

Abstract
Objective
To identify the clinical characteristics and genetic etiology of a family affected with hereditary spastic paraplegia (HSP).
Methods
Clinical, genetic, and functional analyses involving genome-wide linkage coupled to whole-exome sequencing in a consanguineous family with complicated HSP.
Results
A homozygous missense mutation was identified in the ACO2 gene (c.1240T>G p.Phe414Val) that segregated with HSP complicated by intellectual disability and microcephaly. Lymphoblastoid cell lines of homozygous carrier patients revealed significantly decreased activity of the mitochondrial aconitase enzyme and defective mitochondrial respiration. ACO2 encodes mitochondrial aconitase, an essential enzyme in the Krebs cycle. Recessive mutations in this gene have been previously associated with cerebellar ataxia.
Conclusions
Our findings nominate ACO2 as a disease-causing gene for autosomal recessive complicated HSP and provide further support for the central role of mitochondrial defects in the pathogenesis of HSP.
Hereditary spastic paraplegias (HSPs) are a clinically and genetically heterogeneous group of disorders characterized by neuronal degeneration of the corticospinal tracts, typically resulting in progressive weakness in the lower extremities and muscle spasms.1,2 Gait difficulties are the most common presenting symptom with a mean age at onset of 8 years.3 HSP can present as uncomplicated forms, limited to pyramidal tract (plus urinary) dysfunctions, or in complicated forms involving additional neurologic or neuropsychiatric signs and symptoms.4 Complicated HSPs are more often associated with either an autosomal recessive or X-linked pattern of inheritance.5
To date, more than 80 chromosomal loci have been linked to HSP.5–7 Nevertheless, in a substantial percentage of patients (38% in autosomal dominant HSP and 53% in autosomal recessive HSP), no causative mutations are identified.6 Here, we report the identification, through a genome-wide unbiased approach, of a homozygous mutation in the ACO2 gene (c.1240T>G p.Phe414Val) associated with HSP complicated by intellectual disability and microcephaly. ACO2 encodes the mitochondrial isoform of the aconitase enzyme, an iron-sulfur protein that catalyses the stereospecific isomerization of citrate to isocitrate. Lymphoblastoid cell lines (LCLs) derived from patients carrying the ACO2 p.Phe414Val mutation exhibited a marked decrease in aconitase enzyme activity and impaired mitochondrial respiration. Recessive mutations in this gene have been previously associated only with cerebellar ataxia in a very small number of patients. Therefore, our data markedly expand the associated phenotype and nominate ACO2 as responsible for autosomal recessive complicated HSP.
Methods
Family ascertainment
We ascertained a consanguineous Israeli family of Arab-Bedouin descent from Galilee, Northern Israel (figure 1A) with 2 siblings affected with complicated spastic paraplegia. There were no affected relatives in the previous generations, in keeping with an autosomal recessive pattern of disease inheritance.
Figure 1. Family pedigree and MRI.
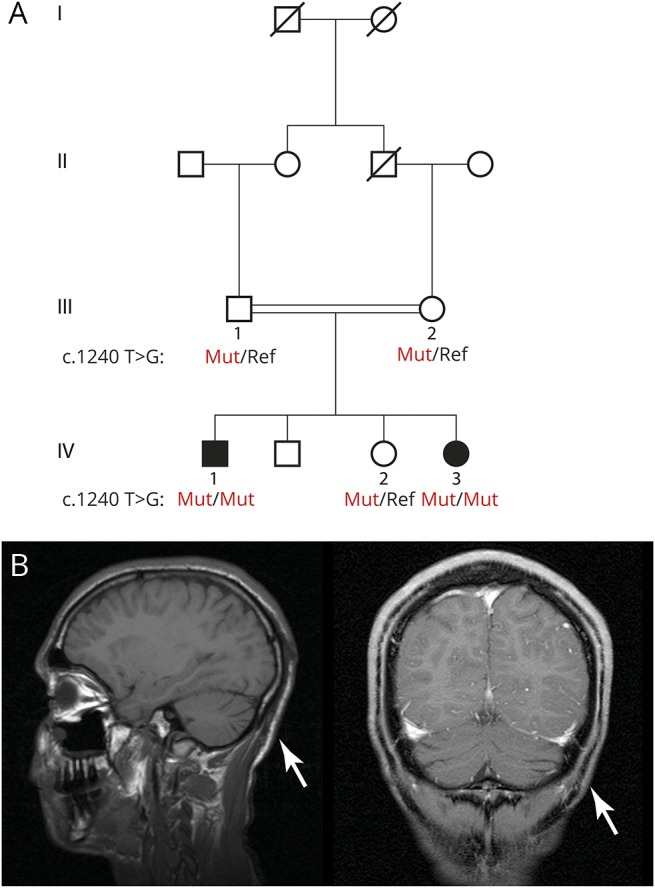
(A) Family pedigree. Shaded symbols indicate family members with complicated hereditary spastic paraplegia (HSP). Subjects of whom DNA was available are numbered. Males are represented with squares and females with circles. (B) Sagittal T1-weighted MRI (bottom left) and coronal T1-weighted MRI (bottom right) demonstrating mild cerebellar atrophy (arrows) in the proband (Ped ID IV-1).
Standard protocol approvals, registrations, and patient consents
This research was performed in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of the Sourasky University Medical Center, Tel-Aviv University, and the Israeli Ministry of Health. Written informed consent was obtained from the adult participants, with assent from the minors and written informed consent provided by their parents.
