsci/kft015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Weston HI, Weston DD, Allen JL, Cory-Slechta DA. Sex-dependent impacts of low-level lead exposure and prenatal stress on impulsive choice behavior and associated biochemical and neurochemical manifestations. Neurotoxicology. 2014;44:169–183. doi: 10.1016/j.neuro.2014.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hattis D, Banati P, Goble R. Distributions of individual susceptibility among humans for toxic effects. How much protection does the traditional tenfold factor provide for what fraction of which kinds of chemicals and effects? Ann N Y Acad Sci. 1999;895:286–316. doi: 10.1111/j.1749-6632.1999.tb08092.x. [DOI] [PubMed] [Google Scholar]
- 116.Renwick AG, Lazarus NR. Human variability and noncancer risk assessment–an analysis of the default uncertainty factor. Regul Toxicol Pharmacol. 1998;27:3–20. doi: 10.1006/rtph.1997.1195. [DOI] [PubMed] [Google Scholar]
- 117.Daston G, Faustman E, Ginsberg G, Fenner-Crisp P, Olin S, Sonawane B, Bruckner J, Breslin W, McLaughlin TJ. A framework for assessing risks to children from exposure to environmental agents. Environ Health Perspect. 2004;112:238–256. doi: 10.1289/ehp.6182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ginsberg G, Slikker W, Jr, Bruckner J, Sonawane B. Incorporating children’s toxicokinetics into a risk framework. Environ Health Perspect. 2004;112:272–283. doi: 10.1289/ehp.6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.OEHHA. Technical Support document for the derivation of noncancer reference exposure levels. In: A.T.a.E. Branch, editor. Air Toxic Hot Spots Risk Assessment Guidelines. Office of Environmental Health Hazard Assessment, California Environmental Protection Agency; 2008. [Google Scholar]
- 120.Chiu WA, Slob W. A Unified Probabilistic Framework for Dose-Response Assessment of Human Health Effects. Environ Health Perspect. 2015;123:1241–1254. doi: 10.1289/ehp.1409385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Smith MT, Guyton KZ, Gibbons CF, Fritz JM, Portier CJ, Rusyn I, DeMarini DM, Caldwell JC, Kavlock RJ, Lambert JPF, Hecht SS, Bucher JR, Stewart BW, Baan RA, Cogliano VJ, Straif K. Key characteristics of carcinogens as a basis for organizing data on mechanisms of carcinogenesis. Environ Health Perspect. 2016;124:713–721. doi: 10.1289/ehp.1509912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Frost G, Darnton A, Harding AH. The effect of smoking on the risk of lung cancer mortality for asbestos workers in Great Britain (1971–2005) Ann Occup Hyg. 2011;55:239–247. doi: 10.1093/annhyg/meq089. [DOI] [PubMed] [Google Scholar]