

Abstract
One of the greatest challenges for mass spectrometry (MS)-based proteomics is the limited ability to analyze small samples. Here we investigate the relative contributions of liquid chromatography (LC), MS instrumentation and data analysis methods with the aim of improving proteome coverage for sample sizes ranging from 0.5 ng to 50 ng. We show that the LC separations utilizing 30-μm-i.d. columns increase signal intensity by >3-fold relative to those using 75-μm-i.d. columns, leading to 32% increase in peptide identifications. The Orbitrap Fusion Lumos MS significantly boosted both sensitivity and sequencing speed relative to earlier generation Orbitraps (e.g., LTQ-Orbitrap), leading to a ~3-fold increase in peptide identifications and 1.7-fold increase in identified protein groups for 2 ng tryptic digests of the bacterium S. oneidensis. The Match Between Runs algorithm of open-source MaxQuant software further increased proteome coverage by ~ 95% for 0.5 ng samples and by ~42% for 2 ng samples. Using the best combination of the above variables, we were able to identify >3,000 proteins from 10 ng tryptic digests from both HeLa and THP-1 mammalian cell lines. We also identified >950 proteins from subnanogram archaeal/bacterial cocultures. The present ultrasensitive LC-MS platform achieves a level of proteome coverage not previously realized for ultra-small sample loadings, and is expected to facilitate the analysis of subnanogram samples, including single mammalian cells.
Keywords: ultrasensitive, nanoLC, Orbitrap Fusion Lumos, match between runs, subnanogram proteomics, small cell populations
Graphical Abstract
1. Introduction
Mass spectrometry (MS)-based proteomics has become an indispensable tool for systems biology research, enabling thousands of proteins to be identified along with their post-translational modifications, interactions, and cellular locations [1,2]. Quantitative and comparative proteomics have greatly enhanced biomarker discovery, therapy evaluation, and pathway analysis [3]. With the rapid progress of biological and medical research, there is a growing demand to analyze much smaller biological samples than have been previously accessible, including rare isolated cells [4], single organs [5], fine needle aspiration biopsies [6], and microbial communities with high spatial resolution [7]. Ultimately, single-cell proteomics (where ‘typical’ mammalian cells contain just 0.05 to 0.2 ng of protein [8]) should lead to important insights regarding heterogeneity of protein expression within populations [9]. However, the extension of proteomics to these small samples is hindered by various limitations in processing and analysis, such that millions of cells containing micrograms protein are still required for most proteomics workflows.
Efforts to extend proteomic analyses to far smaller samples have focused on analytical improvements to increase the efficiency of separation, ionization and MS, as well as advances in sample preparation to effectively minimize the inefficiencies and sample losses associated with traditional bulk workflows. Examples of analytical enhancements include optimization of detector electronics and advanced ion focusing approaches (e.g., the electrodynamic ion funnel [10]) that have enabled mass detection limits as low as 10 zmol for MS and 50 zmol for MS/MS analysis of peptides [4]. Such low detection limits are sufficient to detect many proteins within single mammalian cells [11]. Efficient ionization is also critical, with slower delivery of analytes to the electrospray emitter producing smaller, more readily desolvated droplets [12,13], and increasing ion utilization efficiency. Stable electrosprays have been demonstrated at flow rates as low as several hundred picoliters per minute [14], and flows in the low nanoliter-per-minute range are routinely achieved. Under optimized conditions, >50% of all solution-phase analyte molecules can be converted to gas-phase ions and transmitted to the high-vacuum region of the mass spectrometer [15]. The separation method used in conjunction with MS is also critical for ultrasensitive proteomic analysis. Besides directly determining the flow rate at the ESI source, the liquid-phase separation peak capacity impacts the dynamic range of measurement, the measurement throughput, and the sampling rate required for MS/MS sequencing. Capillary electrophoresis (CE)-MS has enabled ultrasensitive peptide identifications with detection limits as low as 1 zmol using an electrokinetically pumped sheath–flow interface [16]. However, the greater selectivity and peak capacities provided by LC separations (exceeding 1000 for one-dimensional separations in some cases [17]) have maintained the widespread use of LC-MS for proteomic analyses. Several reports have demonstrated the analysis of samples containing protein amounts as small or smaller than single cells [13,16,18].
With the above mentioned analytical advances firmly establishing the ability to analyze samples as small as, or even smaller than, single cells (albeit with very limited coverage), many recent efforts have focused on improving preparation workflows for small samples and delivery of those prepared samples to the analytical platform. Wang et. al. [19] reported a preparation workflow including flow cytometer sorting, ND-40 cell lysis, acetone-based protein precipitation, and tryptic digestion to identify a total of 167 proteins from 500 MCF-7 cells. Wisniewski et. al. [20] developed filter-aided sample preparation (FASP), where centrifugal ultrafiltration tubes were used to remove SDS with high sample recovery, and identified 905 proteins from 500 HeLa cells. Chen et. al. [21] developed a single-step preparation technique, where cells and trypsin were loaded together into a sample loop and incubated at elevated temperature to achieve cell lysis and tryptic digestion. Proteome coverage of 635 protein groups was achieved for 100 DLD cells. A spin-tip-based proteomic processing technique employed strong cation exchange beads for protein preconcentration, reduction, alkylation, and digestion, followed by reversed phase-based sample cleanup and fractionation; 1270 proteins were identified from 2000 HEK 293T cells [22]. Li et. al [4] developed an integrated proteomic platform including adaptive focused acoustics-based cell lysis and protein extraction, one-tube digestion procedures, and porous layer open tube (PLOT) LC-MS (flow rate < 20 nl/min). They reported 1327 proteins identified from lysate equivalent to 50 MCF-7 cells. Our laboratory has previously developed a simplified nano-proteomics platform (SNaPP) by integrating immobilized enzyme reactors, solid phase extraction, and LC-MS to improve enzyme digestion efficiency and operational robustness for sensitive proteomic analysis [23]. SNaPP allowed reproducible profiling of 652–1860 proteins from 500–2000 cells, respectively, that were laser-microdissected from mouse alveolar tissues [24].