Genetic analyses
Genomic DNA was isolated from venous whole blood using standard protocols. Genome-wide search for copy number abnormalities was performed using Illumina HumanOmniExpress-24 BeadChip 700k SNP arrays and NEXUS discovery edition, version 7 (BioDiscovery, El Segundo, CA). Single nucleotide polymorphism (SNP) array data were used to perform a genome-wide linkage scan using Merlin under the assumption of autosomal recessive disease inheritance and full penetrance (penetrance of known HSP genes is estimated at 0.98).
Whole-exome sequencing in the 2 affected siblings was performed at ∼100× average coverage at the Center for Biomics of the Erasmus MC. The exome protocols included in-solution capturing (Agilent SureSelect V4 Human 50 Mb kit; Agilent Technologies) and paired-end sequencing (Illumina Hi-Seq 2000). Reads were aligned to the human reference genome version 19 using Burrows-Wheeler Aligner. SNPs and small insertions or deletions were called using the Genome Analysis Toolkit. Variant filtering was performed using Cartagenia software (Cartagenia Bench lab, Agilent Technologies).
Variants were filtered based on the following criteria: (1) located within the genomic loci supported by linkage analysis under an autosomal recessive model of disease inheritance (Logarithm of the Odds score >0); (2) predicted to affect protein coding (missense, nonsense, frameshift, and splice site); (3) called in both affected individuals; (4) absent from dbSNP129; (5) having a minor allele frequency (MAF) < 0.001 in public databases (Exome Variant Server 6500 [EVS6500], 1000 Genomes, and Exome Aggregation Consortium [ExAC]). The resulting candidate variant was confirmed using direct (Sanger) sequencing (PCR primers—forward: TGCTCACTGTCTCCTCCTGACC and reverse: GGACAATGCCACCCAGATCC) in all participating family members from whom DNA was available.
Lymphoblastoid cell lines
B lymphoblasts were isolated from venous whole blood of 3 family members (Pedigree ID IV-1, IV-2, and IV-3, figure 1A) and 2 unrelated healthy individuals and immortalized with Epstein-Barr virus using a standardized procedure.9 The resulting LCLs were maintained in Roswell Park Memorial Institute 1640 medium containing 15% fetal calf serum, 1% penicillin/streptomycin, 1% GlutaMAX, and 1% Minimum Essential Medium sodium pyruvate (Thermo Fisher Scientific). Functional analyses were performed to quantify aconitase enzyme activity, ACO2 protein levels, and cellular respirometry (e-appendix, links.lww.com/NXG/A35).
Results
Clinical studies
The proband (IV-1) is a 28-year-old man, diagnosed with complicated spastic paraplegia, severe intellectual disability, and microcephaly. He was born following a normal pregnancy with delivery at 40 weeks of gestation. His birth weight was 2,445 g (1st percentile) and head circumference 32 cm (3rd percentile). Throughout infancy, he had failure to thrive and was underweight; however, no vomiting or deterioration related to febrile illness was reported. He had seizures beginning at the age of 3 months, for which he initially received phenobarbital and later valproic acid until the age of 5 years, after which his seizures spontaneously remitted. He underwent surgery for a right inguinal hernia. He had recurrent otitis media as a toddler, and at the age of 4 years, he underwent adenoidectomy and myringotomy. In childhood, he experienced walking difficulties due to progressive spasticity of his lower limbs and did not achieve independent walking. At the age of 6 years, he underwent orthopedic surgeries for bilateral iliopsoas, adductor, hamstring, and Achilles tendon release. At no time in his development did he achieve spoken language other than a few words, but he did acquire the ability to communicate with his family members using vocalized sounds. His cognitive level was evaluated as severely disabled (estimated IQ 40–50). Currently, he is able to walk with assistance albeit with a spastic gait (video 1, links.lww.com/NXG/A36). He is able to eat unassisted, but needs help getting dressed. Hearing and vision are not impaired, including normal fundoscopic examination and auditory event-related potentials. EEG at age 11 was normal. Echocardiography was also normal. At the age of 19 years, MRI of the brain demonstrated mild atrophy of the cerebellum (figure 1B), without marked supratentorial abnormalities. Bilateral lower extremity EMG and nerve conduction velocity studies were normal.
At the most recent neurologic examination (March 2016), his head circumference measured 52 cm (3rd percentile). His pupils were equal and reactive to light, but eye tracking was abnormal. No facial weakness or tongue fasciculation was observed. He did not have scoliosis and had good control of his back. He had normal muscle strength in his upper limbs and normal deep tendon reflexes. However, supination of the upper limbs was limited, right more severely impaired than left. He had lower limb weakness and spasticity and a foot drop (proximal muscle strength 2/5 and distal strength 1/5). Deep tendon reflexes in the lower limbs were brisk with clonus and a bilateral extensor plantar response. He had limited hip adduction with limited range of knee extension and ankle dorsiflexion bilaterally. Pain, touch, and temperature sensation were normal. Vibration test at the ankle was normal. No cerebellar signs were evident, and his manual ability was normal. He had no increased urinary frequency or urgency.
The proband's 14-year-old sister (IV-3) also has complicated spastic paraplegia, moderate intellectual disability, and microcephaly. She was born after a normal pregnancy and unremarkable delivery. She presented with developmental delay evident from age 1 year. During childhood, she experienced episodic attacks of ataxia, tremor, altered consciousness, and behavioral changes related to febrile episodes. She began walking at the age of 2 years and spoke her first words at the age of 3 years. She did not progress to upright ambulation until age 3 years when she started limping on 1 leg and walking on her toes. From the age of 3 years, the proband's sister experienced recurrent episodes of encephalopathy, each followed by successive regression in psychomotor functioning. In the differential diagnosis, the possibility of a lysosomal storage disease was considered. As part of the workup to evaluate this possibility, electron microscopy studies of the liver and rectal biopsy tissue were performed. No specific abnormalities were detected in these specimens. Mild glycogen accumulation was observed in the liver biopsy, a finding that was considered benign and nonspecific. No abnormalities, including glycogen accumulation, were observed in the rectal biopsy tissue.
