

Abstract
Pulmonary hypertension (PH) is characterized by increased vasoconstriction and smooth muscle cell hyperplasia driving pathological vascular remodeling of arterial vessels. In this short review, we discuss the primary source of reactive oxygen species (ROS) and nitric oxide (NO) relevant to PH and the mechanism by which dysregulation of their production contributes to PH. Specifically, hypoxia-induced PH is associated with diminished endothelial nitric oxide synthase (eNOS)-derived NO production and increased production of superoxide (O2.−) through eNOS uncoupling and defective mitochondrial respiration. This drives the inhibition of the NO/soluble guanylate cyclase (sGC) pathway and activation of the transcription factor hypoxia-inducible factor-1α (HIF-1α) with consequential dysregulation of the pulmonary vasculature. Therapeutics aimed at increasing NO or cGMP bioavailabilities are amenable to hypoxia disease-induced PH. Similarly, strategies targeting HIF-1α are now considered. Overall, pulmonary hypertension including hypoxia-induced PH offers unique opportunities for the rational development of therapeutics centered on modulating redox signaling.
Keywords: pulmonary hypertension, vascular remodeling, smooth muscle, hypoxia, nitric oxide, nitrite, mitochondria, reactive oxygen species, superoxide, hydrogen peroxide, HIF-1α
1. Introduction
In general, chronic hypoxia leads to pulmonary artery remodeling driven by smooth muscle proliferation and increase in wall thickness, which causes increase in flow resistance. This phenomenon leads to pressure overload of the heart right ventricle (RV) potentially causing its failure, the main driver of mortality in patients with chronic obstructive disease (Naeije, 2005). The remodeling process is initiated by oxygen sensors present in vascular cells that detect a decrease in partial pressure of oxygen in the blood (pO2), and then activate a signaling system that leads to acute constriction of pulmonary arteries (Prabhakar and Semenza, 2012). Eventually, this acute phase is “consolidated” by architectural remodeling of the vascular wall that perpetuates lumen narrowing (Prabhakar and Semenza, 2012). Interestingly, although RV dysfunction seems to be caused by pressure overload secondary to increase in pulmonary vascular resistance, new evidence suggest that these two phenomena could develop independently in such a way that RV dysfunction occurs even if PH is prevented (Ball et al., 2014).
Among many signaling molecules, the contribution of reactive oxygen species (ROS) and NO to the pathophysiology of PH is complex and partially elucidated in the context of hypoxia-mediated pulmonary hypertension (World Health Organization Class 3). Some of the cellular signals found to participate in the hypoxia-induced PH seem to be also relevant in other models of pulmonary vascular remodeling that occur under normoxia (Bonnet et al., 2006). In this short review, we will outline the salient results that provide a foundation to the delineation of the mechanisms by which aberrant production of NO and ROS may contribute to the pathogenesis of PH.
2. Nitric oxide and pulmonary hypertension
2.1 Relevant Nitric oxide biochemistry
Nitric oxide (NO) is a paracrine and autocrine messenger molecule that is derived from the five electron oxidation of L-arginine (Moncada et al., 1991). This reaction is catalyzed by nitric oxide synthase (NOS; Figure 1), of which 3 isoforms have been described in mammals, neuronal NOS (nNOS, NOS1), inducible NOS (iNOS, NOS2), and endothelial NOS (eNOS, NOS3) (Nathan and Xie, 1994). Tissues and cells conserve NO through nitrosylation and nitrosation of biomolecules and NO itself can be released upon reductive decomposition of functional groups such as S-nitrosothiols (Figure 1; (Feelisch et al., 2002). The reaction of NO with metals to form nitrosyl or nitroso species is also an important step determining many of the functional effects of NO such as the activation of soluble guanylate cyclase (sGC) or inhibition of cytochrome c oxidase (Grisham et al., 1999). In addition, the nitrosation of thiols by reactive species derived from NO serves as a posttranslational modification that modulates protein function such as certain caspases (Foster et al., 2003).
Fig. 1. Biological chemistry of nitric oxide relevant to pulmonary hypertension.
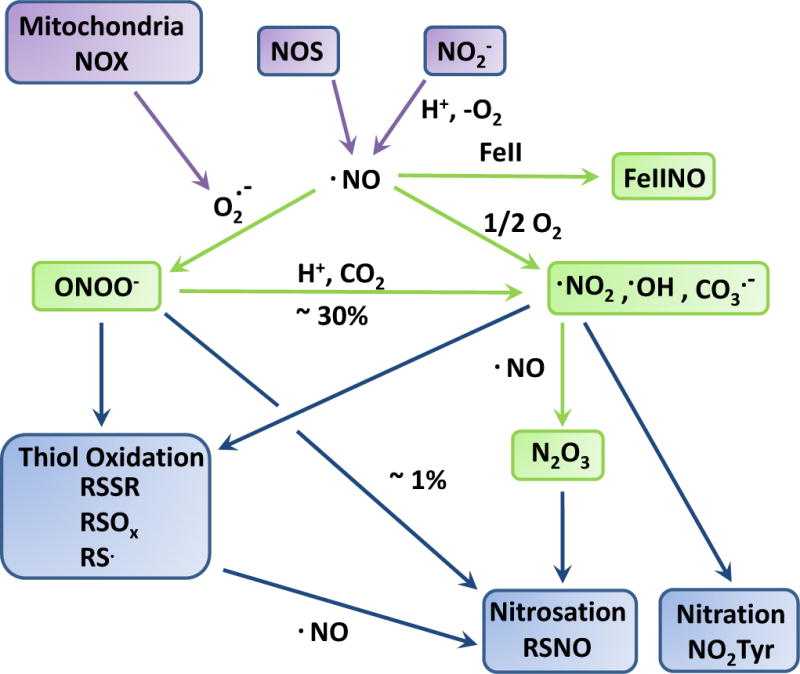
Important sources of nitric oxide (NO) include nitric oxide synthase (NOS) activity and nitrite (NO2−)-reduction under hypoxia and in acidic environments. NO reacts with superoxide (O2. −) to form peroxynitrite (ONOO−). Superoxide may be derived from multiple sources including mitochondrial and NADPH oxidase (NOX) activities. Peroxynitrite - upon protonation or combination with carbon dioxide (CO2) – yields a number of free radicals, including nitrogen dioxide (.NO2), and the hydroxyl (.OH) and carbonate CO3. −) radicals. NO reacts with metals such as iron (FeII) to form a metal-nitrosyls (FeIINO) such as the one found in soluble guanylate cyclase. Nitric oxide also reacts with molecular oxygen to form nitrogen dioxide and dinitrogen trioxide (N2O3). All together these species may be involved in oxidative (such as thiol oxidation), nitrosation (thiol nitrosation, RSNO), and nitration (tyrosine nitration, NO2Tyr) reactions with biological targets. See Text for details.
