

Abstract
Influenza is an annual, global health care concern. Secondary bacterial pneumonia is a severe complication associated with primary influenza virus infection, often resulting in critical morbidity and mortality. Our laboratory has identified influenza-induced suppression of anti-bacterial Type 17 immunity as a mechanism for enhanced susceptibility to bacterial super-infection. We have shown that influenza-induced type I interferon impairs Type 17 activation. STAT1 is a transcription factor involved in interferon signaling, shared by type I, II, and III interferon. In this work, we investigated the role of STAT1 signaling during influenza, methicillin-resistant Staphylococcus aureus (MRSA) super-infection. STAT1−/− mice had increased morbidity and airway inflammation compared to control mice during influenza mono-infection. Despite this worsened anti-viral response, STAT1−/− mice were protected from super-infection bacterial burden and mortality compared to controls. Type 17 immune activation was increased in lymphocytes in STAT1−/− mice during super-infection. The elevation in Type 17 immunity was not related to increased IL-23 production, as type I interferon could inhibit IL-23 expression in a STAT1 independent manner. STAT1−/− antigen presenting cells were inherently biased towards Type 17 polarization compared to control cells. Further, STAT1−/− dendritic cells produced attenuated IL-6 and TNFα upon heat-killed S. aureus stimulation compared to control. Overall, these data indicate that STAT1 signaling plays a detrimental role in influenza, MRSA super-infection by controlling the magnitude of Type 17 immune activation.
Introduction
Influenza infection remains a leading cause of respiratory illness worldwide. During severe seasons, influenza mortality in the United States estimates range up to 50,000 deaths (1). Secondary bacterial pneumonia complicating primary influenza virus infection is an important cause of morbidity and mortality. It is estimated that nearly all influenza-associated deaths during the 1918 Spanish influenza pandemic were due to secondary bacterial super-infection (2), and similarly had a major role in morbidity and mortality during the most recent global pandemic, which occurred in 2008–2009 (3). In recent years, the role of bacterial super-infection with community-acquired methicillin-resistant Staphylococcus aureus (MRSA) during influenza infection has been appreciated as a significant source of mortality (4–6). A more complete understanding of the immune mechanisms that enhance susceptible to secondary bacterial pneumonia during influenza infection may yield improved strategies to prevent or treat human disease.
We have previously demonstrated that influenza infection suppresses Type 17 immunity via type I interferon (IFN), leading to exacerbation of bacterial pneumonia following challenge with both gram-positive and gram-negative bacteria in mice (7–10). The cytokines classically associated with Type 17 immunity are IL-17 and IL-22, which are secreted by T cell subsets and orchestrate inflammation, bacterial clearance, and subsequent airway repair (11, 12). Inhibition of Type 17 immunity during influenza, bacterial super-infection occurred via suppression of IL-23, a critical inducer of IL-17 and IL-22 secretion from T cells in the lung (7).
Type I IFN induces transcriptional activity through the signal transducer of activation and transcription (STAT) pathway. Upon binding of type I IFN α or β to the type I IFN receptor (IFNAR), tyrosine kinase-dependent phosphorylation of STAT1 and STAT2 occurs, forming an activated STAT1, STAT2 heterodimer (13). This heterodimer complexes with a third factor, IRF-9, to form the canonical type I IFN-stimulated multimeric transcription factor, interferon-stimulated gene factor 3 (ISGF3); which then translocates to the nucleus to modulate gene transcription, promoting an antiviral immune state. Of note, type II IFN-γ signals predominantly through formation of activated STAT1 homodimers, while type III IFN-λ signals via IGSF3 but utilizes a distinct surface receptor, IFNLR (14). Modulation of STAT1 activity might impact subsequent susceptibility to bacterial super-infection in the lung due to its effects on interferon response. To further characterize the contribution of STAT1 towards exacerbation of bacterial pneumonia during influenza infection, we subjected wild-type (WT) and STAT1−/− deficient mice to MRSA pulmonary challenge during primary influenza infection.
Materials & Methods
Mice
Eight week old wild-type C57BL/6 mice were purchased from Taconic Farms. STAT1−/− mice on the C57BL/6 background were obtained from Dr. Christian Schindler (Columbia University) and Dr. David Levy (New York University). Mice were maintained under pathogen-free conditions and were co-housed in the same facility prior to the performed studies. All studies were performed using sex matched male mice, except where noted. All of the animal studies were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Viral, Bacterial Super-Infection
Influenza A/PR/8/34 H1N1 was propagated in chicken eggs as previously reported (15). Mice were infected with 50 PFU of influenza virus (in 50 μl of sterile PBS) or vehicle by oropharyngeal aspiration (OP). After 6 days of influenza infection, mice were challenged with 5 x 107 CFU of MRSA, USA300 (in 50 μl of sterile PBS) or vehicle by OP. MRSA, USA300 was obtained from Dr. Alice Prince (Columbia University) and was cultured overnight in modified CCY medium to stationary growth phase. Twenty-four hours after bacterial infection, mouse tissues were harvested for analyses. Mortality studies were performed using a dose of 2.5 x 108 CFU of MRSA via OP.
Analysis of Lung Infection
At the indicated time points, mouse lungs were bronchoalveolar lavaged (BAL) with 1 ml of sterile PBS for inflammatory cell counts. BAL cells were then mounted to slides using a Cytospin (Thermo Fisher) and were stained with Protocol Hema 3 (Fisher Scientific). The cranial lobe of the right lung was homogenized in sterile PBS and used for plating to determine bacterial burden and cytokine Bioplex (Bio-Rad) or Lincoplex (Millipore) analysis. The middle and caudal lobes of the right lung were snap frozen in liquid nitrogen, followed by homogenization and isolation of whole lung RNA using the Absolutely RNA miniprep kit (Agilent Technologies). RNA analysis was performed using commercially available Assay on Demand Taqman probes and primers (Applied Biosystems).
Flow Cytometry
At tissue harvest, the left lung was utilized for flow cytometry analyses. The left lung was digested with collagenase as previously described (16). Resulting single cell preparations were stimulated in vitro with PMA (50 ng/ml) and ionomycin (750 ng/ml) for 4 hours at 37° C. Cell were stained with antibodies against CD4 and γδTCR and then fixed and permeabilized and stained with fluorescent conjugated antibodies against IL-17 and IL-22 (BD Biosciences). Analysis was performed using a FACS Aria (BD Biosciences) apparatus.