As sample preparation methods continue to improve for ultra-small samples, it is important that analytical methods be optimized for trace samples. Here, we have systematically investigated the contributions of LC, MS, and data analysis strategy on the analytical sensitivity and proteome coverage for proteomic samples ranging from 0.5 to 50 ng of total protein. Our investigation showed that LC separations using 30-μm-i.d. columns can increase signal intensities by >3-fold, leading to a 32% increase in peptide identifications compared with those utilizing standard 75-μm-i.d. columns. The Orbitrap Fusion Lumos MS produced ~3× increase in peptide identifications for 2 ng proteomic samples relative to an LTQ-Orbitrap MS. The Match Between Runs (MBR) algorithm of MaxQuant further increased proteome coverage by ~42% for the same samples. Using the best combinations of analytical parameters, we have identified 3172 and 3338 protein groups from 10 ng tryptic digests equivalent to 50 HeLa cells and 100 THP-1 cells, respectively. We have also shown that >950 proteins can be identified from just 0.5 ng microbial cell lysates, indicating the great potential of this platform for subnanogram proteomics.
2. Experimental methods
2.1 Cell culture and proteomic preparation
Both microbial and mammalian samples were used to evaluate the performance of the present LC-MS platform. The bacterium Shewanella oneidensis MR-1 was cultured under fed-batch mode using a Bioflow 3000 fermentor (New Brunswick Scientific, Enfield, USA). HBa MR-1 with 0.5 mL/L of 100 mM ferric NTA, 1 mL/L of 1 mM Na2SeO4, and 1 mL/L of 3 M MgCl2·6H2O as well as vitamins, minerals and amino acids were used as culture media. The cells were harvested at steady state and pelleted at 11,900 ×g for 8 min at 4 °C. Cells were lysed by homogenization with 0.1 mm zirconia/silica beads in the Bullet Blender (Next Advance, Averill Park, USA), speed 8, for 3 min. Protein concentration was determined using the BCA assay (Thermo Scientific, San Jose, USA). Proteins were denatured and reduced with 8 M urea and 5 mM dithiothreitol (DDT) by incubating at 37 °C for 60 min. The sample was then diluted tenfold with 100 mM Na4HCO3 (pH 8) and CaCl2 was added to a final concentration of 1 mM. Tryptic digestion was performed with a trysin/protein ratio of 1:50 and an incubation time of 3 h at 37 °C. The peptide mixture was desalted by DSC-C18 columns (Sigma-Aldrich). The final concentration was measured by peptide BCA assay (Thermo Scientific, San Jose, USA), and aliquoted for-long term storage at −80 °C.
A second microbial sample that was analyzed is an archaeal/bacterial coculture of Methanosarcina acetivorans/Desulfococcus multivorans. 0.5 mL of an established coculture was transferred into 50 mL of anaerobic media as previously described [25] and grown at 28 °C without shaking. Cultures once grown were counted with a hemocytometer and cell numbers were used as a proxy for protein abundance. The cells were pelleted at 11,000 ×g for 10 min at 4 °C and re-suspended at in 1.5× PBS. The cell suspension was mixed with 1:1 methanol/3 × PBS to reach a final concentration of 50% methanol and 1.5× PBS. After overnight incubation, the mixture was centrifuged again at 11,000 ×g for 10 min at 4 °C to remove supernatant. A cocktail containing 0.2% RapiGest SF surfactant (Waters, Milford, USA) in 50 mM Na4HCO3 (pH 8) and 5 mM dithiothreitol (DDT) was added to the pellet. The mixture was then incubated at 75 °C for 1 h to lyse cells and denature and reduce proteins. Iodoacetamide was added to a concentration of 10 mM and allowed to react in dark for 30 min. The protein was pre-digested with Lys-C at 37°C for 4 h with an enzyme-to-protein ratio of ~1/20. Finally, trypsin was added with an enzyme-to-protein ratio of ~1/20 and allowed to digest overnight at 37 °C.
For mammalian samples, THP-1 and HeLa (ATCC, Manassas, USA) cell lines were used. All cells were cultured at 37 °C and 5% CO2 and split every 3 to 4 days following standard protocol. HeLa was grown in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% fetal bovine serum (FBS) and 1× penicillin/streptomycin. THP-1 cells were cultured in ATCC-modified RPMI-1640 medium supplemented with 10% FBS and 1× penicillin streptomycin. Cells were collected in a 10-mL tube and centrifuged at 1,200 rpm for 10 minutes to remove culture media. After washing with PBS 3 times, the cells were counted with a hemocytometer and cell numbers were used as a proxy for protein abundance. RapiGest-based one-tube procedures described above were employed to process the cell for proteomic analysis.