An alternative source of NO is derived from the reduction of nitrite (NO2−; Figure 1) that proceeds at low pH and under hypoxia (Zweier et al., 1995). The significance of this reaction is as an alternative source of NO at sites where NOS might be inhibited due to the lack of molecular oxygen but where hypoxic and acidic reduction of diet or pharmacologically-derived NO2− is possible. Deoxyhemoglobin is an important site upon which NO2− is reduced to NO, although the chemical pathway and mechanism by which NO may escape erythrocytes still need clarification (Huang et al., 2005). Whether non-erythrocytic cells mediate hypoxic NO2− reduction in the lung is also under investigation with multiple intracellular activities identified consisting of additional globins (Myoglobin (Rassaf et al., 2014), Neuroglobin (Tiso et al., 2011), and Cytoglobin (Li et al., 2012)) and molybdenum-containing proteins (xanthine dehydrogenase (Li et al., 2001), sulfite oxidase (Wang et al., 2015), aldehyde oxidase and mitochondria amidoxine reducing component (Sparacino-Watkins et al., 2014)) potentially contributing to this activity in the pulmonary vasculature.
One of the most significant reactions of nitric oxide (NO) is its combination with superoxide (O2. −) at a diffusion-limited rate (Figure 1; (Beckman et al., 1990)). The product of this reaction, peroxynitrite (ONOO−/ONOOH), is a one and two electron oxidant, which modifies DNA, proteins, lipids, and sugars by way of oxidation, nitration, and nitrosation (Beckman et al., 1990, Beckman et al., 1992, Gow et al., 1997). The biochemical reactivity of peroxynitrite in physiologically relevant settings may be dominated by its reaction with thiols and transition metals but also with excess carbon dioxide to yield a nitrosoperoxocarboxylate anion (ONOOCO2−) that partially decomposes to nitrogen dioxide and the carbonate radical (Denicola et al., 1996). Under most conditions, peroxynitrite might not coexist with NO or O2. − because superoxide dismutase and oxyhemoglobin insure limited availability of these molecules in excess of peroxynitrite. However, during conditions characterized by high NO synthase activity and multiple cellular sources of O2. −, peroxynitrite-mediated reactions combined with those of excess NO or O2. − may become important. The formation of peroxynitrite in vivo, inferred from the formation of stable footprints such as 3-nitrotyrosine, represents an important mediator of tissue injury and dysfunction that limits NO bioavailability (Beckman and Koppenol, 1996).
2.2 Nitric oxide signaling in pulmonary hypertension
The bioavailability and signaling of NO is decreased in experimental models and in patients with PH (Xue and Johns, 1995, Kharitonov et al., 1997, Fagan et al., 1999, Quinlan et al., 2000). For example, Giaid and Saleh provided some evidence that the expression of eNOS was decreased in the vascular endothelium of pulmonary arteries in a cohort of patients with pulmonary hypertension with different grades of arteriopathy (Giaid and Saleh, 1995). The dysfunction is usually considered to be decreased vaso-protection including depleted vasodilatory, anti-migratory, and anti-proliferative functions. However, loss of eNOS is also associated with a decrease in muscularization of small pulmonary vessels during chronic hypoxia in the mouse due to a decrease in proliferative capacity (Quinlan et al., 2000). The underlying mechanism has been relatively well-studied in animal models and is usually considered to be multifactorial through changes in eNOS expression and uncoupling (Zhao et al., 2009), alteration in L-arginine metabolism (Block et al., 1995, Xu et al., 2004), and increased NO consumption through O2. −. Although conflicting results exist regarding the levels of eNOS expression during PH, it is possible that – if increased - eNOS in the context of PH is uncoupled, meaning that a fraction of its activity is diverted towards the production of other reactive species such as O2. −. Increased ROS production is associated with endothelial dysfunction and NADPH oxidase (NOX)-derived O2. − limits NO production and downstream signaling through eNOS uncoupling (Landmesser et al., 2003). Overall, this provides conditions conducive to decrease NO bioavailability and increase oxidative and nitrative stress through the formation of peroxynitrite or metal-catalyzed nitration. Insufficient stimulation of sGC by NO reduces cGMP production and downstream effector activation such as cGMP-dependent protein kinase (PKG) (Zhao et al., 2009). In addition, downstream nitration or oxidation of target molecules such as PKG may lead to amplification of the inhibitory effect associated with NO inactivation (Zhao et al., 2009).
2.3 Therapeutics based on direct targeting of cGMP and NO surrogates’ delivery
With a key role for the dysregulation of NO signaling in PH (including WHO Group 3 PH), therapeutic strategies aimed at restoring the NO/cGMP pathway have received increasing attention. A novel class of drugs that directly stimulates sGC independently of NO is now aggressively pursued for the treatment of PH. One such molecule, riociguat (Figure 2), has been approved for the treatment of pulmonary artery hypertension (PAH) and chronic thromboembolic pulmonary hypertension (CTEPH)(Wardle et al., 2016, Tsugu et al., 2016). Riociguat has also shown significant therapeutic effects in patients with other types of PH including interstitial lung disease PH and PH associated with chronic obstructive pulmonary disease (COPD) (Benza et al., 2016). The NO/cGMP pathway may also be targeted by inhibitors of phosphodiesterase type 5 (PDE-5) causing inhibition of the breakdown of cGMP by PDE-5. One such molecule, Tadalafil (Figure 2), has been shown to reduce clinical worsening and improve hemodynamic outcomes in patients with PAH and is use in the clinic in this specific setting (Galie et al., 2009).
Fig. 2. Mechanism of action of NO in the vasculature and therapeutic targets.