At the age of 8 years, she underwent orthopedic surgeries for bilateral hamstrings and Achilles tendon lengthening. At the age of 11 years, she was admitted to the hospital with acute mental change and treated with methylphenidate. At present, she walks with assistance and is able to eat unassisted. She has no history of seizures. She has moderate intellectual disability (estimated IQ 50–60), with basic reading, writing, and mathematics. CT of the head (2005), repeated brain MRI (2005, 2009, 2011), and magnetic resonance spectroscopy (2011) demonstrated a nonspecific isolated subcortical white matter signal abnormality in the left frontal lobe without additional gross abnormal findings. Hearing and visual function were normal, including fundoscopic examination and auditory event-related potentials. Echocardiography was normal. CSF analysis was also normal. Blood tests including blood gases, electrolytes, lactate, ammonia, very-long-chain fatty acids, isotransferrin electrophoresis, amino acids, biotinidase, and thyroid functions were normal. Lysosomal enzyme testing excluded metachromatic leukodystrophy, Krabbe disease, Tay-Sachs disease, and GM1 gangliosidosis. Bilateral lower extremity EMG and nerve conduction velocity studies were normal.
At the most recent neurologic examination (March 2016), she presented with no dysmorphologies other than bilateral syndactyly of the second and third phalanges of the feet. Both feet were in an equinovarus position. Her head circumference measured 50 cm (3rd percentile). Her pupils were equal and reactive to light, with horizontal end point nystagmus. There was no facial weakness. She exhibited stuttering speech. Hyperreflexia was evident in the upper limbs. Pronation and supination were limited in the upper limbs. She had lower limb weakness and spastic scissor gait and walked on her toes (video 2, links.lww.com/NXG/A37). She also had lordosis without crouch gait. Spasticity was present in both legs with limited adduction in both hips and ankle dorsiflexion bilaterally. Deep tendon hyperreflexia, as well as contralateral reflexes, was present in the lower limbs with sustained clonus and extensor plantar responses. Vibration and joint position sense were reduced in the distal lower extremities. Lower limb proximal muscle strength was 3/5, and distal strength was 2/5. No cerebellar signs were evident. She had no increased urinary frequency or urgency.
The other 2 siblings of the proband, a 17-year-old brother and 15-year-old sister, were neurologically intact without any evidence of HSP. Neither the parents nor any known extended family members exhibited signs or symptoms of HSP.
Genetic studies
No homozygous or compound heterozygous copy number variants were identified, which were shared in both affected siblings. Parametric linkage analysis under an autosomal recessive model revealed ∼23 Mb of candidate genomic regions distributed across chromosomes 2, 4, 5, 17, and 22 (table e-1, links.lww.com/NXG/A33).
Whole-exome sequencing revealed only 2 homozygous or compound heterozygous rare variants that were located within the linkage regions shared in both affected siblings and predicted to affect protein coding. The first is a homozygous variant in the gene ACO2 (c.1240T>G p.Phe414Val; figure 1A, table 1, and figure 2A). This missense variant was absent from all public databases (EVS6500, ExAC, and 1000 Genomes) and predicted to be deleterious by the SIFT, PolyPhen-2, likelihood ratio test (LRT), Mutation Taster, and Combined Annotation Dependent Depletion (CADD) (table 1) with strong evolutionary conservation (figure 2B). ACO2 is highly expressed in the human brain throughout neurodevelopment and adulthood (BrainSpan10). Variant genotyping of all available family members was confirmed by Sanger sequencing (figures 1A and 2A).