2.2 LC-MS
Solid phase extraction (SPE) and LC columns were packed with 3-μm C18 packing material (300-Å pore size, Phenomenex, Terrence, USA) as described previously [18]. 60-cm-long LC columns were packed in-house using Self-Pack PicoFrit tubing (New Objective, Woburn, USA) with integrated 10-μm-i.d. ESI emitters. The SPE column was prepared from 5-cm-long, 100-μm-i.d. fused silica capillary, and Kasil frits [26] were prepared at both ends to retain the packing material. The SPE column and LC column were mounted on a 10-port injection/switching valve (VICI Valco Instrument, Houston, USA). Sample was injected into a 2-μL sample loop and then loaded onto the SPE column at a flow rate of 800 nL/min with Buffer A (0.1% formic acid in water) for 10 min. A NCP-3200RS LC pump equipped with a picoflow module (Thermo Scientific, San Jose, USA) was used to deliver gradient flow to the SPE and LC columns. A linear 150-min gradient of 5–28% Buffer B (0.1% formic acid in acetonitrile) was employed for peptide elution. The SPE and LC column was washed by ramping buffer B to 80% in 20 minutes, and finally re-equilibrated with 2% buffer A for another 20 minutes. The volumetric flow rates of 30 μm-i.d. and 75 μm-i.d. LC columns were set as 60 nL/min and 350 nL/min to achieve optimal linear flow rate of 0.15 cm/s [18].
Two mass spectrometers, a LTQ-Orbitrap XL MS and an Orbitrap Fusion Lumos Tribrid MS, were evaluated and compared in this work. Both mass spectrometers are evaluated, calibrated, and maintained routinely by a dedicated instrument team in the MS facility of Environmental Molecular Sciences Laboratory. Standard tuning/calibration solutions specifically for these mass spectrometers were used to evaluate performance and to calibrate as needed. The instrument performance can be accessed based on this evaluation alone. In addition, we used in-house prepared Quality Control (QC) standards for additional assessment. These QC standards were prepared in large batches and included whole cell tryptic digest of Shewanella oneidensis and RAW 264.7 mouse macrophage cells. Because we have been using these QC standards for years, the number of unique peptide identifications observed when the instrument is working at peak performance is well known. Aside from unique peptide identification, a wide range of other metrics are measured as well, including mass error, accurate mass and time tags, chromatographic peak width, etc. We used all these measures to confirm the mass spectrometers were in optimal condition before sample analysis.
For both instruments, a voltage of 1.9 kV was applied at the ESI source, and data-dependent acquisition (DDA) mode was used to trigger precursor isolation and sequencing. For LTQ-Orbitrap MS, the ion transfer capillary was set at 250 °C for desolvation. A full MS scan range was set as 375–1575. The Orbitrap was set for full MS scan with a resolution of 60,000 at m/z 200, an AGC target of 1E6, and a maximum ion accumulation time of 500 ms. The top 10 precursors having intensity values >500 and charges of +2 or greater were isolated at a m/z window of 2 Da and fragmented by CID. The collision energy was 35% and the activation time was 30 ms. An AGC target of 30,000 and a maximum ion accumulation time of 100 ms was utilized for MS/MS scanning. The dynamic exclusion duration was 90 s (mass range of ±1 Da) to reduce repeated sequencing. For the Orbitrap Fusion Lumos, the ion transfer capillary set at 150 °C provided sufficient desolvation with the low-flow 30 μm-i.d LC separations. The S-lens RF level was set at 30, which is the factory recommended setting for boosting ion transmission and reducing in source fragmentation. The Orbitrap was set for full MS scan from 375–1575 with a resolution of 120,000 (at m/z 200), an AGC target of 1E6 and a maximum ion accumulation time of 246 ms. The top 12 precursor ions with charges of +2 to +7 were isolated with an m/z window of 2 and fragmented by high energy dissociation (HCD) with a collision energy of 28%. The signal intensity threshold was set at 6000. MS/MS scans were performed in the Orbitrap, which was set to a resolution of 60,000 at m/z 200, an AGC target of 1E5, and a maximum ion time of 118 ms.
2.3 Data analysis
For Shewanella oneidensis MR-1, THP-1 and HeLa cell samples, MaxQuant (version 1.5.3.30) [27] was used for feature detection, database searching and protein/peptide quantification. For Shewanella oneidensis MR-1, MS/MS spectra were searched against the UniProtKB database (Downloaded in 2/23/2017 and containing 645 reviewed and 3,426 unreviewed sequences). For THP-1 and HeLa cell samples, UniProtKB/Swiss-Prot human database (Downloaded in 12/29/2016 containing 20,129 reviewed sequences) was used. Carbamidomethylation of cysteine residues was set as a fixed modification. For all these three samples, N-terminal protein acetylation and methionine oxidation were selected as variable modifications. Both peptides and proteins were filtered with a maximum false discovery rate (FDR) of 0.01. Match between runs (MBR) with a match window of 0.7 min and alignment window of 20 min was activated to increase peptide/protein identification of small samples. Other unmentioned parameters were the default settings of the MaxQuant software. Perseus [28] was used to perform data filtering and extraction. The extracted data were further processed and visualized using Excel and OriginLab.
For D. multivorans/M. acetivorans co-cultures, MSGFPlus [29] was used for MS/MS spectra searches against two UniProtKB databases containing 3,912 sequences of D. multivorans DSM 2059 and 4,721 sequences of M. acetivorans C2A. Carbamidomethylation of cysteine residues was set as a fixed modification and methionine oxidation were set as a variable modification. Both peptides and proteins were filtered with a maximum FDR of 0.01.
3. Results and discussion
3.1 Comparison of 30-μm and 75-μm LC columns
In this work, a 30-μm-i.d. LC column was selected as one of the separation formats based on the following considerations: (1) The optimized operational flow rate is ~60 nL/min, which is in mass-flux sensitive zone of electrospray [30], providing a high ionization efficiency. (2) No external emitter was required using the integrated self-pack columns, and thus high-performance and reliable LC separations were maintained at low flow rates. (3) State-of-the-art nanoLC gradient pumps are capable of delivering stable flow at flow rates as low as 50 nL/min, eliminating the need for a flow-splitting interface. However, additional gains in sensitivity will likely be realized by further miniaturizing the LC separation.