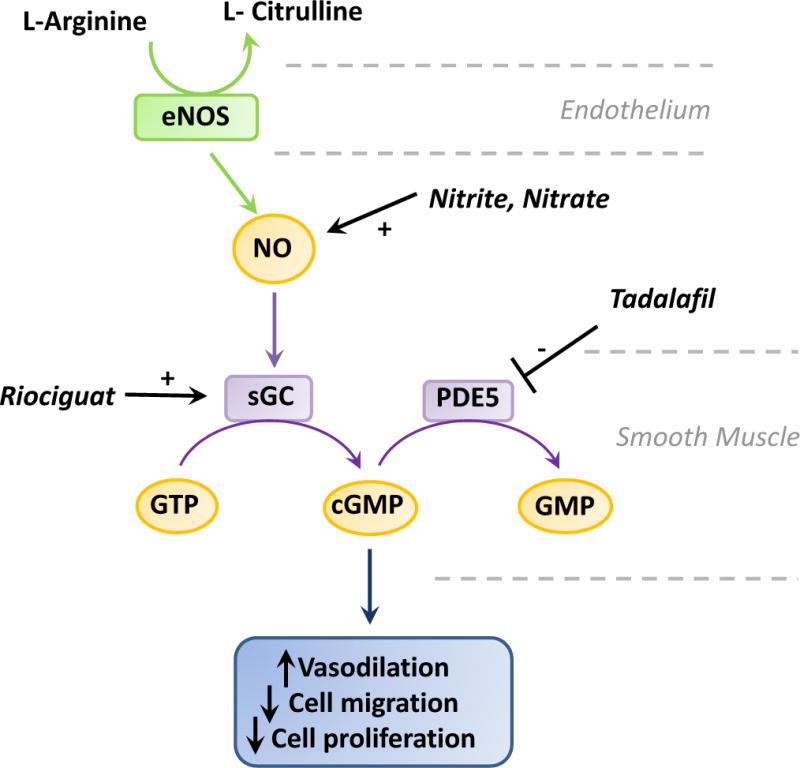
Nitric oxide (NO) is generated from the oxidation of L-arginine to L-citrulline by endothelium Nitric Oxide Synthase (eNOS). NO diffuses into target cells such as smooth muscle cells to bind and activate soluble guanylate cyclase (sGC), which in turn generates cyclic GMP (cGMP) from GTP to promote vasodilation, and inhibit cell migration and proliferation. The signal is turned off upon cGMP hydrolysis to GMP by phosphodiesterase 5 (PDE5). Inhibition of this pathway is thought to contribute to the pathogenesis of pulmonary hypertension (PH). Increase NO biovailability through nitrite or nitrate delivery, stimulating sGC with Riociguat, or inhibiting PDE5 with Tadalafil all provides therapeutic means for the treatment of certain type of PH. See Text for details.
A number of preclinical studies have also indicated the beneficial effect of providing an alternate source of NO in the form of pharmacological delivery of NO2− or nitrate (NO3−) to alleviate PH (Zuckerbraun et al., 2010, Baliga et al., 2012, Pankey et al., 2012). In the context of hypoxia-induced PH in the mouse, inhaled nebulized NO2− inhibits and reverse pre-establish PH and high right ventricular pressure. In this case, the effect of NO2− has been shown to be inhibited by a xanthine oxidase inhibitor or through diet-mediated inhibition of molybdenum-containing enzymes (Zuckerbraun et al., 2010). Dietary NO3− (which can be reduced to NO2−through the entero-salivary cycle) also reduced pulmonary vascular remodeling in mouse exposed to hypoxia for three weeks (Baliga et al., 2012). Interestingly, this effect required eNOS in addition to xanthine oxidase, suggesting a role for eNOS as a nitrite reductase. In a recent early phase II pilot study, Simon and coworkers have shown that inhaled NO2− provides some hemodynamics improvement in a small group (n=6) of patients with PH due to lung disease or hypoxia, although these effects were less than those observed in patients with WHO Group 2 PH (Simon et al., 2016).
3. Reactive oxygen species and pulmonary hypertension
3.1 Significant sources of ROS
While signaling pathways centered on NO bioavailability are key therapeutic targets for the treatment of PH, the production of ROS is also an essential contributor to hypoxia-induced PH. In this case, cellular respiration is an important source of ROS (Waypa et al., 2016). Accordingly, ROS role in the development of PH is strongly suggested by studies showing profound alteration in mitochondrial structure and function in that context. Using human and rodent models, Ryan and colleagues found evidence of mitochondrial fragmentation (Ryan et al., 2013), which is associated with a decrease in the expression of mitofusin-2 (MFN2), a molecular regulator that promotes the fusion of mitochondria into long tubular structures. Also, PGC1α, a transcriptional activator of mitochondrial biogenesis was found to be downregulated in that context. Adenoviral overexpression of MFN2 increased mitochondrial fusion, decreased proliferation, lessened the severity of PH, and improved exercise capacity in the rodent model (Ryan et al., 2013). These results suggest that decreases in MFN2 and PGC1α contribute to pulmonary vascular remodeling and provide indirect evidence of a potential role of mitochondrial-derived ROS in the pathophysiology of PH.
Although not completely elucidated in one single model, compelling evidence from different laboratories indicates that low oxygen stimulation leads to mitochondrial production of ROS, which serves as an activator of prolyl-4-hydroxylases which in turn induce activation of Hypoxia-inducible factor 1 (HIF-1), a necessary event that triggers vascular remodeling and narrowing of the pulmonary arteries (Figure 3). We present, in the following paragraphs, a concise review of the current evidence supporting this mechanism.
Fig. 3. Potential mechanisms of hypoxia-induced pulmonary hypertension.
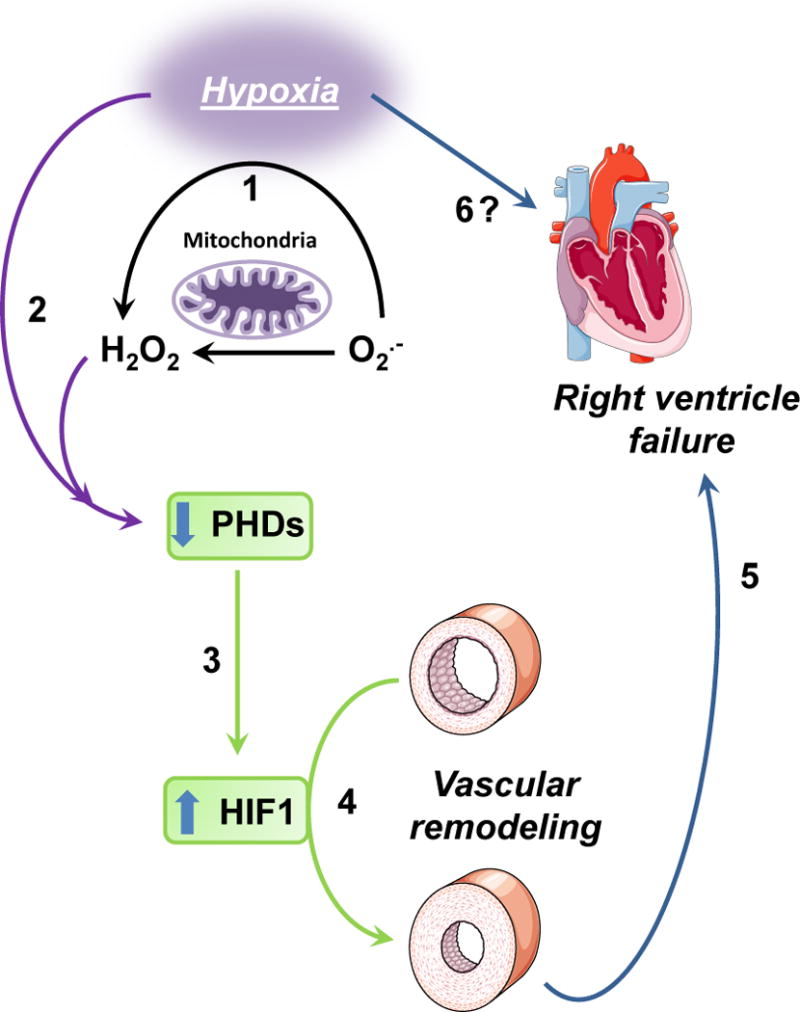
1: hypoxia causes cells to release superoxide (O2.−) which is converted to hydrogen peroxide (H2O2). 2: Both low oxygen and ROS production cause a reduction in hydroxylase activity of PHDs. 3: Lower PHDs activity causes a reduction of HIF-1α proline-402 and proline-564 hydroxylation, which leads to dissociation from VHL and stabilization of the transcriptional activity of HIF-1α. 4: Increase in the transcriptional activity of HIF-1α causes pulmonary vascular remodeling. 5: Increased pulmonary vascular resistance leads to right ventricle failure. 6: Hypoxia could also contribute to right ventricle remodeling and failure independent of HIF-1α (see Ball et.al.).