Table 1.
Identified exonic variants (GRCh37/hg19)
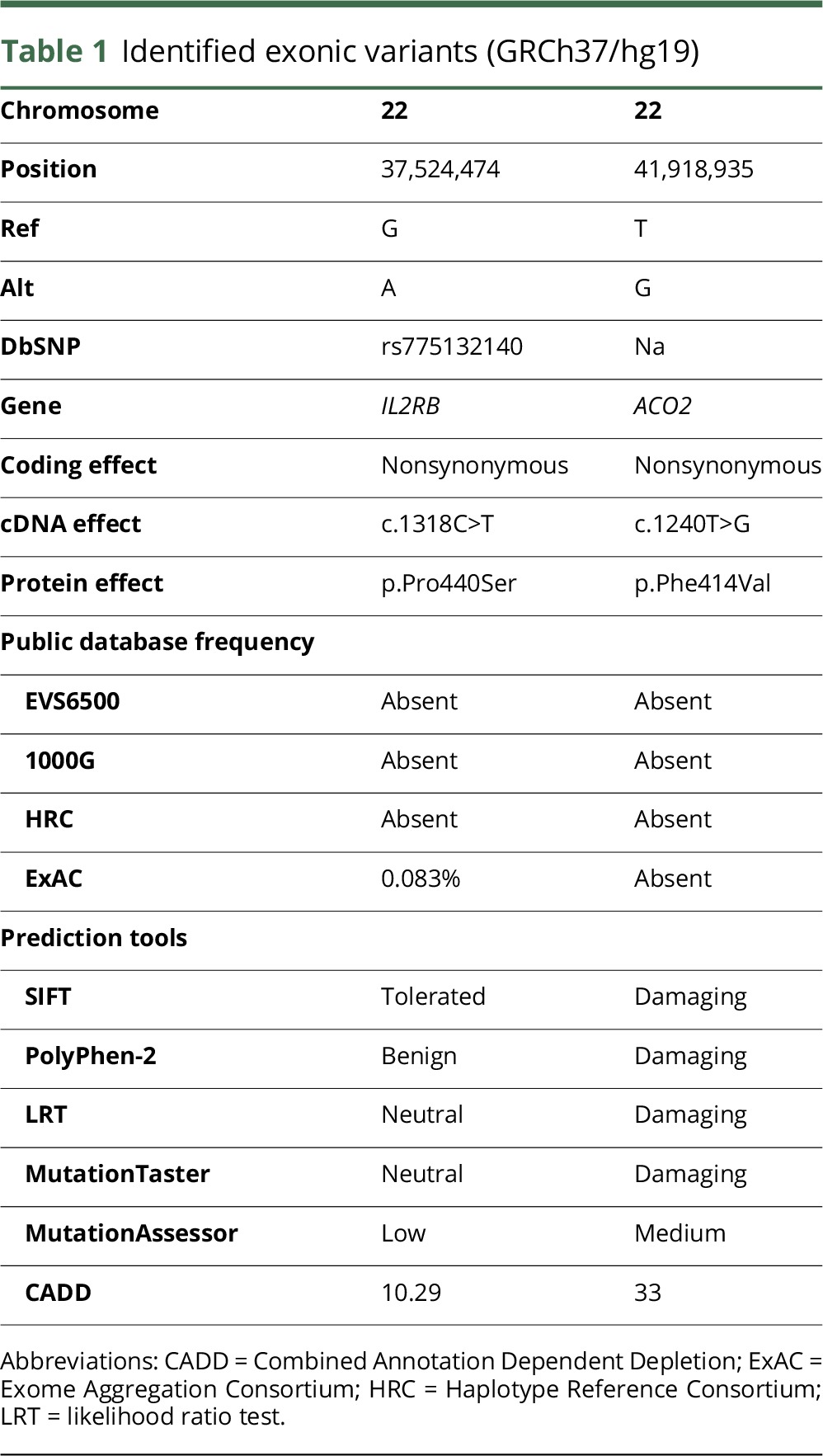
Figure 2. Sanger sequencing, conservation, and summary of known ACO2 mutations.

(A) Electropherograms indicating the homozygous ACO2 mutation (affected family members), the heterozygous mutation (both parents and unaffected sibling), and the reference sequence (unaffected, unrelated subject). (B) Amino acid conservation within the ACO2 protein homologs across species. (C) Homozygous (top) and compound heterozygous (bottom) ACO2 mutations identified to date in patients with neurodegenerative phenotypes.
The other segregating variant was in the IL2RB gene (c.1318C>T p. Pro440Ser), with an MAF of 8 × 10−5 (ExAC). This variant is predicted as benign by the SIFT, PolyPhen-2 LRT, MutationTaster, MutationAssessor, and CADD (table 1). IL2RB exhibits very low expression across all developmental ages in the human brain (BrainSpan10).
No additional cases of homozygous or compound heterozygous ACO2 mutations were identified by exome sequencing among subjects with recessive ataxia (n = 319), neurodegenerative disease (n = 2,000), or spastic paraplegia (n = 144).
Functional studies
LCLs were established from the homozygous carrier patients (Pedigree IDs IV-1 and IV-3, ACO2F414V/F414V), a heterozygous sibling (Pedigree ID IV-2, ACO2F414V/c), and 2 unrelated controls (ACO2c/c). Aconitase enzyme activity was significantly reduced in LCLs derived from the homozygous carriers, despite normal protein levels of ACO2 (figure 3, A and B). Given that multiple proteins, including ACO2, contribute to the aconitase enzymatic activity measured in whole-cell lysates, we sought to perform subcellular fractionation to examine mitochondrial aconitase activity, which is determined exclusively by ACO2. Consistent with selective impairment of ACO2 enzymatic activity, cellular fractionation revealed a highly specific defect in mitochondrial aconitase activity, whereas the cytoplasmic fraction that does not contain ACO2 revealed intact aconitase function (figure 3, C and D).
Figure 3. Mutation carrier–derived lymphoblastoid cell lines (LCLs) show decreased aconitase 2 activity and mitochondrial respiration deficiency compared with control LCLs.
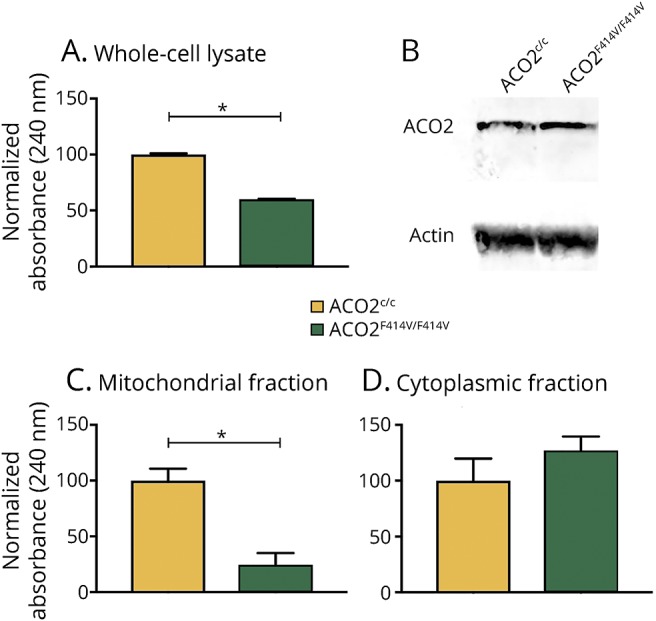
(A) Aconitase enzyme activity in LCL lysates of healthy controls (ACOc/c) and homozygous patient carriers (ACO2F414V/F414V) (p = 1.6 × 10−8). (B) Western blot of corresponding LCL lysates showing equal ACO2 protein levels in healthy controls and homozygous carrier patients. (C) Mitochondrial fraction of LCL lysates show a significant decrease in aconitase enzyme activity in homozygous patient carrier samples (p = 7.4 × 10−7). (D) ACO2 enzyme activity is unchanged in the cytoplasmic fraction of corresponding LCL lysates (p = 0.11). Data in (A, C and D) are expressed as mean ± SEM, representative experiment of 3 independent experiments, n = 6 time curve measurements per condition; results of the paired t test are indicated. *p < 0.0001.