Since 75-μm-i.d. is the most commonly used LC column sizes for current proteomics studies, we compared the analytical performance between 30-μm and 75-μm-i.d. columns for analysis of small samples. As expected, an evident signal gain for separations using 30-μm columns was observed in base peak chromatograms using 10-ng tryptic digest of S. oneidensis as a model sample (Figure 1a). The peptides observed in both column systems were extracted and quantified using MaxQuant algorithms and the intensities of the 30-μm-column separated peptides over those separated with the 75-μm column were plotted as log2 ratios (Figure 1b). For the 5,689 quantifiable peptides, the median log2 intensity ratio was 1.67, corresponding to a signal gain of 3.2 fold for the 30-μm column. 69% of peptides were found to have signal enhancement in the range of 2–4 fold. The use of 30-μm-i.d columns instead of 75-μm-i.d columns also led to a modest increase of total MS/MS events from 17507 to 20592, corresponding to an increase of 15%. However, the identified MS/MS spectra increased by 32% from 9249 to 12164, which could be ascribed to the improved signal intensity and MS/MS spectra quality. Unique peptide and protein identifications were ultimately increased by 32% and 13%, respectively (Figure 1c).
Figure 1.

Comparison of LC column size on the performance of proteomic analysis. (a1–a2) Base peak chromatograms of 10-ng tryptic digest of Shewanella oneidensis with (a1) 75-μm and (a2) 30-μm i.d. LC columns and a LTQ-Orbitrap MS. Y axis was fixed at 8E6 to show the signal gain for the 30-μm LC. (b) Peptide intensity ratio between 30-μm and 75-μm i.d. LC. Each point in the chart corresponds to a peptide identified from both LC columns. (c) The number of unique peptides and protein groups identified with 75-μm and 30-μm LC columns. In each condition, two replicates were analyzed and averaged to generate results. LC conditions: 30-μm i.d. column operated at 60 nL/min; 75-μm i.d. column operated at 350 nL/min; a 150-min gradient from 5% to 28% Buffer B was used
We evaluated the sensitivity of 30-μm column LC and LTQ-Orbitrap MS by analyzing 0.5-ng to 50-ng tryptic lysate. As shown in Figure 2a, both protein and peptide identification rates significantly relied on the sample input in the range of 0.5 ng to 10 ng, indicating LC-MS sensitivity controlled the proteomic coverage in low nanogram samples. It should be noted that, although LTQ-Orbitrap was a relatively dated MS, it still identified 812 peptides and 494 protein groups with a sample loading amount of 0.5 ng. From 10 ng to 50 ng, only a 20% increase in peptides and 3.7% increase in protein groups were observed, indicating that MS sequencing speed limited the proteomic coverage above 10 ng. We next used the MBR algorithm [27], which, as with the Accurate Mass and Time (AMT) tag approach [31], can identify unsequenced peptides based on accurate intact mass and LC retention times [32], to determine its impact on the number of identified peptides. As shown in Figure 2b, peptide identifications increased by 247% and 92% for 0.5 ng and 2 ng samples, leading to protein identifications increasing by 95% and 42%, respectively. The results demonstrate that approaches utilizing accurate mass and retention time are a promising means of improving proteome coverage for ultra-small samples and will likely play an indispensable role in subnanogram proteomics.
Figure 2.
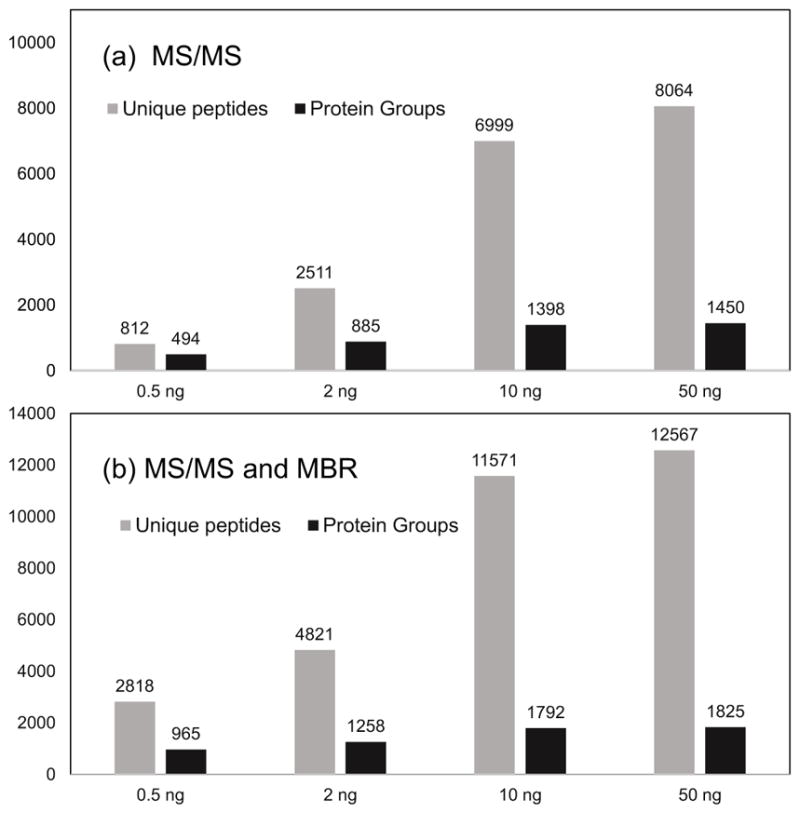
Evaluation of the sensitivity of 30-μm i.d. column LC and LTQ-Orbitrap MS with various sample loading amounts from 0.5 ng to 50 ng tryptic digest of S. oneidensis. (a–b) Peptide and protein identification results with (a) MS/MS only and (b) combined MS/MS with match between runs (MBR). For each condition, at least two replicates were analyzed and averaged to generate the results. LC conditions: 30-μm i.d. column operated at 60 nL/min and a 150-min gradient from 5% to 28% Buffer B
3.2 Comparison of LTQ-Orbitrap and Orbitrap Fusion Lumos MS