During hypoxia, mitochondria from vascular cells release superoxide (O2. −) from complex III to the intermembrane space, where it is converted to hydrogen peroxide (H2O2) by superoxide dismutase (Thompson, 2016, Waypa et al., 2016). The H2O2 then enters the cytosol, where it activates multiple responses contributing to smooth muscle contraction and remodeling. The mechanism of hypoxia-driven ROS generation was first suspected using pulmonary artery cell homogenates, which suggested that superoxide generation increased during hypoxia in an effect that was inhibited by diphenylene iodonium, a flavoptrotein inhibitor of NADPH oxidase but not by the mitochondrial inhibitor myxothiazol, which blocks electron entry into complex III (Marshall et al., 1996). Chandel et al first demonstrated that mitochondrial ROS signals control gene transcription in hypoxia (Chandel et al., 1998). Specifically, using mitochondrial inhibitors, ρ0 cells lacking a functional electron transport chain (ETC), and ROS-sensitive fluorescent chemical probes, they showed that under hypoxia ETC is required for ROS-dependent stabilization of the HIF-1α transcription factor subunit (Chandel et al., 1998). Also, the same group found that even though anoxia is the most extreme form of hypoxia, these two stimulations operate differently on downstream signaling: the activation of HIF-1 under hypoxia requires mitochondrial ETC whereas, under anoxia, HIF-1 is activated without involvement of ETC (Schroedl et al., 2002).
The specific mechanisms linking hypoxia and ROS generation are not completely understood, but evidence indicates that they involve oxidation of complex III, which requires cytochrome c and leads to the formation of O2. − that is later ejected to the intermembrane space due to electrical gradient. Indeed, when cytochrome c is absent, complex III remains fully reduced, which prevents ROS generation under hypoxia and are unable to stabilize HIF-1α (Mansfield et al., 2005). These findings implicate electron flux through complex III as a critical event in the detection of hypoxia in cells (Mansfield et al., 2005). Waypa et al. demonstrated that hypoxic pulmonary vasoconstriction required electron flux through complex III, and that increases in ROS generation were responsible for eliciting the hypoxic response (Waypa et al., 2010, Waypa et al., 2013). Specifically, these authors demonstrated that acute production of ROS during hypoxia pulmonary artery smooth muscle cells depends on the Rieske iron-sulfur protein subunit of complex III, as reflected by PH attenuation in animals with deletion of this gene using a Cre/loxP system (Waypa et al., 2013).
3.2 Signaling pathways associated with increased ROS in pulmonary hypertension
3.2.1 ROS cause HIF-1 activation
HIF-1 is a highly conserved transcription factor present in almost all cell types (Prabhakar and Semenza, 2012). It is tightly regulated by O2 availability, and modulates the expression of hundreds of genes. HIF-1 exists as a heterodimer, consisting of HIF-1α and HIF-1β subunits. HIF-1β is constitutively expressed, whereas HIF-1α is found at very low levels under normoxic conditions (Shimoda and Semenza, 2011). In this context, HIF-1α protein is ubiquitinated and degraded by the proteasomal pathway; however, acute exposure of pulmonary arterial smooth muscle cells (PASMCs) or endothelial cells (ECs) to hypoxia (1% O2) causes increased HIF-1α protein levels and HIF-1 DNA-binding activity. Thus, HIF-1α confers sensitivity and specificity for hypoxic induction of HIF-1 transcriptional activity (Prabhakar and Semenza, 2012, Shimoda and Semenza, 2011).
Under normoxia, HIF-1 is associated to von Hippel-Lindau protein (VHL), which recruits an E3-ubiquitin protein ligase Elongin 2 and 3, Cullin 2, and RBX1(Kamura et al., 2000, Maxwell et al., 1999). Binding of VHL depends on hydroxylation of HIF-1 proline-402 and 564 in well-oxygenated cells (Ivan et al., 2001). Three prolyl-4-hydroxylase domain proteins (PHDs) that hydroxylate proline-402 and 564 in an O2-dependent manner are identified in mammalian cells (Ivan et al., 2002, Epstein et al., 2001). These proteins, known as PHD1, PHD2, and PHD3 are members of a superfamily of dioxygenases that contain Fe(II) in their catalytic center and utilize O2 and α-ketoglutarate as substrates. Reduction of Fe(III) to Fe(II) in the catalytic center by ascorbate is required for a subsequent catalytic cycle. The observed reduction in hydroxylase activity under hypoxic has been proposed to be due to substrate (O2) limitation (Epstein et al., 2001, Chua et al., 2010) and/or by an increase in mitochondrial production of ROS that may oxidize Fe(II) and inactivate the PHDs (Brunelle et al., 2005, Guzy et al., 2005, Mansfield et al., 2005). Thus, hypoxia can lead to PDHs deactivation and HIF-1 stabilization via a direct effect of either low oxygen, or also ROS on PDHs (Figure 3). Importantly, at least in the acute phase, the generation of ROS appears to be a necessary step in the process of PH under hypoxia (Waypa et al., 2013).