Bone Marrow Dendritic Cell Culture
Bone marrow was isolated from the femurs of C57BL/6 and STAT1−/− mice as previously reported (17). Following 7 days in culture with DMEM containing GM-CSF, the non-adherent cells (bone marrow dendritic cells) were collected and used for assays. Dendritic cells were stimulated overnight with peptidoglycan (PGN) or lipoteichoic acid (LTA) from S. aureus (Sigma Aldrich) in the presence or absence of IFNβ or IFNγ (PBL Technology; 10 U/mL, 10 ng/mL, respectively).
In vitro T cell Polarization
Naïve mouse T cells were isolated from the spleens of OTII mice using CD4+ CD62L+ selection beads (Miltenyi) (18). Bone marrow dendritic cells from C57BL/6 or STAT1−/− mice were pulsed with heat-killed MRSA or vehicle for 24 hours. Following this, naïve OTII T cells were treated with IL-2 (40U/ml) and OTII ovalbumin peptide (OVA323-339, 5μM) and were incubated with the bone marrow dendritic cells for 5 days. Cytokine production was assessed by Bioplex assay.
Statistical Analyses
The data presented herein represent individual animals (or replicates) with an overlay of the mean ± standard error of the mean. Significance was tested by one-way or two-way ANOVA as appropriate followed by Tukey post-hoc test. For comparison of two groups, unpaired t test was utilized. Mouse survival data were analyzed by Mann-Whitney test. All statistical tests were performed using GraphPad Prism software. All studies were repeated a minimum of twice in individual animal cohorts.
Results
Influenza-induced pulmonary inflammation is enhanced in STAT1−/− mice
It has been previously reported that STAT1−/− mice are deficient in interferon signaling and have altered susceptibility to influenza infection (19). In order to study secondary bacterial infection in this mouse model, it was necessary to evaluate the impact of STAT1 on influenza mono-infection. To confirm the published phenotype in our mice, WT or STAT1−/− mice were infected with influenza A/PR/8/34 for 7 days. We chose a dose of influenza virus that would induce moderate disease in WT animals due to the expectation that STAT1−/− mice would have a worse disease phenotype. As expected, STAT1−/− mice lost significantly more weight than control animals at day 6 post-infection (Figure 1A). This increased morbidity did not correlate with a consistent increase in viral titers on day 7, similar to previous findings (Figure 1B) (19). Next, airway inflammation was assessed by BAL cell analysis. STAT1−/− mice had elevated BAL cell counts and specifically, neutrophils were increased compared to WT mice, similar to previous reports (Figure 1C, D) (20). Consistent with these findings, the concentration of the pro-neutrophilic chemokines IL-17 (44.2 ± 22.1 pg/mL vs. 7.7 ± 2.2 pg/mL, p = 0.11) and G-CSF (3527 ± 1534 pg/mL vs. 119.9 pg/mL, p = 0.04) were increased in whole lung homogenate from STAT1−/− mice compared to WT mice.
Figure 1.
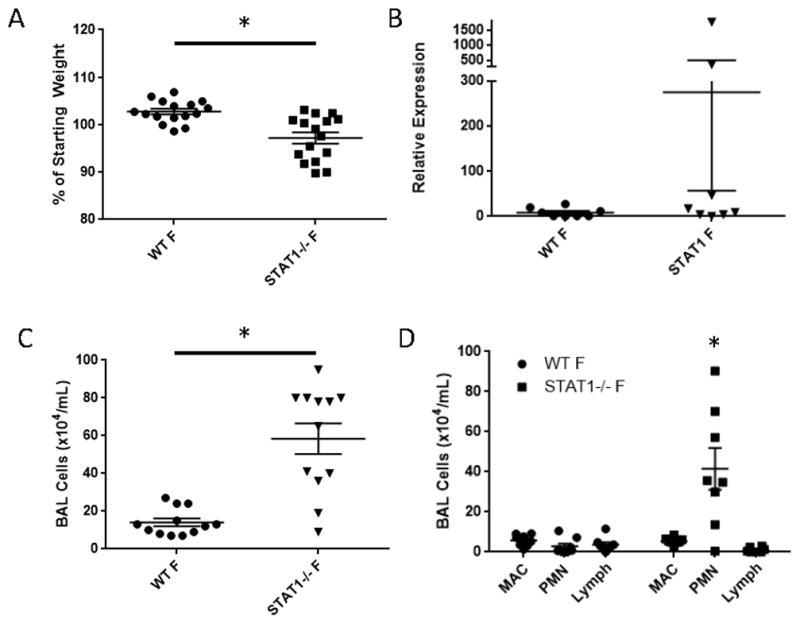
Loss of STAT1 signaling results in increased influenza-induced morbidity and inflammation. WT or STAT1−/− mice were infected with influenza A/PR/8/34 and morbidity, viral burden, and inflammation was assessed 6 or 7 days post-infection. A) Influenza-induced morbidity by weight loss on day 6 post-infection (n = 16). B) Influenza burden by M protein RT-PCR on day 7 post-infection (n = 8). C) BAL inflammation (n = 12). D) Differential BAL cell counts (n = 8). F = influenza infected, MAC = macrophage, PMN = polymorphonuclear cells, Lymph = lymphocytes. *p < 0.05
Due to the elevated inflammation, we then confirmed that STAT1−/− mice are deficient in known STAT1 signaling. CXCL9 and CXCL10 are well known type II IFN-induced genes, which are known to signal exclusively through STAT1. STAT1−/− mice lacked production of CXCL9 and CXCL10, confirming our STAT1−/− phenotype during influenza infection (Figure 2A, B). Further, we assessed expression of known interferon stimulated genes (ISG). Consistent with protein data, Cxcl10 expression was inhibited in STAT1−/− mice compared to controls (approximately 41 fold) (Figure 2C). Induction of the Type I IFN target gene, Mx1 was significantly attenuated in STAT1−/− mice, but to a lesser extent (approximately 5 fold) than Cxcl10 (Figure 2D). Expression of the type I and II IFN stimulated gene, Ccl12 was not altered by STAT1 deletion (Figure 2E). These data indicate that STAT1−/− mice have increased morbidity and airway inflammation compared to WT that is unrelated to viral burden, consistent with previous reports, and that ISG responses are differentially impacted.