Analysis of cellular respiration in isolated mitochondria revealed that LCLs derived from homozygous carriers have a significant reduction in complex I–linked adenosine diphosphate (ADP)-coupled (state 3) respiration and an attenuated maximal respiration following uncoupling with carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) (state 4u) (figure 4, A and B). Moreover, the respiratory control ratio (RCR), defined as the ADP-activated flux reflecting coupled oxidative phosphorylation capacity, was attenuated in the mitochondria of LCLs derived from homozygous and heterozygous carriers (figure 4, C and D).
Figure 4. Mitochondrial respiration is affected in lymphoblastoid cell lines (LCLs) of ACO2 mutation carriers.

Mitochondrial respiration was measured in isolated mitochondria from homozygous patient carriers (ACO2F414V/F414V), heterozygous ACO2 mutation carrier (ACO2F414V/c), and healthy control cell lines (ACO2c/c) by high-resolution respirometry. (A) State 3 (complex I–linked ADP-coupled respiration) with pyruvate and malate as substrates and (B) State 4u (maximum uncoupled respiration induced by FCCP after ATP synthase inhibition by oligomycin) are depicted. Respiratory efficiency is shown as (C) the respiratory control ratio of ADP-activated flux measuring coupled OXPHOS capacity (state 3/state 4) and (D) the efficiency of FCCP (maximum uncoupled respiration efficiency) (state 4u/state 4). Data are expressed as mean ± SD, n = 3–6 independent experiments per condition, with in total 6–11 measurements per condition; Tukey post hoc results for the one-way analysis of variance are indicated. *p < 0.05, **p < 0.01.
Discussion
We performed genome-wide linkage analysis and exome sequencing in a consanguineous family with 2 siblings affected by HSP, complicated by intellectual disability and microcephaly. This led to the identification of 2 candidate homozygous missense variants. On the basis of the variant frequency in public databases, prior association with neurodegenerative disease, in silico pathogenicity prediction, and expression in human brain, we considered the ACO2 variant (c.1240T>G, p.Phe414Val) as the likely causal mutation. The ACO2 p.Phe414Val mutation was absent from 319 patients with recessive ataxia, 2,000 patients with neurodegenerative disease, and 144 patients with spastic paraplegia, indicating the rareness of this form of cHSP. ACO2 encodes the protein aconitase 2, a critical enzyme in the tricarboxylic acid cycle (TCA) cycle. Through its enzymatic function, aconitase 2 catalyses the isomerization of citrate to isocitrate. The TCA cycle is the primary source of cellular metabolic energy and therefore strongly conserved evolutionarily. TCA cycle enzymopathies have previously been reported as the cause of a variety of cerebral encephalopathies, also often with muscular hypotonia and developmental delay as prominent presenting features.11
The corticospinal tract contains the longest neurons of the human body, posing strong metabolic demands including the requirement for long-distance transport of proteins and organelles to distal axon terminals. Genes responsible for the functional integrity of this complex machinery, including mitochondrial proteins, have been implicated in diverse forms of HSP.1 The finding that a homozygous mutation in ACO2 is associated with HSP provides further support for the central role of mitochondrial defects in the pathogenesis of spastic paraplegia. Notably, several genetic mutations associated with HSP have been functionally demonstrated to impair mitochondrial function: SPG7 (PGN), SPG13 (HSPD1), SPG28 (DDHD1), SPG31 (REEP1), SPG55 (C12ORF65), mitochondrial adenosine triphosphate (ATP) synthase 6 (mt-ATP6), IBA57, and OPA3. SPG7 encodes an m-AAA metalloprotease localized to the inner mitochondrial membrane where it functions to guard the integrity of proteins within the respiratory pathway. A mutation in SPG7 was found to cosegregate in a multiplex family with HSP.12 Patients with SPG7 have reduced mitochondrial respiration rates and are more sensitive to oxidative stress.13 Patients with mutations in HSPD1 have a pure HSP phenotype14 and impaired mitochondrial integrity.15 Patients with SPG28 can be affected with pure or complicated HSP. Mutations in REEP1 can cause reduced mitochondrial respiration and ATP production.16,17 Patients with SPG31 can have a pure or complicated form of HSP and have a dysfunction in mitochondrial energy production.18–20 A homozygous nonsense mutation in C12ORF65 (SPG55) has been described in a family from Japan with 2 patients affected by HSP complicated by optic atrophy and polyneuropathy. This mutation caused a reduction in mitochondrial protein synthesis and respiratory function.21 Familial maternally inherited late-onset HSP was described in association with a homoplasmic mutation in mt-ATP6 whereby the clinical severity of symptoms was strongly correlated with biochemical functioning of mt-ATP6.22 Recessive mutations in IBA57 associated with HSP complicated by optic atrophy and peripheral neuropathy have been found to cause a reduction in mitochondrial 4FE-4S protein expression.23 Moreover, induced pluripotent stem cell–derived neurons from patients with HSP resulting from SPG3A mutations were reported to have reduced mitochondrial motility.24 Lastly, exome sequencing revealed a segregating homozygous missense mutation in OPA3 segregating with optic atrophy, chorea, cerebellar ataxia, dystonia, and lower limb pyramidal symptoms.25 Heterozygous mutations in this gene are known to affect mitochondrial functioning.26
Although we cannot exclude that the c.1318C>T p.Pro440Ser variant in IL2RB plays a role in the phenotype, the very low gene expression in the human brain, functional prediction as benign, and weak evolutionary conservation argue strongly against the pathogenicity of this variant. Gene-level burden analysis previously demonstrated an association of rare coding variation in IL2RB with rheumatoid arthritis.27 However, to our knowledge, no known associations between IL2RB and diseases of the central or peripheral nervous system have been reported. Furthermore, functional evidence from LCLs demonstrates that the ACO2 p.Phe414Val mutation is responsible for a severe defect in mitochondrial aconitase activity and respiratory function. Notably, basal, maximum, and complex I–linked ADP-coupled (state 3) respiration were all affected only in homozygous carriers. By contrast, the RCR was also decreased in heterozygous carrier LCLs, indicating a subtle intermediate phenotype conferred by heterozygous carriership.