3.2.2 HIF-1 mediates hypoxia-driven pulmonary hypertension
Seminal work by Shimoda and coworkers (Shimoda et al., 2001) established the effects of chronic hypoxia (CH) on heterozygous mice lacking one copy of the HIF-1 gene (homozygous animals could not be used due to intrauterine lethality). Compared with wild-type control animals, heterozygous HIF-1a mice demonstrated impaired lung vascular remodeling in chronic hypoxia and attenuated RV hypertrophic responses (Shimoda et al., 2001, Kline et al., 2002, Yu et al., 1999, Shimoda et al., 2006). This was associated with lower level of vascular smooth muscle hypertrophy, attenuated up-regulation of transient potential receptor proteins and Na+/H+ exchanger-isoform 1, and failure to suppress the expression of plasma membrane K1 channels during CH (Shimoda et al., 2001). Recently, using a Cre/loxP system smooth muscle-specific conditional deletion of HIF-1, Ball et al demonstrated that HIF-1 is critical as a mediator of pulmonary arterial remodeling under hypoxia. Interestingly, they also found that loss of HIF-1 function in smooth muscle did not affect hypoxic cardiac remodeling (Ball et al., 2014); suggesting that the cardiac hypertrophy response is not directly coupled to the increase in pulmonary artery pressure (Figure 3). This last finding challenges the “hemodynamic dogma” that states that the right ventricular hypertrophy and eventual failure depend purely on pressure overload due to increase of pulmonary vascular resistance and suggests that ventricular and vascular remodeling are distinct and somewhat independent processes. Similar challenges are emerging regarding the left ventricular remodeling in connection to systemic hypertension (Popov et al., 2014).
3.2.3 Mechanisms of HIF1-driven vascular wall remodeling
Chronic hypoxia induces functional and structural changes in the endothelial and smooth muscle cells, and fibroblasts that make up the intima, media, and adventitia of pulmonary arterial wall thus contributing to pulmonary hypertension (Morrell et al., 2009). The effect of hypoxia on vascular remodeling has been mostly studied in PASMC. Acute hypoxia leads to an increase in intracellular calcium [Ca2+]i that is reversible upon reoxygenation. In contrast, chronic hypoxia causes a sustained increase on [Ca2+]i which remains elevated even after return to normoxia (Shimoda et al., 2000). This effect is mediated by store-operated Ca2+ channels, which are activated by depletion of intracellular Ca2+ stores during chronic hypoxia (Wang et al., 2006). These channels are composed of transient receptor potential (TRP) proteins, which are under the control of HIF-1 (Wang et al., 2006). Indeed, infection with an adenovirus encoding a constitutively active form of HIF-1α increases TRPC1 and TRPC6 expression under non-hypoxic conditions.
Hypoxia inhibits opening of voltage-gated K+ channels Kv1.5 and Kv2.1, which contributes to PASMC depolarization (Archer et al., 1998). The expression of these channels is also decreased in PASMCs subjected to chronic hypoxia in vivo or ex vivo (Reeve et al., 2001, Yuan et al., 1998), and these changes in gene expression are also HIF-1α dependent (Bonnet et al., 2006). In addition to increased [Ca2+]i, chronic hypoxia also results in increased intracellular pH (pHi) in PASMCs, an effect that is due to HIF-1-dependent expression of the sodium-hydrogen exchanger NHE1 (Shimoda et al., 2006). Increased [Ca2+]i and pHi contribute to the activation of signal transduction pathways that promote PASMC hypertrophy and hyperplasia, which leads to the medial thickening of pulmonary arterioles, which is the pathological hallmark of hypoxia-induced PAH. Indeed, exposure of WT but not Hif-1−/− mice to chronic hypoxia induces PASMC alkalinization and hypertrophy (Shimoda et al., 2006, Shimoda et al., 2001).
The demonstration that mice partially deficient in HIF-1α expression are protected against the development of PAH suggests that pharmacological inhibition of HIF-1 may be of therapeutic benefit in this clinical context. This hypothesis was tested by the daily administration of digoxin, a cardiac glycoside that has been used to treat congestive heart failure and cardiac arrhythmias for decades, which inhibits the synthesis of HIF-1α protein (Zhang et al., 2008). Treatment with digoxin attenuated the development of right ventricular hypertrophy and prevented the changes in pulmonary vascular [Ca2+]i and pHi, remodeling, and pressure that occur in mice exposed to chronic hypoxia (Abud et al., 2012).
The role of HIF-1 in the pathogenesis of PH is not restricted to hypoxia-induced PH. The spontaneous development of PAH in fawn-hooded rats is associated with increased HIF-1α expression, HIF-dependent reductions in K+ currents and Kv1.5 expression, increased PDK1 expression, and a switch from oxidative to glycolytic metabolism in pulmonary artery smooth muscle cells (Bonnet et al., 2006). These metabolic changes appear to play a critical role in the pathogenesis of PAH because treatment of animals with dichloroacetate, an inhibitor of PDK1, leads to regression of PAH (Michelakis et al., 2002).
4. Conclusion
Dysregulation in NO and ROS production have been established in the context of PH including hypoxia/disease-induced PH and there is now sufficient evidence to indicate that decreased NO bioavailability and increased mitochondrial-derived O2.−/H2O2 production are central to the pathogenesis of PH. Whether these two arms of redox biology converge on common molecular pathways and pathologies is still debatable and will require additional investigation. What is clear is that the elucidation of the signaling pathways downstream of NO and ROS and their pathophysiological alterations will continue to provide a foundation for the rational design and clinical implementation of new therapies targeting PH.
Acknowledgments
This work was supported in part by National Institutes of health grant K01HL130704 (A. Jaitovich) and American Heart Association grant AHA-16GRNT31280002 (D. Jourd’heuil). Figures were produced using Servier Medical Art (www.servier.com).