Figure 2.

STAT1 deletion impairs the influenza-induced ISG response. WT or STAT1−/− mice were infected with influenza A/PR/8/34 for 7 days. The ISG response was measured by Lincoplex on lung homogenate (A, B, n = 8) or RT-PCR on lung RNA (C – E, n = 3–15). Naïve mice are untreated WT mice. F = influenza infected. *p < 0.05
STAT1−/− mice are protected from influenza, MRSA super-infection
Our previously published data suggested that a mechanism by which influenza increases susceptibility to bacterial pneumonia is suppression of Type 17 immune activation by IFN (7). Specifically, we have shown that type I IFN, which signals through STAT1 and STAT2, impairs IL-23 production by dendritic cells resulting in abrogated Type 17 host defense. To examine the role of STAT1 in this pathway, WT or STAT1−/− mice were infected with influenza A/PR/8/34 or vehicle for 6 days followed by MRSA infection for 24 hours. Preceding influenza infection resulted in significantly increased MRSA burden in the lung in WT mice, but STAT1−/− mice were protected from exacerbation of secondary MRSA pneumonia by influenza infection (Figure 3A). To then determine if the protection from bacterial burden was relevant, WT and STAT1−/− mice were challenged with a combination of influenza A/PR/8/34 and MRSA known to be lethal in WT mice. As expected, WT mice succumbed to super-infection by 24 hours post-challenge, however STAT1−/− mice survived longer with only 50% mortality at 24 hours post-infection (Figure 3B). This protection was observed in the context of STAT1−/− mice having worse morbidity due to influenza infection alone. Similar to influenza mono-infection, STAT1−/− super-infected mice had increased BAL cells, predominantly neutrophils, in their lungs when compared to WT mice (Figure 3C, D). These data demonstrate that STAT1 is detrimental during influenza, MRSA super-infection and deletion results in protection from bacterial burden and mortality.
Figure 3.
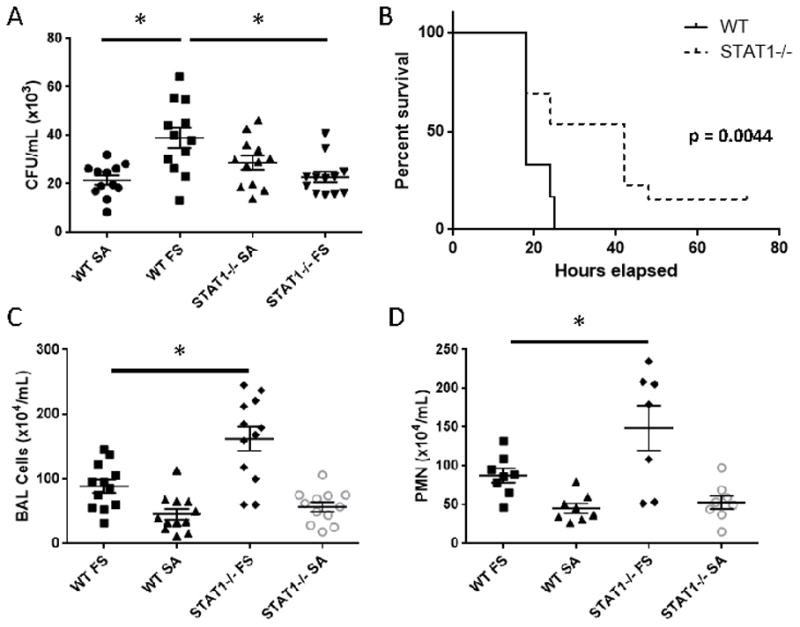
STAT1−/− mice are protected from influenza, MRSA super-infection. WT or STAT1−/− mice were infected with influenza A/PR/8/34 or vehicle for 6 days prior to infection with MRSA (USA300) or vehicle for 24 hours. A) Bacterial burden in the lungs of infected mice (n = 12). B) Mortality plot for super-infected mice (n = 12–13). C) BAL inflammation (n = 12). D) Differential BAL cell counts (n = 7–8). SA = MRSA infected, FS = influenza MRSA super-infected, PMN = polymorphonuclear cells. *p < 0.05
STAT1−/− mice have accentuated Type 17 immune activation compared to WT mice
Type 17 cytokines are known to induce neutrophilia and promote host defense against S. aureus (7, 9–11). The observation that STAT1−/− mice have elevated neutrophilia and improved MRSA clearance during super-infection suggests increased Type 17 immune activation. To test this hypothesis, we measured IL-17 and IL-22 gene expression in whole lung from influenza, MRSA, and super-infected mice. Indeed, STAT1−/− mice had significantly increased Il17 expression compared to WT mice during influenza mono-infection, MRSA mono-infection, and super-infection (Figure 4A). Further, STAT1−/− mice had increased Il22 expression during influenza mono-infection and super-infection (Figure 4B). To better define the impact of STAT1 deletion on Type 17 immune activation, we performed flow cytometry on the lungs from mono- and super-infected animals. We have previously shown that S. aureus induces IL-17 production from CD4+ and γδTCR+ cells, which is attenuated by preceding influenza infection (7). Flow cytometry revealed that STAT1−/− mice had increased numbers of CD4+ IL-17+ cells compared to WT mice (Figure 4C). Further, STAT1−/− mice had increased numbers of γδTCR+ IL-17+ cells during MRSA mono-infection (Figure 4D). Similarly, STAT1−/− mice displayed increased CD4+ IL-22+ cells (Figure 4E) and increased γδTCR+ IL-22+ cells (Figure 4F) compared to WT during super-infection. These data confirm a bias towards Type 17 immune activation in STAT1−/− mice compared to WT and suggest a primary effect on CD4+ T cells.
Figure 4.