Compared with LTQ-Orbitrap, the state-of-art Orbitrap Fusion Lumos incorporates multiple features expected to enhance small sample analyses. The Fusion Lumos has an electrodynamic ion funnel [10] and a high capacity transfer tube, which significantly improve ion transmission efficiency to high vacuum. The high-field Orbitrap analyzer acquires spectra 3× faster than the LTQ-Orbitrap, enabling highly sensitive measurements without compromising sequencing speed. The sequencing speed was further doubled with parallel operations of ion accumulation and ion detection in the Fusion Lumos. In a typical setup of full MS at a resolution of 60,000 and top 10 for MS/MS sequencing, the cycle time of Fusion Lumos is ~1s, which is more than 3 times faster than that of the LTQ-Orbitrap (~3.6 s). To make a direct comparison, 2 ng and 10 ng tryptic digests of Shewanella oneidensis were analyzed by the two mass spectrometers with the same 30-μm-i.d. LC separation. As shown in Figure 3 and Table 1, significant increases in proteome coverage were observed for both samples when using the Lumos Fusion MS compared with the LTQ-Orbitrap. A 2.9-fold increase in peptide identifications and a 1.7-fold increase in identified protein groups were observed for 2-ng samples. We mainly attributed the gain to the enhanced sensitivity of Lumos Fusion, because it has > 4 times more observable isotope patterns (z>1) and >2 times more MS/MS events, than that of LTQ-Orbitrap (Table 1). When MBR was used, the observable unique peptide numbers were 4,821 and 9,725 for LTQ-Orbitrap and Lumos Fusion, respectively, which further demonstrated the enhanced sensitivity of Lumos Fusion. For 10-ng samples, both speed and sensitivity contributed the gain. With a 150-min gradient, 45,502 precursors were sequenced by the Lumos and 41% of the sequence events resulted in peptide identifications. The present results demonstrated the combination of 30-μm-i.d. LC column with Lumos Fusion MS can provide unprecedented capability for proteomic profiling of ultra-small biological samples.
Figure 3.

Proteomic profiling of (a) 2 ng and (b) 10 ng tryptic digestion of Shewanella oneidensis with LTQ-Orbitrap and Lumos Fusion MS. LC conditions: 30-μm i.d. column operated at 60 nL/min and a 150-min gradient from 5% to 28% buffer B.
Table 1.
Comparison of LTQ-Orbitrap and Orbitrap Fusion Lumos MS on the performance of proteomic profiling of 2 ng and 10 ng tryptic digest of Shewanella oneidensis. LC conditions: 30-μm i.d. column operated at 60 nL/min and a 150-min gradient from 5% to 28% buffer B.
| Isotope Patterns (z>1) | MS/MS | MS/MS Identified | Unique peptides/With MBR | Protein Groups/With MBR | |
|---|---|---|---|---|---|
| 2 ng with LTQ Orbitrap | 11,785 | 9,991 | 3,638 | 2,511/4,821 | 885/1,258 |
| 2 ng with Lumos Fusion | 48,005 | 28,707 | 8,699 | 7,225/9,725 | 1,476/1,731 |
| 10 ng with LTQ Orbitrap | 45,275 | 20,592 | 12,165 | 6,999/11,571 | 1,398/1,792 |
| 10 ng with Lumos Fusion | 107,048 | 45,502 | 18,630 | 14,471/15,354 | 1,946/1,970 |
3.3 Proteomic profiling of small amounts of mammalian cell lysate
To assess the performance of the present LC-MS platform on mammalian samples, two cultured human cell lines, HeLa and THP-1, were used as model samples. Cell lysates were processed according to the Rapigest protocol [33] and diluted to for injection. Based on the volumes of these cells, each injection contained ~ 10 ng proteins. An average 2989 and 2575 protein groups were identified from triplicate analysis of cell lysate equivalent to 100 THP-1 cells and 50 HeLa cells, respectively. The number of identified protein groups increased to 3338 and 3172, respectively, when MBR was used (Figure 4a). Overlap analysis showed over 76% of the protein groups were found in triplicate runs, indicating high reproducibility of the present platform (Figure 4b and 4c). Compared with other platforms [4,19–22], the present result represents a >500-fold decrease in required cell numbers to achieve >3000 protein identifications.
Figure 4.