Homozygous and compound heterozygous mutations in ACO2 have been previously associated with varying combinations of cerebellar ataxia, retinopathy, and developmental delay (figure 2C and table e-2, links.lww.com/NXG/A34). A p.Ser112Arg missense mutation was first identified in 2 families with infantile cerebellar-retinal degeneration.28 Affected individuals exhibited developmental delay including severe psychomotor retardation, with an age at onset between 2 and 6 months. Brain MRI revealed cerebellar degeneration and white matter abnormalities (dysmyelination). Optic atrophy and retinal degeneration were readily identifiable in the setting of progressively severe visual impairment. Mitochondrial aconitase enzymatic activity was significantly reduced in lymphoblasts.
Through exome sequencing of sporadic cases of complicated optic neuropathy, 4 patients with homozygous or compound heterozygous mutations in ACO2 were identified. Two patients from 1 family had a compound heterozygous mutation (p.Leu74Val and p.Gly661Arg) and presented with decreased visual acuity in childhood and progression of ophthalmologic symptoms into the fourth decade of life. Brain MRI was not performed. A third patient (homozygous p.Gly259Asp mutation) was born with a low APGAR score, intermittent episodes of central apnea, and moderate cerebellar atrophy. The fourth patient (compound heterozygous mutation, p.Leu736Asn and p.Leu776Asnfs*49) exhibited ophthalmologic impairments with developmental delay and moderate cerebellar atrophy. Mitochondrial aconitase enzymatic activity was significantly reduced in patient fibroblasts.29
Furthermore, a 3-year-old sporadic patient with developmental delay, cerebellar dysfunction, and mild auditory neuropathy in the absence of retinal degeneration was reported with compound heterozygous mutations in ACO2 (c.1819C>T p.Arg607Cys and c.2135C>T p.Pro712Leu). Mitochondrial aconitase activity was also significantly reduced in patient fibroblasts.30
Lastly, an 18-year-old sporadic patient was described with childhood-onset ataxia, profound intellectual disability, intractable epilepsy starting at the age of 2 years, cerebellar atrophy peripheral neuropathy, and childhood-onset optic atrophy and pigmentary retinopathy. Metabolic and comparative genomic hybridization array screening did not identify causative genetic mutations. Using exome sequencing, a compound heterozygous mutation in ACO2 (c.2328-2331delGGAA p.Lys776Asnfs*49 and c.1091T>C p.Val364Ala) was identified.31
In contrast to previous reports of cases with ACO2 mutations, the patients in the family described in the current article developed severe HSP without cerebellar signs or retinal abnormalities, despite a similar decrease in mitochondrial aconitase activity and respiration. Previous reports of ACO2 mutations highlight an important degree of phenotypic heterogeneity (table e-2, links.lww.com/NXG/A34), with distinct genotypes being associated with optic neuropathy and/or cerebellar ataxia. The presently reported mutation (c.1240T>G p.Phe414Val) was associated with distinct phenotypic characteristics (HSP, microcephaly) in the absence of optic neuropathy or cerebellar ataxia. Notably, the proband exhibited mild atrophy of the cerebellar vermis and hemispheres, confirming previous reports of discordance between cerebellar atrophy and cerebellar ataxia among carriers of recessive ACO2 mutations. Moreover, the absence of optic neuropathy and cerebellar ataxia in recessive carriers of the p.Phe414Val mutation may result from its unique localization to the second alpha helix domain, which is distinct from any of the other previously reported ACO2 mutations (figure 2C).
Our findings nominate ACO2 as a disease-causing gene for autosomal recessive complicated HSP. Genetic screening of ACO2 should be considered for patients with complicated HSP in an effort to obtain a molecular diagnosis. Moreover, these results provide additional support for the central role of mitochondrial defects in the pathogenesis of HSP.