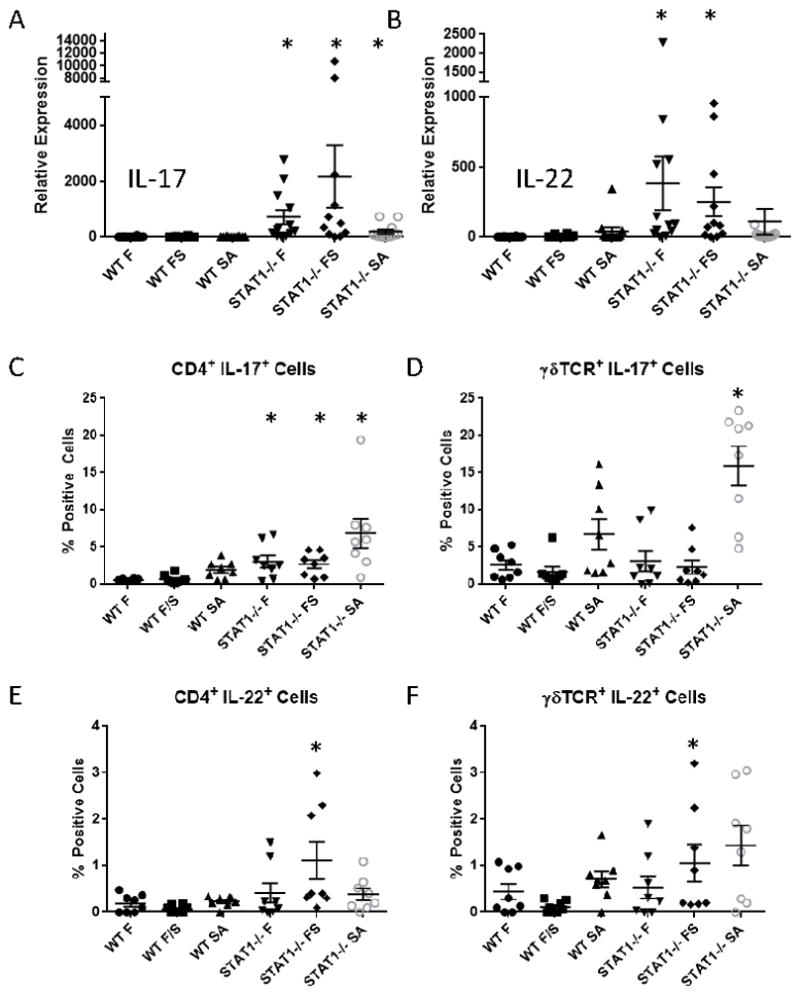
Type 17 immunity is increased in STAT1−/− mice. WT or STAT1−/− mice were infected with influenza A/PR/8/34 or vehicle for 6 days prior to infection with MRSA (USA300) or vehicle for 24 hours. A, B) Type 17 cytokine gene expression by RT-PCR on lung RNA (n = 11–12). C – F) Type 17 cytokine producing cells by flow cytometry (n = 8). F = influenza infected, SA = MRSA infected, FS = influenza MRSA super-infected. *p < 0.05 vs. WT for each treatment
Increased Type 17 immune activation in STAT1−/− mice is not IL-23 dependent
Our previous data demonstrated that preceding influenza inhibits S. aureus induced IL-23 production, thus limiting Type 17 immunity (7). We hypothesized that STAT1 deletion would result in increased IL-23 production compared to WT mice. Surprisingly, STAT1−/− mice did not display increased expression of IL-23 compared to WT mice in the lung (Figure 5A). To further investigate the relationship between STAT1 signaling and IL-23 production, bone marrow dendritic cells were isolated from WT and STAT1−/− mice and stimulated with type I and II IFN. As we have previously shown, type I IFN suppressed PGN-induced IL-23 production (Figure 5B). Type II IFN did not have a similar effect on this pathway. In the absence of STAT1 signaling, type I IFN was still able to inhibit IL-23 production similar to its effect in WT cells. In additional studies using S. aureus–derived LTA (which, similar to PGN is a TLR2 ligand), we confirmed that the effect of type I IFN on IL-23 production by dendritic cells does not require STAT1 (Figure 5C). Interestingly, type II IFN was able to inhibit IL-23 production in this context in a STAT1 dependent manner. These data indicate that enhanced Type 17 immunity in STAT1−/− mice is not due to increased IL-23 production.
Figure 5.
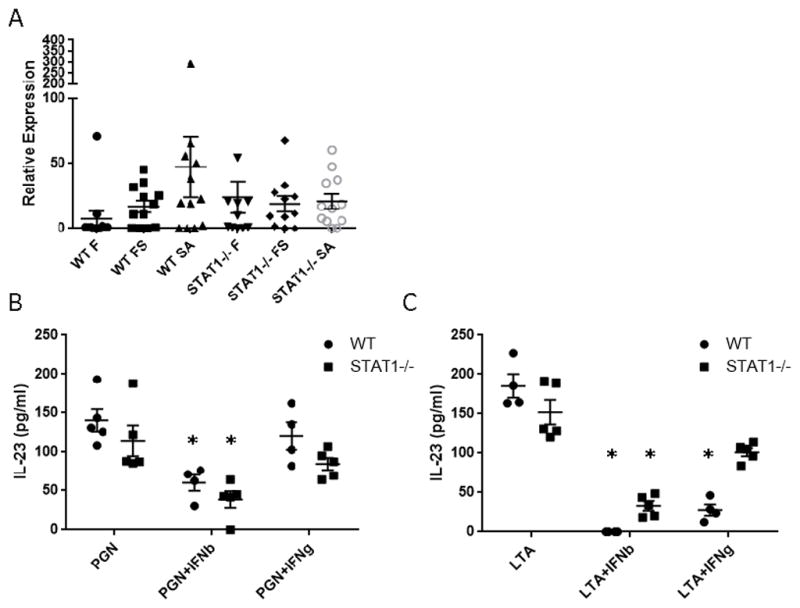
IL-23 production is not impacted by STAT1 deletion. WT or STAT1−/− mice were infected with influenza A/PR/8/34 or vehicle for 6 days prior to infection with MRSA (USA300) or vehicle for 24 hours. A) IL-23 gene expression by RT-PCR on lung RNA (n = 11–12). WT or STAT1−/− bone marrow derived dendritic cells were stimulated with the Tlr2 ligands peptidoglycan (PGN) or lipoteichoic acid (LTA) for 24 hours in the presence or absence of IFNs. B, C) IL-23 production by ELISA (n = 4–5). F = influenza infected, SA = MRSA infected, FS = influenza MRSA super-infected. *p < 0.05 vs. WT in the absence of IFN
STAT1−/− antigen presenting cells are biased towards Type 17 immune polarization
Next, we examined whether Type 17 immune polarization is inherently enhanced in STAT1−/− mice compared to controls. To do this, we isolated bone marrow dendritic cells from WT and STAT1−/− mice. These cells were then pulsed with heat-killed MRSA or vehicle for 24 hours and were then incubated with naïve T cells (OVA-specific) from OTII mice in the presence of OVA peptide. Heat-killed MRSA treatment inhibited antigen-specific T cell production of IL-17, but increased IL-6 and TNFα levels compared to control cultures in both WT and STAT1−/− mice (Figure 6). T cells polarized with STAT1−/− dendritic cells produced increased IL-17 compared to T cells polarized with WT dendritic cells in both control and MRSA-stimulated settings (Figure 6A). Interestingly heat-killed MRSA stimulated lower levels of IL-6 and TNFα in STAT1−/− cultures (Figure 6B, C). Collectively, these data indicate that STAT1−/− dendritic cells favor Type 17 polarization of WT naïve T cells and have impaired MRSA-mediated induction of pro-inflammatory cytokines.