Reference List
- ABUD EM, MAYLOR J, UNDEM C, PUNJABI A, ZAIMAN AL, MYERS AC, SYLVESTER JT, SEMENZA GL, SHIMODA LA. Digoxin inhibits development of hypoxic pulmonary hypertension in mice. Proc Natl Acad Sci U S A. 2012;109:1239–44. doi: 10.1073/pnas.1120385109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARCHER SL, SOUIL E, DINH-XUAN AT, SCHREMMER B, MERCIER JC, EL YAAGOUBI A, NGUYEN-HUU L, REEVE HL, HAMPL V. Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulmonary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest. 1998;101:2319–30. doi: 10.1172/JCI333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALIGA RS, MILSOM AB, GHOSH SM, TRINDER SL, MACALLISTER RJ, AHLUWALIA A, HOBBS AJ. Dietary nitrate ameliorates pulmonary hypertension: cytoprotective role for endothelial nitric oxide synthase and xanthine oxidoreductase. Circulation. 2012;125:2922–32. doi: 10.1161/CIRCULATIONAHA.112.100586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALL MK, WAYPA GB, MUNGAI PT, NIELSEN JM, CZECH L, DUDLEY VJ, BEUSSINK L, DETTMAN RW, BERKELHAMER SK, STEINHORN RH, SHAH SJ, SCHUMACKER PT. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1alpha. Am J Respir Crit Care Med. 2014;189:314–24. doi: 10.1164/rccm.201302-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BECKMAN JS, BECKMAN TW, CHEN J, MARSHALL PA, FREEMAN BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BECKMAN JS, ISCHIROPOULOS H, ZHU L, VAN DER WOERD M, SMITH C, CHEN J, HARRISON J, MARTIN JC, TSAI M. Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys. 1992;298:438–445. doi: 10.1016/0003-9861(92)90432-v. [DOI] [PubMed] [Google Scholar]
- BECKMAN JS, KOPPENOL WH. Nitric oxide, superoxide, and peroxynitrite; the good, the bad, and the ugly. Am J Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- BENZA R, MATHAI S, NATHAN SD. sGC stimulators: Evidence for riociguat beyond groups 1 and 4 pulmonary hypertension. Respir Med. 2016 doi: 10.1016/j.rmed.2016.11.010. [DOI] [PubMed] [Google Scholar]
- BLOCK ER, HERRERA H, COUCH M. Hypoxia inhibits L-arginine uptake by pulmonary artery endothelial cells. Am J Physiol. 1995;269:L574–80. doi: 10.1152/ajplung.1995.269.5.L574. [DOI] [PubMed] [Google Scholar]
- BONNET S, MICHELAKIS ED, PORTER CJ, ANDRADE-NAVARRO MA, THEBAUD B, BONNET S, HAROMY A, HARRY G, MOUDGIL R, MCMURTRY MS, WEIR EK, ARCHER SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–41. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- BRUNELLE JK, BELL EL, QUESADA NM, VERCAUTEREN K, TIRANTI V, ZEVIANI M, SCARPULLA RC, CHANDEL NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 2005;1:409–14. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- CHANDEL NS, MALTEPE E, GOLDWASSER E, MATHIEU CE, SIMON MC, SCHUMACKER PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–20. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHUA YL, DUFOUR E, DASSA EP, RUSTIN P, JACOBS HT, TAYLOR CT, HAGEN T. Stabilization of hypoxia-inducible factor-1alpha protein in hypoxia occurs independently of mitochondrial reactive oxygen species production. J Biol Chem. 2010;285:31277–84. doi: 10.1074/jbc.M110.158485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DENICOLA A, FREEMAN BA, TRUJILLO M, RADI R. Peroxynitrite reaction with carbon dioxide/bicarbonate kinetics and influence on peroxynitrite-mediated oxidations. Arch Biochem Biophys. 1996;333:49–58. doi: 10.1006/abbi.1996.0363. [DOI] [PubMed] [Google Scholar]
- EPSTEIN AC, GLEADLE JM, MCNEILL LA, HEWITSON KS, O’ROURKE J, MOLE DR, MUKHERJI M, METZEN E, WILSON MI, DHANDA A, TIAN YM, MASSON N, HAMILTON DL, JAAKKOLA P, BARSTEAD R, HODGKIN J, MAXWELL PH, PUGH CW, SCHOFIELD CJ, RATCLIFFE PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- FAGAN KA, FOUTY BW, TYLER RC, MORRIS KG, JR, HEPLER LK, SATO K, LECRAS TD, ABMAN SH, WEINBERGER HD, HUANG PL, MCMURTRY IF, RODMAN DM. The pulmonary circulation of homozygous or heterozygous eNOS-null mice is hyperresponsive to mild hypoxia. J Clin Invest. 1999;103:291–9. doi: 10.1172/JCI3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEELISCH M, RASSAF T, MNAIMNEH S, SINGH N, BRYAN NS, JOURD’HEUIL D, KELM M. Concomitant S-, N-, and heme-nitros(yl)ation in biological tissues and fluids: implications for the fate of NO in vivo. FASEB J. 2002;16:1775–1785. doi: 10.1096/fj.02-0363com. [DOI] [PubMed] [Google Scholar]
- FOSTER MW, MCMAHON TJ, STAMLER JS. S-nitrosylation in health and disease. Trends in Molecular Medicine. 2003;9:160–168. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- GALIE N, BRUNDAGE BH, GHOFRANI HA, OUDIZ RJ, SIMONNEAU G, SAFDAR Z, SHAPIRO S, WHITE RJ, CHAN M, BEARDSWORTH A, FRUMKIN L, BARST RJ, PULMONARY ARTERIAL, H. & RESPONSE TO TADALAFIL STUDY, G Tadalafil therapy for pulmonary arterial hypertension. Circulation. 2009;119:2894–903. doi: 10.1161/CIRCULATIONAHA.108.839274. [DOI] [PubMed] [Google Scholar]
- GIAID A, SALEH D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–21. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- GOW AJ, BUERK DG, ISCHIROPOULOS H. A novel reaction mechanism for the formation of S-nitrosothiol in vivo. J Biol Chem. 1997;272:2841–2845. doi: 10.1074/jbc.272.5.2841. [DOI] [PubMed] [Google Scholar]
- GRISHAM MB, JOURD’HEUIL D, WINK DA. Nitric oxide. I. Physiological chemistry of nitric oxide and its metaolites: implication in inflammation. Am J Physiol. 1999;39:G315–G321. doi: 10.1152/ajpgi.1999.276.2.G315. [DOI] [PubMed] [Google Scholar]