Glossary
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- ExAC
Exome Aggregation Consortium
- FCCP
carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- HSP
hereditary spastic paraplegia
- MAF
minor allele frequency
- LCL
lymphoblastoid cell line
- LRT
likelihood ratio test
- CADD
Combined Annotation Dependent Depletion
- RCR
respiratory control ratio
- SNP
single nucleotide polymorphism
- TCA
tricarboxylic acid cycle
Author contributions
Dr. Bouwkamp—study concept and design; acquisition, analysis, and interpretation of data; and manuscript writing. Dr. Afawi—study concept and design; acquisition and interpretation of data; and critical revision of the manuscript. Dr. Fattal-Valevski—acquisition and interpretation of data and critical revision of the manuscript. Dr. Krabbendam, Dr. Rivetti, and Dr. Masalha—acquisition, analysis, and interpretation of data. Dr. Quadri and Dr. Breedveld—analysis and interpretation of data. Dr. Mandel, Dr. Abu Tailakh, and Dr. Beverloo—acquisition of data. Dr. Stevanin, Dr. Brice, Dr. Van IJcken, and Dr. Vernooij—acquisition, analysis, and interpretation of data. Dr. Dolga and Dr. De Vrij—analysis and interpretation of data and critical revision of the manuscript. Dr. Bonifati—study concept and design; analysis and interpretation of data; and manuscript writing. Dr. Kushner—study concept and design; acquisition, analysis, and interpretation of data; and manuscript writing.
Study funding
This study was funded in part by the Netherlands Organization for Scientific Research (NWO) and Netherlands Organisation for Health Research and Development (ZonMw) to S.A.K. and V.B., the Royal Netherlands Academy of Arts and Sciences (KNAW) Sara van Dam Foundation to C.G.B. and Z.A., the NeuroBasic-PharmaPhenomics consortium to S.A.K., and Stichting ParkinsonFonds (The Netherlands) to V.B.
Disclosure
Dr. Bouwkamp and Dr. Afawi report study funding provided by the Royal Netherlands Academy of Arts and Sciences, Sara van Dam Foundation. Dr. Fattal-Valevski, Dr. Krabbendam, Dr. Rivetti, and Dr. Masalha report no disclosures. Dr. Quadri has received funding for travel from the International Parkinson & Movement Disorder Society and has served as a consultant of Stichting Centre for Human Drug Research. Dr. Breedveld, Dr. Mandel, Dr. Abu Tailakh, and Dr. Beverloo report no disclosures. Dr. Stevanin has received funding for travel and/or speaker honoraria from the Movement Disorder Society; holds patents with regard to Diagnosis of HSPs by identification of a mutation in the KIAA1840 gene or protein and diagnosis of HSPs by identification of a mutation in the ZFYVE26 gene or protein; and has received research support from European Community's Seventh Framework Program FP7/2007–2013 under agreement 2012-2017-305121 “Integrated European-omics research project for diagnosis and therapy in rare neuromuscular and neurodegenerative diseases (NEUROMICS),” French Agency for Research, Erare Programme 2013–2018, Erare programme 2016–2019, “Investissements d'avenir” ANR-10-IAIHU-06, Association Strumpell-Lorrain France 2016–2018, and Association Connaitre les Syndromes Cérébelleux 2015–2017. Dr. Brice has served on the scientific advisory boards of the Research Foundation—Flanders (FWO), European Research Council (ERC), and Bundesministeriums für Bildung und Forschung (BMBF) Berlin, Germany; has served on the editorial boards of Neurology and Clinical Neuroscience, Parkinsonism and Related Disorders, Brain, Neurodegenerative Diseases, The Cerebellum, and Neurogenetics; and has received research support from the French Research Agency, France Parkinson Association, Roger de Spoelberch Foundation (RDS), Fondation de France (FDF), and Fondation pour la Recherche Médicale (FRM). Dr. Van IJcken reports no disclosures. Dr. Vernooij has received research support from the Erasmus MC University Medical Center, Rotterdam, the Netherlands, and Netherlands Organization of Scientific Research (NWO)/ZonMW. Dr. Dolga and Dr. De Vrij report no disclosures. Dr. Bonifati has served on a scientific advisory board or Data Safety Monitoring Board of Stichting Parkinson Fonds, International Association of Parkinsonism and Related Disorders (IAPRD), and Erasmus MC Medical Research Advisory Committee (Mrace); has received travel funding and/or speaker honoraria from Sun Pharmaceutical Laboratories Limited; has served on the editorial board of Parkinsonism & Related Disorders, Current Neurology and Neuroscience Reports, Neurogenetics, Journal of Parkinson's, and Movement Disorders Clinical Practice; has served as a consultant of the International Parkinson and Movement Disorder Society and LSP Life Sciences Fund N.V., The Netherlands; and has received research support from the Erasmus Medical Center Rotterdam, The Netherlands, ZonMw, The Netherlands (under the aegis of the EU Joint Programme—Neurodegenerative Disease Research [JPND]), Stichting Parkinson Fonds (The Netherlands), and the Centre for Human Drug Research (CHDR), Leiden, The Netherlands. Dr. Kushner has received study funding and/or speaker honoraria from the Netherlands Organization for Scientific Research (NWO), Netherlands Ministry of Economic Affairs: Technology Foundation STW, European Research Council (ERC FP7 Marie Curie International Fellowship), and Netherlands Ministry of Finance. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol 2008;7:1127–1138. [DOI] [PubMed] [Google Scholar]
- 2.Deluca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol 2004;30:576–584. [DOI] [PubMed] [Google Scholar]
- 3.de Bot ST, van de Warrenburg BP, Kremer HP, Willemsen MA. Child Neurology: hereditary spastic paraplegia in children. Neurology 2011;26:75–79. [DOI] [PubMed] [Google Scholar]