Figure 6.
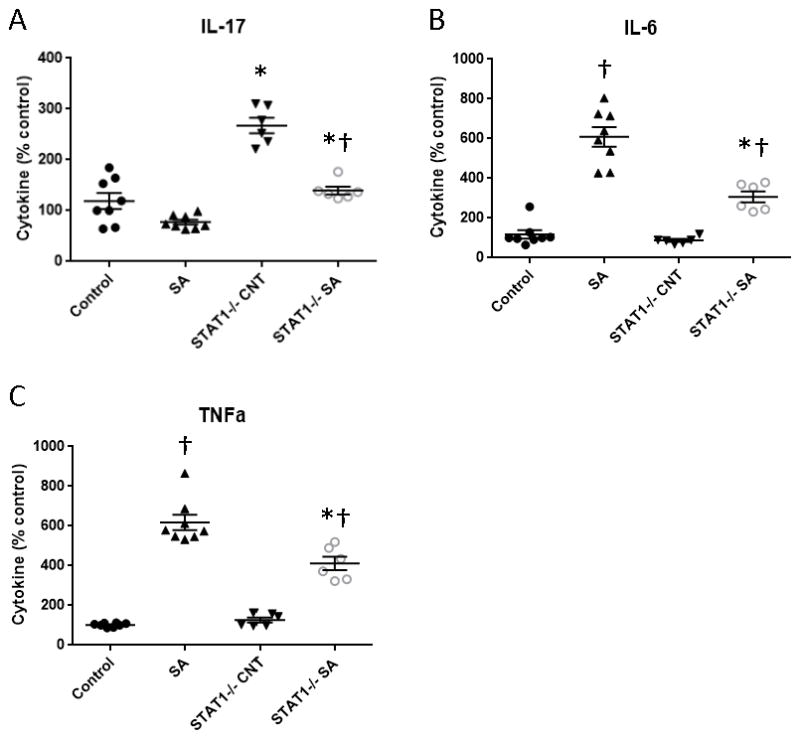
STAT1−/− antigen presenting cells favor Type 17 immune polarization. WT or STAT1−/− bone marrow derived dendritic cells were incubated with OTII naïve ovalbumin-specific T cells for 5 days in the presence of ovalbumin peptide with or without preceding challenge with heat-killed MRSA. Cytokine levels were determined by Bioplex assay (n = 6–8). SA = heat-killed MRSA, CNT = control. *p < 0.05 WT vs. STAT1, †p < 0.05 SA vs. CNT
Discussion
In this study, we have demonstrated that STAT1 activity is associated with exacerbation of secondary MRSA pneumonia during influenza infection. STAT1−/− mice had decreased bacterial burden and improved survival compared to WT mice during influenza, MRSA super-infection, despite increased morbidity and inflammation due to influenza infection alone. Further, we have shown that this protection from super-infection was associated with increased Type 17 immune activation in STAT1−/− mice compared to WT mice. These findings are consistent with our previous studies and further support a critical role for suppression of Type 17 immunity in the pathogenesis of influenza, S. aureus super-infection (7–10). In previous studies, impaired secretion of IL-23, which helps activate and maintain Type 17 immunity, was identified as a key mechanism. Interestingly, we found in this study that type I IFN-mediated IL-23 suppression was not dependent on STAT1. Our previous finding that IL-23 production in the lung was dependent on the presence of IFNAR during influenza, S. aureus super-infection suggests that type I IFN regulates IL-23 through alternative signaling mechanism(s) (7). Together, these findings suggest the presence of IFNAR-dependent, STAT1-independent mechanisms for IL-23 suppression. Our finding that bacterial clearance was rescued despite IL-23 suppression in the setting of STAT1 deficiency indicates the existence of additional pathways leading to the activation of Type 17 immunity independent of IL-23.
STAT1−/− mice demonstrated increased morbidity during primary influenza infection, consistent with previous reports (19–22). In prior studies, the degree of susceptibility varied with the strain of influenza used and the genetic background of the mice. In one study, infection with influenza A/PR/8/34 in C57BL/6 mice, as were used here, resulted in increased morbidity and marked granulocytic pulmonary infiltration, which were unrelated to viral burden (20). We report similar findings herein. That study also demonstrated reduced LD50 to influenza compared to WT mice and demonstrated a bias towards Type 2 polarized T cells in the absence of IFN signaling, but did not study the effects of influenza on Type 17 immune responses. The morbidity we observed during influenza mono-infection was associated with increased inflammatory cells, particularly neutrophils, in BAL, increased lung expression of IL-17 and IL-22, and increased proportions of IL-17+ and IL-22+ T cells in the lung. These findings all suggest increased neutrophilia and inflammation due to enhanced Type 17 immune activation, which has previously been shown to be associated with increased lung pathology during primary influenza infection (16). In humans, the role of IL-17 and influenza severity remains unclear. Much of our knowledge on the subject was generated during the most recent H1N1 influenza pandemic, where results may not be generalizable to the pathogenesis of seasonal influenza infections. In general, those studies have shown that severe pandemic H1N1 influenza was associated with increased peripheral IL-17, but that decreased IL-17 was a risk factor for mortality and more severe disease (23–27). Given its recognized role in the pathogenesis of a spectrum of auto-immune and inflammatory conditions, it is plausible that increased Type 17 activity would also be associated with increased inflammatory lung disease during human influenza infections. While tempting to speculate, it is unclear from these studies if inappropriately decreased IL-17 was a marker of inadequate host immune response or was associated with secondary bacterial infection, either of which could have increased mortality risk.