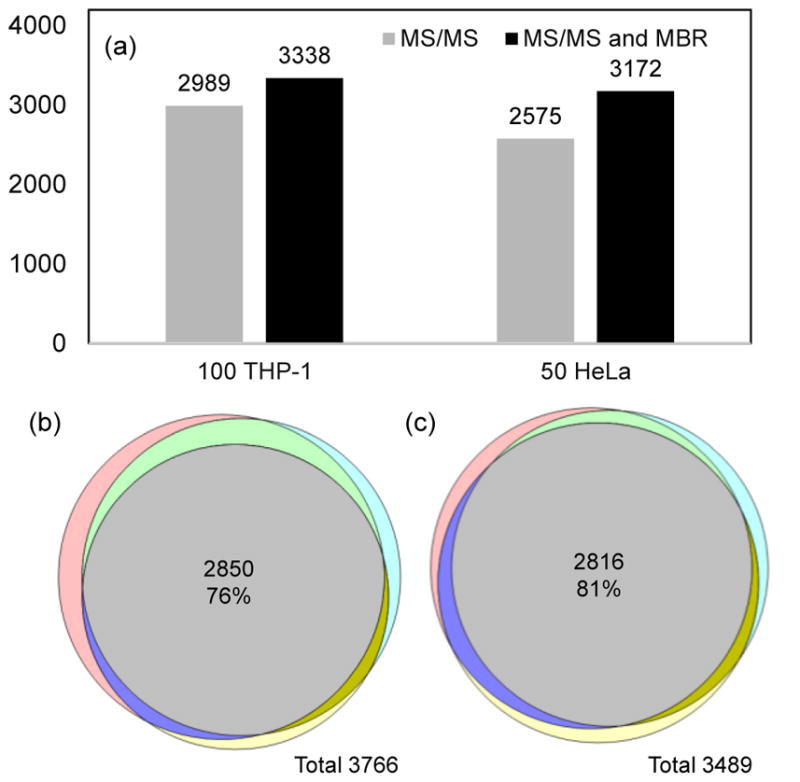
(a) Protein identification from tryptic digestions equivalent to 100 THP-1 and 50 HeLa cells with or without MBR algorithm. (b–c) Venn diagrams showing the protein overlaps in triplicate runs of cell lysates equivalent to (b) 100 THP-1 and (c) 50 HeLa cells. LC conditions: 30-μm i.d. column operated at 60 nL/min and a 150-min gradient from 5% to 28% buffer B. Lumos Fusion MS was used for data acquisition.
3.4 Proteomic profiling of subnanogram cell lysates of Methanosarcina acetivorans/Desulfococcus multivorans co-cultures
We next applied the present platform to study protein expression in an established archaeal/bacterial co-culture that was developed as a model for environmental syntrophic consortia mediating sulfate-based anaerobic oxidation of methane in deep-sea methane seep sediments [34,35]. The co-culture consisted of a methanogenic archaeon (Methanosarcina acetivorans) and a sulfate-reducing deltaproteobacterium (Desulfococcus multivorans), which aggregated together when supplied with methanol and lactate as carbon sources. These organisms are phylogenetically-related to the uncultured methanotrophic ANME-2 archaea and syntrophic sulfate-reducing bacterial partner SEEP-SRB1a, within the Desulfococcus/Desulfosarcina clade and were used here to test the efficiency of the protein extraction protocol and protein identification prior to the analysis of environmental methane-oxidizing ANME-SRB consortia. Proteins were successfully identified from as little as 0.75 ng and 1.5 ng of cell lysate, recovering 3499 and 4719 unique peptides, respectively, and 1031 and 1425 respective protein identifications. Overlap analysis indicated 95% of the protein groups identified in the 0.75 ng sample were also included in the 1.5 ng sample. Of all the 1031 proteins found in the 0.75 ng sample, corresponding to approximately 4950 of cells, 592 proteins were from Desulfococcus multivorans and 439 were from Methanosarcina acetivorans. This translated to a coverage of 15.1% and 9.3% of the proteome for Desulfococcus and Methanosarcina respectively, and included multiple proteins involved in methylotrophic methanogenesis with methanol (e.g. methanol cobalamin methyltransferase, alpha, beta, gamma subunits of methyl coenzyme M reductase, and F420-dependent methylenetetrahydromethanopterin dehydrogenase), as well as those required for dissimilatory sulfate reduction (e.g. alpha, beta, and gamma subunits of dissimilatory-type sulfite reductase, adenylylsulfate reductase, ATP sulfurylase) and lactate utilization (lactate dehydrogenase). The present result demonstrated the feasibility of subnanogram proteomics for profiling protein expression from small numbers of microbial consortia recovered from the environment.
4. Conclusion
In this work, we have investigated the impact of LC, MS and data analysis contributions to proteome coverage for samples containing 0.5 ng to 50 ng of protein. Past efforts to analyze subnanogram amounts of protein have resulted in extremely low coverage, e.g., <100 proteins. Here, we show that the combination of low flow rate provided by a 30-μm-i.d. LC column, Orbitrap Fusion Lumos Tribrid mass spectrometer, and the MBR algorithm of MaxQuant provided unprecedented proteome coverage of nearly 1,000 proteins from 0.5 ng samples, and >3,000 proteins for ~10 ng samples of mammalian cell lysate, a level of coverage typically achieved only with much larger samples. We have identified >1000 proteins from 0.75 ng co-cultures of M. acetivorans/D. multivorans, paving the way for analysis of individual microaggregates. We found modest gains (35% increase in peptide identifications) by reducing the LC column i.d. from 75 μm to 30 μm, with a 7-fold flow rate reduction. This may indicate that further efforts to miniaturize the LC separation will result in diminishing returns with respect to proteome coverage, although this should be verified experimentally. In contrast, the improved speed, resolution and sensitivity of the latest generation Orbitrap Fusion Lumos mass spectrometer relative to the LTQ Orbitrap increased peptide identifications by at least a factor of two. However, even with the Lumos, proteome coverage continues to be limited by MS/MS undersampling. While this can be addressed to some extent by implementation of AMT tag or MBR strategies, the implementation of alternatives to LC separations such as ultrahigh resolution ion mobility/MS enabled by, e.g., Structures for Lossless Ion Manipulations (SLIM) [36,37] may prove well suited to resolving the complexity of subnanogram proteomes. We envision that with further development, subnanogram proteomics will find broad applications and enable discoveries in the analysis of circulating tumor cells and mini organs, understanding of tissue microenvironments by spatial proteomics, embryonic development, and single cell analysis.
Highlight.