- GUZY RD, HOYOS B, ROBIN E, CHEN H, LIU L, MANSFIELD KD, SIMON MC, HAMMERLING U, SCHUMACKER PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005;1:401–8. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- HUANG Z, SHIVA S, KIM-SHAPIRO DB, PATEL RP, RINGWOOD LA, IRBY CE, HUANG KT, HO C, HOGG N, SCHECHTER AN, GLADWIN MT. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. Journal of Clinical Investigation. 2005 doi: 10.1172/JCI24650. JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IVAN M, HABERBERGER T, GERVASI DC, MICHELSON KS, GUNZLER V, KONDO K, YANG H, SOROKINA I, CONAWAY RC, CONAWAY JW, KAELIN WG., JR Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci U S A. 2002;99:13459–64. doi: 10.1073/pnas.192342099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IVAN M, KONDO K, YANG H, KIM W, VALIANDO J, OHH M, SALIC A, ASARA JM, LANE WS, KAELIN WG., JR HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- KAMURA T, SATO S, IWAI K, CZYZYK-KRZESKA M, CONAWAY RC, CONAWAY JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A. 2000;97:10430–5. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHARITONOV SA, CAILES JB, BLACK CM, DU BOIS RM, BARNES PJ. Decreased nitric oxide in the exhaled air of patients with systemic sclerosis with pulmonary hypertension. Thorax. 1997;52:1051–5. doi: 10.1136/thx.52.12.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLINE DD, PENG YJ, MANALO DJ, SEMENZA GL, PRABHAKAR NR. Defective carotid body function and impaired ventilatory responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. Proc Natl Acad Sci U S A. 2002;99:821–6. doi: 10.1073/pnas.022634199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANDMESSER U, DIKALOV S, PRICE SR, MCCANN L, FUKAI T, HOLLAND SM, MITCH WE, HARRISON DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–9. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI H, HEMANN C, ABDELGHANY TM, EL-MAHDY MA, ZWEIER JL. Characterization of the mechanism and magnitude of cytoglobin-mediated nitrite reduction and nitric oxide generation under anaerobic conditions. J Biol Chem. 2012;287:36623–33. doi: 10.1074/jbc.M112.342378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI H, SAMOUILOV A, LIU X, ZWEIER JL. Characterization of the magnitude and kinetics of xanthine oxidase-catalyzed nitrite reduction. J Biol Chem. 2001;276:24482–24489. doi: 10.1074/jbc.M011648200. [DOI] [PubMed] [Google Scholar]
- MANSFIELD KD, GUZY RD, PAN Y, YOUNG RM, CASH TP, SCHUMACKER PT, SIMON MC. Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab. 2005;1:393–9. doi: 10.1016/j.cmet.2005.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARSHALL C, MAMARY AJ, VERHOEVEN AJ, MARSHALL BE. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol. 1996;15:633–44. doi: 10.1165/ajrcmb.15.5.8918370. [DOI] [PubMed] [Google Scholar]
- MAXWELL PH, WIESENER MS, CHANG GW, CLIFFORD SC, VAUX EC, COCKMAN ME, WYKOFF CC, PUGH CW, MAHER ER, RATCLIFFE PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- MICHELAKIS ED, MCMURTRY MS, WU XC, DYCK JR, MOUDGIL R, HOPKINS TA, LOPASCHUK GD, PUTTAGUNTA L, WAITE R, ARCHER SL. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation. 2002;105:244–50. doi: 10.1161/hc0202.101974. [DOI] [PubMed] [Google Scholar]
- MONCADA S, PALMER RMJ, HIGGS EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43(2):109–142. [PubMed] [Google Scholar]
- MORRELL NW, ADNOT S, ARCHER SL, DUPUIS J, JONES PL, MACLEAN MR, MCMURTRY IF, STENMARK KR, THISTLETHWAITE PA, WEISSMANN N, YUAN JX, WEIR EK. Cellular and molecular basis of pulmonary arterial hypertension. J Am Coll Cardiol. 2009;54:S20–31. doi: 10.1016/j.jacc.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAEIJE R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2:20–2. doi: 10.1513/pats.200407-037MS. [DOI] [PubMed] [Google Scholar]
- NATHAN C, XIE QW. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- PANKEY EA, BADEJO AM, CASEY DB, LASKER GF, RIEHL RA, MURTHY SN, NOSSAMAN BD, KADOWITZ PJ. Effect of chronic sodium nitrite therapy on monocrotaline-induced pulmonary hypertension. Nitric Oxide. 2012;27:1–8. doi: 10.1016/j.niox.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POPOV S, TAKEMORI H, TOKUDOME T, MAO Y, OTANI K, MOCHIZUKI N, PIRES N, PINHO MJ, FRANCO-CERECEDA A, TORIELLI L, FERRANDI M, HAMSTEN A, SOARES-DA-SILVA P, ERIKSSON P, BERTORELLO AM, BRION L. Lack of salt-inducible kinase 2 (SIK2) prevents the development of cardiac hypertrophy in response to chronic high-salt intake. PLoS One. 2014;9:e95771. doi: 10.1371/journal.pone.0095771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRABHAKAR NR, SEMENZA GL. Adaptive and maladaptive cardiorespiratory responses to continuous and intermittent hypoxia mediated by hypoxia-inducible factors 1 and 2. Physiol Rev. 2012;92:967–1003. doi: 10.1152/physrev.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUINLAN TR, LI D, LAUBACH VE, SHESELY EG, ZHOU N, JOHNS RA. eNOS-deficient mice show reduced pulmonary vascular proliferation and remodeling to chronic hypoxia. Am J Physiol Lung Cell Mol Physiol. 2000;279:L641–50. doi: 10.1152/ajplung.2000.279.4.L641. [DOI] [PubMed] [Google Scholar]
- RASSAF T, TOTZECK M, HENDGEN-COTTA UB, SHIVA S, HEUSCH G, KELM M. Circulating nitrite contributes to cardioprotection by remote ischemic preconditioning. Circ Res. 2014;114:1601–10. doi: 10.1161/CIRCRESAHA.114.303822. [DOI] [PubMed] [Google Scholar]
- REEVE HL, MICHELAKIS E, NELSON DP, WEIR EK, ARCHER SL. Alterations in a redox oxygen sensing mechanism in chronic hypoxia. J Appl Physiol (1985) 2001;90:2249–56. doi: 10.1152/jappl.2001.90.6.2249. [DOI] [PubMed] [Google Scholar]
- RYAN JJ, MARSBOOM G, FANG YH, TOTH PT, MORROW E, LUO N, PIAO L, HONG Z, ERICSON K, ZHANG HJ, HAN M, HANEY CR, CHEN CT, SHARP WW, ARCHER SL. PGC1alpha-mediated mitofusin-2 deficiency in female rats and humans with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013;187:865–78. doi: 10.1164/rccm.201209-1687OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHROEDL C, MCCLINTOCK DS, BUDINGER GR, CHANDEL NS. Hypoxic but not anoxic stabilization of HIF-1alpha requires mitochondrial reactive oxygen species. Am J Physiol Lung Cell Mol Physiol. 2002;283:L922–31. doi: 10.1152/ajplung.00014.2002. [DOI] [PubMed] [Google Scholar]