While STAT1 deficiency had a detrimental effect on the host response to influenza mono-infection, it had a protective effect when STAT1−/− mice were subsequently challenged with MRSA on day 6 of influenza infection. STAT1−/− mice had significantly decreased lung bacterial burden 24 hours following bacterial challenge; the burden of bacteria in the lung was equivalent to that seen following MRSA mono-infection, suggesting complete rescue from the impairment in bacterial clearance from the lung observed during influenza, MRSA super-infection in WT mice. This improvement in bacterial clearance was also associated with a survival benefit, as STAT1−/− mice demonstrated prolonged survival following a combination of influenza and MRSA that was rapidly lethal to WT mice. During super-infection, the total number of inflammatory cells, specifically neutrophils, in the lung was again elevated compared to WT mice. Furthermore, similar to influenza mono-infection, lung expression of IL-17 and IL-22 and the proportion of IL-17+ and IL-22+ T cells in the lung were increased compared to WT mice. This suggests that the improved outcomes during influenza, MRSA super-infection could be due to rescue of Type 17 immunity.
The specific effector(s) responsible for these findings remain unclear. Type 17 immunity is generally thought to exert anti-bacterial effects by the proliferation and recruitment of inflammatory cells, particularly neutrophils, and by the induction of antimicrobial peptides at the mucosal surface. Enhanced inflammatory cell infiltrate present in the lung at the time of bacterial challenge is thus one possibility: STAT1−/− mice had significant neutrophilia induced by influenza alone while WT mice did not, indicating that a large additional population of phagocytic cells were present in the lung at the time of bacterial challenge. Previous studies demonstrated that neutrophil depletion did not affect S. aureus lung bacterial burden (9, 28), and that antimicrobial peptides may have a more significant role (9). While neutrophils might be dispensable for ultimate S. aureus clearance if alternate effector mechanisms remain intact, increased neutrophilia at the time of bacterial challenge could have a significant impact on early bacterial clearance, affecting the overall kinetics of MRSA clearance from the lung. When WT and STAT1−/− mice (n=8) were subjected to super-infection for 120 hours, STAT1−/− mice had complete clearance of MRSA by 120 hours, while WT mice continued to have elevated bacterial burden (P=0.04, data not shown). This kinetic response is identical to what was previously described for the clearance of S. aureus from the lung during bacterial mono-infection (10). Bacterial clearance was also associated with normalization of neutrophil counts in BAL from STAT1−/− mice, while they continued to remain elevated in WT mice, suggesting a primary role for neutrophils in this response. To further test the hypothesis that neutrophils were the primary effector responsible for improved bacterial clearance in STAT1−/− mice, we attempted neutrophil depletion in STAT−/− mice using 1A8 neutralizing antibodies, but we were unable to generate adequate neutrophil depletion. Depletion using RB6 neutralizing antibodies, which also affects monocyte populations in addition to neutrophils, proved lethal in this model (data not shown).
When we assessed antimicrobial peptide expression in the lung, we found that expression of regenerating islet-derived (REG) 3β was significantly increased in the lung in STAT1−/− mice compared to WT (n=8) during influenza mono-infection (p = 0.003), MRSA mono-infection (p = 0.08), and super-infection (p = 0.05) (data not shown). REG3β belongs to the C-type lectin family antimicrobial peptides, and mediates antimicrobial activity via binding of bacterial peptidoglycan (29). We previously demonstrated that Type 17 cytokines induce up-regulation of REG3β in mouse respiratory epithelial cells, and influenza, S. aureus super-infection decreased expression of REG3β in the lung of WT mice (10). We did not see a similar effect on expression of lipocalin 2, which we had previously demonstrated to be an important mediator of bacterial clearance in influenza, S. aureus super-infection (10). This finding provides further evidence of the rescue of Type 17 immunity in the lung during influenza, MRSA super-infection in the absence of STAT1 activity and suggests a role for antimicrobial peptides in bacterial clearance.
Surprisingly, we found that type I IFN inhibited S. aureus-induced IL-23 production from dendritic cells independently of STAT1. This was unexpected considering our previous finding that IL-23 production by CD11c+ monocytes in the lung was dependent on IFNAR (7). These data suggest an IFNAR-dependent, STAT1-independent mechanism of IL-23 suppression. Others have reported different responses to influenza infection in IFNAR−/− vs. STAT1−/− mice, with IFNAR−/− mice showing intact interferon-stimulated gene expression, presumably due to signaling redundancy via type III IFN, while STAT1−/− had no such induction (21). The finding that IL-23 suppression was rescued in IFNAR−/− mice but not STAT1−/− is curious; any signaling redundancy mediated by type III IFN would also be abrogated in STAT1−/− deficiency, since signaling occurs for both IFNs via the same transcription factor, ISGF3, containing STAT1. Complicating matters further is the observation that the specific bacterial ligand used (PGN or LTA) had a differential effect on IL-23 suppression in the presence of type II IFN, with WT dendritic cells cultured with type II IFN showing impairment of IL-23 production in the presence of LTA, but not PGN. Finally, we found no effect on IL-23 secretion in the presence of type III IFN (data not shown). It is unclear if the presence of S. aureus-derived PGN in vivo would be sufficient to induce IL-23 despite the concurrent inhibitory effect of LTA in the presence of type II IFN. Others have shown that type II IFN has an important role in influenza, Streptococcus pneumonia super-infection (30). However, in the setting of influenza, S. aureus model, we have demonstrated that type II IFN deficiency did not protect mice from impaired bacterial lung clearance (7), suggesting that the primary effect in vivo is mediated by type I IFN.