In-depth proteome analysis was achieved for low- and subnanogram biological samples
NanoLC with 30-μm-i.d. column and Orbitrap Fusion Lumos MS provided greatest coverage
This is a promising analytical platform for proteomic analysis of single mammalian cells
Acknowledgments
A portion of this research was performed under the Facilities Integrating Collaborations for User Science (FICUS) initiative and used resources at the DOE Joint Genome Institute and the Environmental Molecular Sciences Laboratory, which are DOE Office of Science User Facilities. Both facilities are sponsored by the Office of Biological and Environmental Research and operated under Contract Nos. DE-AC02-05CH11231 (JGI) and DE-AC05-76RL01830 (EMSL). This work was also supported by the NIH National Institute of General Medical Sciences (P41 GM103493) and the NIH National Institute of Biomedical Imaging and Bioengineering (1R21EB020976-01A1). Y.Z., R.Z., P.D.P., R.J.M., L.P.-T. W.-J. Q. and R.T.K would like to thank R.D.S. for many years of inspiration and friendship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mann M, Kulak NA, Nagaraj N, Cox J. The coming age of complete, accurate, and ubiquitous proteomes. Mol Cell. 2013;49:583–590. doi: 10.1016/j.molcel.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 2.Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 3.Wulfkuhle JD, Liotta La, Petricoin EF. Proteomic applications for the early detection of cancer. Nat Rev Cancer. 2003;3:267–275. doi: 10.1038/nrc1043. [DOI] [PubMed] [Google Scholar]
- 4.Li S, Plouffe BD, Belov AM, Ray S, Wang X, Murthy SK, Karger BL, Ivanov AR. An integrated platform for isolation, processing, and mass spectrometry-based proteomic profiling of rare cells in whole blood. Mol Cell Proteomics. 2015;14:1672–1683. doi: 10.1074/mcp.M114.045724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waanders LF, Chwalek K, Monetti M, Kumar C, Lammert E, Mann M. Quantitative proteomic analysis of single pancreatic islets. Proc Natl Acad Sci USA. 2009;106:18902–18907. doi: 10.1073/pnas.0908351106. 0908351106 [pii]\n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guo T, Kouvonen P, Koh CC, Gillet LC, Wolski WE, Röst HL, Rosenberger G, Collins BC, Blum LC, Gillessen S, Joerger M, Jochum W, Aebersold R. Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat Med. 2015;21:407–13. doi: 10.1038/nm.3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ram RJ, VerBerkmoes NC, Thelen MP, Tyson GW, Baker BJ, Blake RC, Shah M, Hettich RL, Banfield JF. Community proteomics of a natural microbial biofilm. Science. 2005;308:1915–1920. doi: 10.1126/science.1109070. [DOI] [PubMed] [Google Scholar]
- 8.Hu S, Zhang L, Newitt R, Aebersold R, Kraly JR, Jones M, Dovichi NJ. Identification of proteins in single-cell capillary electrophoresis fingerprints based on comigration with standard proteins. Anal Chem. 2003;75:3502–3505. doi: 10.1021/ac034154j. [DOI] [PubMed] [Google Scholar]
- 9.Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol Cell. 2015;58:610–620. doi: 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Kelly RT, Tolmachev AV, Page JS, Tang K, Smith RD. The ion funnel: Theory, implementations, and applications. Mass Spectrom Rev. 2010;29:294–312. doi: 10.1002/mas.20232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Z, Krylov S, Arriaga EA, Polakowski R, Dovichi NJ. One-dimensional protein analysis of an HT29 human colon adenocarcinoma cell. Anal Chem. 2000;72:318–322. doi: 10.1021/ac990694y. [DOI] [PubMed] [Google Scholar]
- 12.Wilm M, Mann M. Analytical properties of the nanoelectrospray ion source. Anal Chem. 1996;68:1–8. doi: 10.1021/ac9509519. [DOI] [PubMed] [Google Scholar]
- 13.Smith RD, Shen Y, Tang K. Ultrasensitive and quantitative analyses from combined separations-mass spectrometry for the characterization of proteomes. Acc Chem Res. 2004;37:269–278. doi: 10.1021/ar0301330. [DOI] [PubMed] [Google Scholar]
- 14.Marginean I, Tang K, Smith RD, Kelly RT. Picoelectrospray ionization mass spectrometry using narrow-bore chemically etched emitters. J Am Soc Mass Spectrom. 2014;25:30–36. doi: 10.1007/s13361-013-0749-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marginean I, Page JS, Tolmachev AV, Tang K, Smith RD. Achieving 50% ionization efficiency in subambient pressure ionization with nanoelectrospray. Anal Chem. 2010;82:9344–9349. doi: 10.1021/ac1019123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun L, Zhu G, Zhao Y, Yan X, Mou S, Dovichi NJ. Ultrasensitive and fast bottom-up analysis of femtogram amounts of complex proteome digests. Angew Chemie - Int Ed. 2013;52:13661–13664. doi: 10.1002/anie.201308139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen Y, Zhao R, Berger SJ, Anderson GA, Rodriguez N, Smith RD. High-efficiency nanoscale liquid chromatography coupled on-line with mass spectrometry using nanoelectrospray ionisation for proteomics. Anal Chem. 2002;74:4235–4249. doi: 10.1021/ac0202280. [DOI] [PubMed] [Google Scholar]
- 18.Shen Y, Tolić N, Masselon C, Paša-Tolić L, Camp DG, Hixson KK, Zhao R, Anderson GA, Smith RD. Ultrasensitive proteomics using high-efficiency on-line micro-SPE-nanoLC-nanoESI MS and MS/MS. Anal Chem. 2004;76:144–154. doi: 10.1021/ac030096q. [DOI] [PubMed] [Google Scholar]