- SHIMODA LA, FALLON M, PISARCIK S, WANG J, SEMENZA GL. HIF-1 regulates hypoxic induction of NHE1 expression and alkalinization of intracellular pH in pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol. 2006;291:L941–9. doi: 10.1152/ajplung.00528.2005. [DOI] [PubMed] [Google Scholar]
- SHIMODA LA, MANALO DJ, SHAM JS, SEMENZA GL, SYLVESTER JT. Partial HIF-1alpha deficiency impairs pulmonary arterial myocyte electrophysiological responses to hypoxia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L202–8. doi: 10.1152/ajplung.2001.281.1.L202. [DOI] [PubMed] [Google Scholar]
- SHIMODA LA, SEMENZA GL. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med. 2011;183:152–6. doi: 10.1164/rccm.201009-1393PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMODA LA, SHAM JS, SHIMODA TH, SYLVESTER JT. L-type Ca(2+) channels, resting [Ca(2+)](i), and ET-1-induced responses in chronically hypoxic pulmonary myocytes. Am J Physiol Lung Cell Mol Physiol. 2000;279:L884–94. doi: 10.1152/ajplung.2000.279.5.L884. [DOI] [PubMed] [Google Scholar]
- SIMON MA, VANDERPOOL RR, NOURAIE M, BACHMAN TN, WHITE PM, SUGAHARA M, GORCSAN J, 3RD, PARSLEY EL, GLADWIN MT. Acute hemodynamic effects of inhaled sodium nitrite in pulmonary hypertension associated with heart failure with preserved ejection fraction. JCI Insight. 2016;1:e89620. doi: 10.1172/jci.insight.89620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPARACINO-WATKINS CE, TEJERO J, SUN B, GAUTHIER MC, THOMAS J, RAGIREDDY V, MERCHANT BA, WANG J, AZAROV I, BASU P, GLADWIN MT. Nitrite reductase and nitric-oxide synthase activity of the mitochondrial molybdopterin enzymes mARC1 and mARC2. J Biol Chem. 2014;289:10345–58. doi: 10.1074/jbc.M114.555177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMPSON CB. Into Thin Air: How We Sense and Respond to Hypoxia. Cell. 2016;167:9–11. doi: 10.1016/j.cell.2016.08.036. [DOI] [PubMed] [Google Scholar]
- TISO M, TEJERO J, BASU S, AZAROV I, WANG X, SIMPLACEANU V, FRIZZELL S, JAYARAMAN T, GEARY L, SHAPIRO C, HO C, SHIVA S, KIM-SHAPIRO DB, GLADWIN MT. Human neuroglobin functions as a redox-regulated nitrite reductase. J Biol Chem. 2011;286:18277–89. doi: 10.1074/jbc.M110.159541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TSUGU T, MURATA M, KAWAKAMI T, KATAOKA M, NAGATOMO Y, TSURUTA H, ITABASHI Y, MAEKAWA Y, FUKUDA K. Amelioration of right ventricular function after hybrid therapy with riociguat and balloon pulmonary angioplasty in patients with chronic thromboembolic pulmonary hypertension. Int J Cardiol. 2016;221:227–9. doi: 10.1016/j.ijcard.2016.06.331. [DOI] [PubMed] [Google Scholar]
- WANG J, KRIZOWSKI S, FISCHER-SCHRADER K, NIKS D, TEJERO J, SPARACINO-WATKINS C, WANG L, RAGIREDDY V, FRIZZELL S, KELLEY EE, ZHANG Y, BASU P, HILLE R, SCHWARZ G, GLADWIN MT. Sulfite Oxidase Catalyzes Single-Electron Transfer at Molybdenum Domain to Reduce Nitrite to Nitric Oxide. Antioxid Redox Signal. 2015;23:283–94. doi: 10.1089/ars.2013.5397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG J, WEIGAND L, LU W, SYLVESTER JT, SEMENZA GL, SHIMODA LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res. 2006;98:1528–37. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- WARDLE AJ, SEAGER MJ, WARDLE R, TULLOH RM, GIBBS JS. Guanylate cyclase stimulators for pulmonary hypertension. Cochrane Database Syst Rev. 2016:CD011205. doi: 10.1002/14651858.CD011205.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAYPA GB, MARKS JD, GUZY R, MUNGAI PT, SCHRIEWER J, DOKIC D, SCHUMACKER PT. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res. 2010;106:526–35. doi: 10.1161/CIRCRESAHA.109.206334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAYPA GB, MARKS JD, GUZY RD, MUNGAI PT, SCHRIEWER JM, DOKIC D, BALL MK, SCHUMACKER PT. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am J Respir Crit Care Med. 2013;187:424–32. doi: 10.1164/rccm.201207-1294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAYPA GB, SMITH KA, SCHUMACKER PT. O2 sensing, mitochondria and ROS signaling: The fog is lifting. Mol Aspects Med. 2016;47–48:76–89. doi: 10.1016/j.mam.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU W, KANEKO FT, ZHENG S, COMHAIR SA, JANOCHA AJ, GOGGANS T, THUNNISSEN FB, FARVER C, HAZEN SL, JENNINGS C, DWEIK RA, ARROLIGA AC, ERZURUM SC. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004;18:1746–8. doi: 10.1096/fj.04-2317fje. [DOI] [PubMed] [Google Scholar]
- XUE C, JOHNS RA. Endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:1642–4. doi: 10.1056/NEJM199512143332416. [DOI] [PubMed] [Google Scholar]
- YU AY, SHIMODA LA, IYER NV, HUSO DL, SUN X, MCWILLIAMS R, BEATY T, SHAM JS, WIENER CM, SYLVESTER JT, SEMENZA GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest. 1999;103:691–6. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YUAN XJ, WANG J, JUHASZOVA M, GAINE SP, RUBIN LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet. 1998;351:726–7. doi: 10.1016/S0140-6736(05)78495-6. [DOI] [PubMed] [Google Scholar]
- ZHANG H, QIAN DZ, TAN YS, LEE K, GAO P, REN YR, REY S, HAMMERS H, CHANG D, PILI R, DANG CV, LIU JO, SEMENZA GL. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105:19579–86. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO YY, ZHAO YD, MIRZA MK, HUANG JH, POTULA HH, VOGEL SM, BROVKOVYCH V, YUAN JX, WHARTON J, MALIK AB. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. 2009;119:2009–18. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZUCKERBRAUN BS, SHIVA S, IFEDIGBO E, MATHIER MA, MOLLEN KP, RAO J, BAUER PM, CHOI JJ, CURTIS E, CHOI AM, GLADWIN MT. Nitrite potently inhibits hypoxic and inflammatory pulmonary arterial hypertension and smooth muscle proliferation via xanthine oxidoreductase-dependent nitric oxide generation. Circulation. 2010;121:98–109. doi: 10.1161/CIRCULATIONAHA.109.891077. [DOI] [PubMed] [Google Scholar]
- ZWEIER JL, WANG P, SAMOUILOV A, KUPPUSAMY P. Enzyme-independent formation of nitric oxide in biological tissues. Nature Medicine. 1995;1:804–809. doi: 10.1038/nm0895-804. [DOI] [PubMed] [Google Scholar]