Despite suppression of dendritic cell-derived IL-23 at levels similar to those seen in WT mice, Type 17 immunity was rescued nevertheless in STAT1−/− mice. Thus, the rescue of Type 17 immunity we observed appeared to be independent of IL-23 activation. The precise pathways for the development, polarization, activation, and maintenance of Type 17 immune cells remain incompletely understood. It is known that mediators other than IL-23, including IL-1β, TGF-β, IL-6, and IL-21 also modulate Type 17 immune cells (31, 32). Thus, in the setting of STAT1 deficiency, alternate IL-23 independent pathways may play important roles in regulating Type 17 immune cell activity. The reason for these observations is unclear and merit further investigation. Naïve antigen presenting cells from STAT1−/− mice induced higher levels of IL-17 production by WT naïve T cells compared to WT antigen presenting cells with or without priming by heat-killed MRSA. T cells from both WT and STAT1−/− mice secreted IL-6 and TNFα following polarization with MRSA-primed dendritic cells, but cytokine levels were significantly lower in STAT1−/− mice. IL-6 and TNFα are classically associated with Type 1 responses. These results thus suggest that antigen presenting cells from STAT1−/− are inherently biased towards Type 17 T cell polarization. The inherent bias towards Type 17 T cell polarization in the absence of STAT1−/− may contribute to the rescue of Type 17 immunity during influenza, MRSA super-infection and obviate the need for IL-23 production.
In summary, we have demonstrated that STAT1 signaling has a protective effect during influenza mono-infection, but a detrimental effect during influenza, MRSA super-infection. Diminished anti-viral activity due to loss of interferon response during influenza was associated with increased morbidity, but during pulmonary bacterial challenge this deficiency led to improved bacterial clearance and prolonged survival associated with rescue of Type 17 cells, gene transcription, and effectors in the lung. Our data indicate that STAT1 signaling impairs Type 17 T cell polarization through effects on antigen presenting cells. Further studies are required to more fully characterize the mechanisms involved. Ultimately, greater understanding of how interferon responses and Type 17 immunity can be modulated may lead to interventions and therapies to reduce morbidity and mortality associated with influenza and influenza, bacterial super-infections.
Acknowledgments
Support – NIH R01HL107380 (JFA), Parker B. Francis Foundation Fellowship (JFA, KMR, MLM), T32 HD071834 (BL), K08 HL133445 (KMR)
We would like to thank our late friend and colleague Alison Logar, who assisted with analysis of the flow cytometry studies presented herein.
References
- 1.Estimates of deaths associated with seasonal influenza --- United States, 1976–2007. MMWR Morb Mortal Wkly Rep. 2010;59:1057–1062. [PubMed] [Google Scholar]
- 2.Morens DM, Taubenberger JK, Fauci AS. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rice TW, Rubinson L, Uyeki TM, Vaughn FL, John BB, Miller RR, 3rd, Higgs E, Randolph AG, Smoot BE, Thompson BT. Critical illness from 2009 pandemic influenza A virus and bacterial coinfection in the United States. Crit Care Med. 2012;40:1487–1498. doi: 10.1097/CCM.0b013e3182416f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finelli L, Fiore A, Dhara R, Brammer L, Shay DK, Kamimoto L, Fry A, Hageman J, Gorwitz R, Bresee J, Uyeki T. Influenza-associated pediatric mortality in the United States: increase of Staphylococcus aureus coinfection. Pediatrics. 2008;122:805–811. doi: 10.1542/peds.2008-1336. [DOI] [PubMed] [Google Scholar]
- 5.Hall MW, Geyer SM, Guo CY, Panoskaltsis-Mortari A, Jouvet P, Ferdinands J, Shay DK, Nateri J, Greathouse K, Sullivan R, Tran T, Keisling S, Randolph AG I. Pediatric Acute Lung, P. S. I. Sepsis Investigators Network. Innate immune function and mortality in critically ill children with influenza: a multicenter study. Crit Care Med. 2013;41:224–236. doi: 10.1097/CCM.0b013e318267633c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rubinstein E, Kollef MH, Nathwani D. Pneumonia caused by methicillin-resistant Staphylococcus aureus. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2008;46(Suppl 5):S378–385. doi: 10.1086/533594. [DOI] [PubMed] [Google Scholar]
- 7.Kudva A, Scheller EV, Robinson KM, Crowe CR, Choi SM, Slight SR, Khader SA, Dubin PJ, Enelow RI, Kolls JK, Alcorn JF. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J Immunol. 2011;186:1666–1674. doi: 10.4049/jimmunol.1002194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee B, Robinson KM, McHugh KJ, Scheller EV, Mandalapu S, Chen C, Di YP, Clay ME, Enelow RI, Dubin PJ, Alcorn JF. Influenza-induced Type I Interferon Enhances Susceptibility to Gram-negative and Gram-positive Bacterial Pneumonia in Mice. Am J Physiol Lung Cell Mol Physiol. 2015;309:L158–67. doi: 10.1152/ajplung.00338.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson KM, Choi SM, McHugh KJ, Mandalapu S, Enelow RI, Kolls JK, Alcorn JF. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. J Immunol. 2013;191:5153–5159. doi: 10.4049/jimmunol.1301237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robinson KM, McHugh KJ, Mandalapu S, Clay ME, Lee B, Scheller EV, Enelow RI, Chan YR, Kolls JK, Alcorn JF. Influenza A Virus Exacerbates Staphylococcus aureus Pneumonia in Mice by Attenuating Antimicrobial Peptide Production. J Infect Dis. 2014;209:865–75. doi: 10.1093/infdis/jit527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khader SA, Gaffen SL, Kolls JK. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal immunology. 2009;2:403–411. doi: 10.1038/mi.2009.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aujla SJ, Alcorn JF. T(H)17 cells in asthma and inflammation. Biochim Biophys Acta. 2011;1810:1066–1079. doi: 10.1016/j.bbagen.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Decker T, Stockinger S, Karaghiosoff M, Muller M, Kovarik P. IFNs and STATs in innate immunity to microorganisms. J Clin Invest. 2002;109:1271–1277. doi: 10.1172/JCI15770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lopusna K, Rezuchova I, Betakova T, Skovranova L, Tomaskova J, Lukacikova L, Kabat P. Interferons lambda, new cytokines with antiviral activity. Acta virologica. 2013;57:171–179. doi: 10.4149/av_2013_02_171. [DOI] [PubMed] [Google Scholar]