- 19.Wang N, Xu M, Wang P, Li L. Development of mass spectrometry-based shotgun method for proteome analysis of 500 to 5000 cancer cells. Anal Chem. 2010;82:2262–2271. doi: 10.1021/ac9023022. [DOI] [PubMed] [Google Scholar]
- 20.Wiśniewski JR, Ostasiewicz P, Mann M. High recovery FASP applied to the proteomic analysis of microdissected formalin fixed paraffin embedded cancer tissues retrieves known colon cancer markers. J Proteome Res. 2011;10:3040–3049. doi: 10.1021/pr200019m. [DOI] [PubMed] [Google Scholar]
- 21.Chen Q, Yan G, Gao M, Zhang X. Ultrasensitive proteome profiling for 100 living cells by direct cell injection, online digestion and nano-LC-MS/MS analysis. Anal Chem. 2015;87:6674–6680. doi: 10.1021/acs.analchem.5b00808. [DOI] [PubMed] [Google Scholar]
- 22.Chen W, Wang S, Adhikari S, Deng Z, Wang L, Chen L, Ke M, Yang P, Tian R. Simple and integrated spintip-based technology applied for deep proteome profiling. Anal Chem. 2016;88:4864–4871. doi: 10.1021/acs.analchem.6b00631. [DOI] [PubMed] [Google Scholar]
- 23.Huang EL, Piehowski PD, Orton DJ, Moore RJ, Qian WJ, Casey CP, Sun X, Dey SK, Burnum-Johnson KE, Smith RD. Snapp: Simplified nanoproteomics platform for reproducible global proteomic analysis of nanogram protein quantities. Endocrinology. 2016;157:1307–1314. doi: 10.1210/en.2015-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clair G, Piehowski PD, Nicola T, Kitzmiller JA, Huang EL, Zink EM, Sontag RL, Orton DJ, Moore RJ, Carson JP, Smith RD, Whitsett JA, Corley RA, Ambalavanan N, Ansong C. Spatially-resolved proteomics: rapid quantitative analysis of laser capture microdissected alveolar tissue samples. Sci Rep. 2016;6:39223. doi: 10.1038/srep39223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dawson KS, Osburn MR, Sessions AL, Orphan VJ. Metabolic associations with archaea drive shifts in hydrogen isotope fractionation in sulfate-reducing bacterial lipids in cocultures and methane seeps. Geobiology. 2015;13:462–477. doi: 10.1111/gbi.12140. [DOI] [PubMed] [Google Scholar]
- 26.Savas JN, De Wit J, Comoletti D, Zemla R, Ghosh A, Yates JR. Ecto-Fc MS identifies ligand-receptor interactions through extracellular domain Fc fusion protein baits and shotgun proteomic analysis. Nat Protoc. 2014;9:2061–74. doi: 10.1038/nprot.2014.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11:2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 28.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 29.Kim S, Pevzner PA. MS-GF+ makes progress towards a universal database search tool for proteomics. Nat Commun. 2014;5:5277. doi: 10.1038/ncomms6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bateman KP, White RL, Thibault P. Disposable emitters for on-line capillary zone electrophoresis/nanoelectrospray mass spectrometry. Rapid Commun Mass Spectrom. 1997;11:307–315. doi: 10.1002/(SICI)1097-0231(19970215)11:3<307::AID-RCM850>3.0.CO;2-M. [DOI] [Google Scholar]
- 31.Conrads TP, Anderson GA, Veenstra TD, Paša-Tolić L, Smith RD. Utility of accurate mass tags for proteome-wide protein identification. Anal Chem. 2000;72:3349–3354. doi: 10.1021/ac0002386. [DOI] [PubMed] [Google Scholar]
- 32.Zimmer JSD, Monroe ME, Qian WJ, Smith RD. Advances in proteomics data analysis and display using an accurate mass and time tag approach. Mass Spectrom Rev. 2006;25:450–482. doi: 10.1002/mas.20071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanca A, Biosa G, Pagnozzi D, Addis MF, Uzzau S. Comparison of detergent-based sample preparation workflows for LTQ-Orbitrap analysis of the Escherichia coli proteome. Proteomics. 2013;13:2597–2607. doi: 10.1002/pmic.201200478. [DOI] [PubMed] [Google Scholar]
- 34.Boetius A, Ravenschlag K, Schubert CJ, Rickert D, Widdel F, Gieseke A, Amann R, Jørgensen BB, Witte U, Pfannkuche O. A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature. 2000;407:623–626. doi: 10.1038/35036572. [DOI] [PubMed] [Google Scholar]
- 35.Orphan VJ, Taylor LT, Hafenbradl D, Delong EF. Culture-dependent and culture-independent characterization of microbial assemblages associated with high-temperature petroleum reservoirs. Appl Environ Microbiol. 2000;66:700–711. doi: 10.1128/AEM.66.2.700-711.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Deng L, Ibrahim YM, Hamid AM, Garimella SVB, Webb IK, Zheng X, Prost SA, Sandoval JA, Norheim RV, Anderson GA, Tolmachev AV, Baker ES, Smith RD. Ultra-high resolution ion mobility separations utilizing traveling waves in a 13 m serpentine path length structures for lossless ion manipulations module. Anal Chem. 2016;88:8957–8964. doi: 10.1021/acs.analchem.6b01915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng L, Ibrahim YM, Baker ES, Aly NA, Hamid AM, Zhang X, Zheng X, Garimella SVB, Webb IK, Prost SA, Sandoval JA, Norheim RV, Anderson GA, Tolmachev AV, Smith RD. Ion mobility separations of isomers based upon long path length structures for lossless ion manipulations combined with mass spectrometry. Chemistry Select. 2016;1:2396–2399. doi: 10.1002/slct.201600460. [DOI] [PMC free article] [PubMed] [Google Scholar]