- 4.Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet 1983;321:1151–1155. [DOI] [PubMed] [Google Scholar]
- 5.Lo Giudice T, Lombardi F, Santorelli FM, Kawarai T, Orlacchio A. Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp Neurol 2014;261:518–539. [DOI] [PubMed] [Google Scholar]
- 6.Tesson C, Koht J, Stevanin G. Delving into the complexity of hereditary spastic paraplegias: how unexpected phenotypes and inheritance modes are revolutionizing their nosology. Hum Genet 2015;134:511–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fink JK. Hereditary spastic paraplegia: clinical principles and genetic advances. Semin Neurol 2014;34:293–305. [DOI] [PubMed] [Google Scholar]
- 8.Fink JK. Hereditary Spastic Paraplegia Overview. Seattle: University of Washington; 2014. Available at: ncbi.nlm.nih.gov/books/NBK1509/. Accessed March 14, 2016. [Google Scholar]
- 9.Neitzel H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet 1986;73:320–326. [DOI] [PubMed] [Google Scholar]
- 10.Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, et al. . An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 2012;489:391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munnich A. Casting an eye on the Krebs cycle. Nat Genet 2008;40:1148–1149. [DOI] [PubMed] [Google Scholar]
- 12.Casari G, De Fusco M, Ciarmatori S, et al. . Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell 1998;93:973–983. [DOI] [PubMed] [Google Scholar]
- 13.Atorino L, Silvestri L, Koppen M, et al. . Loss of m-AAA protease in mitochondria causes complex I deficiency and increased sensitivity to oxidative stress in hereditary spastic paraplegia. J Cell Biol 2003;163:777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fontaine B, Davoine C-S, Dürr A, et al. . A new locus for autosomal dominant pure spastic paraplegia, on chromosome 2q24-q34. Am J Hum Genet 2000;66:702–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bross P, Naundrup S, Hansen J, et al. . The hsp60-(p.V98I) mutation associated with hereditary spastic paraplegia SPG13 compromises chaperonin function both in vitro and in vivo. J Biol Chem 2008;283:15694–15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bouslam N, Benomar A, Azzedine H, et al. . Mapping of a new form of pure autosomal recessive spastic paraplegia (SPG28). Ann Neurol 2005;57:567–571. [DOI] [PubMed] [Google Scholar]
- 17.Tesson C, Nawara M, Salih MAM, et al. . Alteration of fatty-acid-metabolizing enzymes affects mitochondrial form and function in hereditary spastic paraplegia. Am J Hum Genet 2012;91:1051–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beetz C, Schüle R, Deconinck T, et al. . REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain 2008;131:1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goizet C, Depienne C, Benard G, et al. . REEP1 mutations in SPG31: frequency, mutational spectrum, and potential association with mitochondrial morpho-functional dysfunction. Hum Mutat 2011;32:1118–1127. [DOI] [PubMed] [Google Scholar]
- 20.Battini R, Fogli A, Borghetti D, et al. . Clinical and genetic findings in a series of Italian children with pure hereditary spastic paraplegia. Eur J Neurol 2011;18:150–157. [DOI] [PubMed] [Google Scholar]
- 21.Shimazaki H, Takiyama Y, Ishiura H, et al. . A homozygous mutation of C12orf65 causes spastic paraplegia with optic atrophy and neuropathy (SPG55). J Med Genet 2012;49:777–784. [DOI] [PubMed] [Google Scholar]
- 22.Verny C, Guegen N, Desquiret V, et al. . Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion 2011;11:70–75. [DOI] [PubMed] [Google Scholar]
- 23.Lossos A, Stümpfig C, Stevanin G, et al. . Fe/S protein assembly gene IBA57 mutation causes hereditary spastic paraplegia. Neurology 2015;84:659–667. [DOI] [PubMed] [Google Scholar]
- 24.Zhu PP, Denton KR, Pierson TM, Li XJ, Blackstone C. Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum Mol Genet 2014;23:5638–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arif B, Kumar KR, Seibler P, et al. . A novel OPA3 mutation revealed by exome sequencing. JAMA Neurol 2013;70:783. [DOI] [PubMed] [Google Scholar]
- 26.Grau T, Burbulla LF, Engl G, et al. . A novel heterozygous OPA3 mutation located in the mitochondrial target sequence results in altered steady-state levels and fragmented mitochondrial network. J Med Genet 2013;50:848–858. [DOI] [PubMed] [Google Scholar]
- 27.Diogo D, Kurreeman F, Stahl EA, et al. . Rare, low-frequency, and common variants in the protein-coding sequence of biological candidate genes from GWASs contribute to risk of rheumatoid arthritis. Am J Hum Genet 2013;92:15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spiegel R, Pines O, Ta-Shma A, et al. . Infantile cerebellar-retinal degeneration associated with a mutation in mitochondrial aconitase, ACO2. Am J Hum Genet 2012;90:518–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metodiev MD, Gerber S, Hubert L, et al. . Mutations in the tricarboxylic acid cycle enzyme, aconitase 2, cause either isolated or syndromic optic neuropathy with encephalopathy and cerebellar atrophy. J Med Genet 2014;51:834–838. [DOI] [PubMed] [Google Scholar]
- 30.Sadat R, Barca E, Masand R, et al. . Functional cellular analyses reveal energy metabolism defect and mitochondrial DNA depletion in a case of mitochondrial aconitase deficiency. Mol Genet Metab 2016;118:28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Srivastava S, Gubbels CS, Dies K, et al. . Increased survival and partly preserved Cognition in a patient with ACO2 -related disease secondary to a novel variant. J Child Neurol 2017;32:840–845. [DOI] [PMC free article] [PubMed] [Google Scholar]