- 15.Braciale TJ. Immunologic recognition of influenza virus-infected cells. I. Generation of a virus-strain specific and a cross-reactive subpopulation of cytotoxic T cells in the response to type A influenza viruses of different subtypes. Cell Immunol. 1977;33:423–436. doi: 10.1016/0008-8749(77)90170-8. [DOI] [PubMed] [Google Scholar]
- 16.Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, Ross TM, Witztum JL, Kolls JK. Critical role of IL-17RA in immunopathology of influenza infection. J Immunol. 2009;183:5301–5310. doi: 10.4049/jimmunol.0900995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin Y, Ritchea S, Logar A, Slight S, Messmer M, Rangel-Moreno J, Guglani L, Alcorn JF, Strawbridge H, Park SM, Onishi R, Nyugen N, Walter MJ, Pociask D, Randall TD, Gaffen SL, Iwakura Y, Kolls JK, Khader SA. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity. 2009;31:799–810. doi: 10.1016/j.immuni.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manni ML, Mandalapu S, McHugh KJ, Elloso MM, Dudas PL, Alcorn JF. Molecular Mechanisms of Airway Hyperresponsiveness in a Murine Model of Steroid-Resistant Airway Inflammation. J Immunol. 2016;196:963–977. doi: 10.4049/jimmunol.1501531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garcia-Sastre A, Durbin RK, Zheng H, Palese P, Gertner R, Levy DE, Durbin JE. The role of interferon in influenza virus tissue tropism. J Virol. 1998;72:8550–8558. doi: 10.1128/jvi.72.11.8550-8558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Durbin JE, Fernandez-Sesma A, Lee CK, Rao TD, Frey AB, Moran TM, Vukmanovic S, Garcia-Sastre A, Levy DE. Type I IFN modulates innate and specific antiviral immunity. Journal of immunology (Baltimore, Md : 1950) 2000;164:4220–4228. doi: 10.4049/jimmunol.164.8.4220. [DOI] [PubMed] [Google Scholar]
- 21.Davidson S, Crotta S, McCabe TM, Wack A. Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun. 2014;5:3864. doi: 10.1038/ncomms4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jewell NA, Cline T, Mertz SE, Smirnov SV, Flano E, Schindler C, Grieves JL, Durbin RK, Kotenko SV, Durbin JE. Lambda interferon is the predominant interferon induced by influenza A virus infection in vivo. J Virol. 2010;84:11515–11522. doi: 10.1128/JVI.01703-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.To KK, Hung IF, Li IW, Lee KL, Koo CK, Yan WW, Liu R, Ho KY, Chu KH, Watt CL, Luk WK, Lai KY, Chow FL, Mok T, Buckley T, Chan JF, Wong SS, Zheng B, Chen H, Lau CC, Tse H, Cheng VC, Chan KH, Yuen KY. Delayed clearance of viral load and marked cytokine activation in severe cases of pandemic H1N1 2009 influenza virus infection. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2010;50:850–859. doi: 10.1086/650581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Almansa R, Socias L, Ramirez P, Martin-Loeches I, Valles J, Loza A, Rello J, Kelvin DJ, Leon C, Blanco J, Andaluz D, Micheloud D, Maravi E, Ortiz de Lejarazu R, Bermejo-Martin JF. Imbalanced pro- and anti-Th17 responses (IL-17/granulocyte colony-stimulating factor) predict fatal outcome in 2009 pandemic influenza. Crit Care. 2011;15:448. doi: 10.1186/cc10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee N, Wong CK, Chan PK, Chan MC, Wong RY, Lun SW, Ngai KL, Lui GC, Wong BC, Lee SK, Choi KW, Hui DS. Cytokine response patterns in severe pandemic 2009 H1N1 and seasonal influenza among hospitalized adults. PloS one. 2011;6:e26050. doi: 10.1371/journal.pone.0026050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arankalle VA, Lole KS, Arya RP, Tripathy AS, Ramdasi AY, Chadha MS, Sangle SA, Kadam DB. Role of host immune response and viral load in the differential outcome of pandemic H1N1 (2009) influenza virus infection in Indian patients. PloS one. 2010:5. doi: 10.1371/journal.pone.0013099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bermejo-Martin JF, Ortiz de Lejarazu R, Pumarola T, Rello J, Almansa R, Ramirez P, Martin-Loeches I, Varillas D, Gallegos MC, Seron C, Micheloud D, Gomez JM, Tenorio-Abreu A, Ramos MJ, Molina ML, Huidobro S, Sanchez E, Gordon M, Fernandez V, Del Castillo A, Marcos MA, Villanueva B, Lopez CJ, Rodriguez-Dominguez M, Galan JC, Canton R, Lietor A, Rojo S, Eiros JM, Hinojosa C, Gonzalez I, Torner N, Banner D, Leon A, Cuesta P, Rowe T, Kelvin DJ. Th1 and Th17 hypercytokinemia as early host response signature in severe pandemic influenza. Crit Care. 2009;13:R201. doi: 10.1186/cc8208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iverson AR, Boyd KL, McAuley JL, Plano LR, Hart ME, McCullers JA. Influenza virus primes mice for pneumonia from Staphylococcus aureus. The Journal of infectious diseases. 2011;203:880–888. doi: 10.1093/infdis/jiq113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kolls JK, McCray PB, Jr, Chan YR. Cytokine-mediated regulation of antimicrobial proteins. Nature reviews Immunology. 2008;8:829–835. doi: 10.1038/nri2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun K, Metzger DW. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat Med. 2008;14:558–564. doi: 10.1038/nm1765. [DOI] [PubMed] [Google Scholar]
- 31.Burkett PR, Meyer zu Horste G, Kuchroo VK. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J Clin Invest. 2015;125:2211–2219. doi: 10.1172/JCI78085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]