

Abstract
Our quest to isolate and characterize natural products with in vitro solid tumor selectivity is driven by access to repositories of Indo-Pacific sponge extracts. In this project an extract of a species of Haplosclerida sponge obtained from the US NCI Natural Products Repository displayed, by in vitro disk diffusion assay (DDA) and IC50 determinations, selective cytotoxicity with modest potency to a human pancreatic cancer cell line (PANC-1) relative to the human lymphoblast leukemia cell line (CCRF-CEM). Two brominated indoles, the known 6-bromo conicamin (1) and the new derivative, 6-Br-8-keto-conicamin A (2), were identified and 2 (IC50 1.5 μM for the natural product vs 4.1 μM for the synthetic material) was determined to be responsible for the cytotoxic activity of the extract against the PANC-1 tumor cell line. The new natural product and ten additional analogs were prepared for further SAR testing.
Keywords: PANC-1 cytotoxicity, Sponge-derived halogenated indoles, Synthetic halogenated indoles
Graphical abstract
1. Introduction
We believe that the study of bioactive natural products from sponges obtained within the Indonesian Coral Triangle, called by some the “biodiversity bullseye,” represents an underexplored, rich opportunity. One example of a significant result obtained from exploration of the region is that manzamine A, a complex sponge-derived antimalarial alkaloid1 (also a putative sponge-associated bacterial product)2 and subject of sustained investigation,3 can be isolated in large amounts from several Indonesian sponges. Motivated by this outcome, we continue to evaluate sponge extracts from repositories rich in Indonesian sponge taxa. Currently, these resources consist of: 558 alcohol preserved sponge samples collected by the UC Santa Cruz team, and 230 Indonesian marine sponge extracts provided to UCSC from the NCI-DTP repository.4 An important filter to prioritize work on these samples utilizes a primary in vitro disk diffusion assay (DDA) developed at the Henry Ford Cancer Institute. This assay flags samples based on selectivity and potency against five solid tumor cell lines (human pancreatic cancer PANC-1, human ovarian OVCAR-5, human lung H125, human hepatoma HepG2, and human brain U251N) compared to responses from two normal cell lines (murine and human hematopoietic progenitor cells, CFU-GM) and a leukemia cell line (CEM).5 The five cancers represented by these cell lines account for 74% of all solid tumor cancers (1,658,000 cases per year in the US) and 70% of solid tumor mortality (589,000 deaths per year in the US).6 The output from our primary screen is quantified by measuring the radius of cell death after exposure to extract or compounds, where selectivity is defined by a differential zone of inhibition between cell lines.
The first phase of our effort to evaluate the sample library described above has been to pinpoint extracts and compounds with solid tumor selectivity in the DDA5 (those with a > 6.5 mm difference in zone of inhibition for solid tumor cell lines vs. either bone marrow progenitor or leukemia cells). During the last two years, 120 of the UCSC sponges along with 136 organic extracts of the NCI-DTP sponge samples have been extracted, pre-fractionated, and tested. The overall priority hit rate is low (≅ 1%), but there have been a number of positive outcomes using this approach with Indonesian sponges, including our recently published finding that makaluvamine J can be isolated from both UCSC and DTP collections of Zyzzya fuliginosa. This compound exhibits solid tumor selectivity for both PANC-1 (IC50 = 54 nM) and OVCAR-5 (IC50 = 120 nM) cells.7 The general steps involved in the discovery process (Chart 1) have now been applied to the 256 samples in this assemblage and one chosen for detailed analysis has provided the results reported herein, while outcomes from the remaining sponge active extracts will be published elsewhere.
Chart 1.
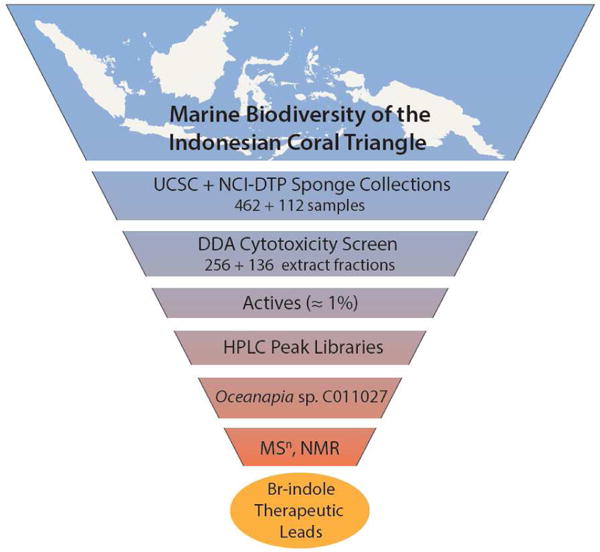
Overview of the campaign involving: sample acquisition, extraction, prefractionation, and Disk Diffusion Assay (DDA) for solid tumor selectivity against five human tumor cell lines. The work flow also involved dereplication and structure elucidation by accurate MSn and NMR spectroscopy along with SAR biological evaluation of natural products and synthetic analogues.
2. Results and discussion
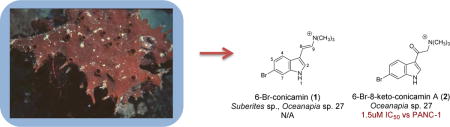
The extract of Oceanapia sp. C011027 (from the NCI-DTP repository) was prioritized for investigation in this study using the soft agar disk diffusion assay (DDA) whose results are shown in Table 1. The DDA measures a zone of inhibition (in mm) for a sample diffusing from a disk placed at the edge of the petri dish containing tumor cells embedded in a soft agar matrix. Samples considered to have high cytotoxicity exhibit a zone of inhibition ≥ 20 mm in any of the cell lines, which included five solid tumor cell lines, a leukemia line (CEM), and bone marrow progenitor cells (CFU-GM). Some benchmark results are provided by the activities for two clinical drugs, gemcitabine and 5-fluorouracil, which are powerful cytotoxins against almost all of the eight cell lines screened. Both drugs also displayed selectivity between one or more solid tumor cell lines and either the leukemia or a normal cell surrogate (zone of inhibition difference > 7mm, indicated by bold numbers in Table 1). The best response for the Oceanapia sp. extract was against PANC-1 (zone of inhibition = 23 mm) which represented selective cytotoxicity relative to both CEM and murine CFU-GM cells (Δinhibition zone = 10 mm and 7 mm, respectively). These results prompted creation of an HPLC peak library that was screened in the DDA and the active fractions were further evaluated by MSn and NMR. Subsequent isolation afforded the known Suberites sponge compound, 6-Br-concamin (1)8 and an unreported compound 6-Br-8-keto-concamin A (2) shown in Fig. 1. While 1 was not cytotoxic by the DDA, synthetic 2 (2a) at 112 nmol/disk exhibited a zone of inhibition = 28 mm against PANC-1 and 33 mm against MCF-7, each with a greater than 20 mm differential between CEM and CFU-GM cell lines. We also synthesized a family of bromoindole compounds bearing quaternary ammonium substituents including the 5-Br analog of 2 (3), which was also active in the DDA with an inhibition zone of 20 mm against PANC-1 and 33 mm against MCF-7, again selective between these solid tumor cell lines and CEM and CFU-GM. Eight other synthetic intermediates employed in the syntheses were inactive in the DDA. Additional elements of the study included two comprehensive evaluations: (a) unraveling NMR δ values to facilitate the placement of a halogen at the 5 vs. 6 position of an indole ring, and (b) using the PANC-1 IC50’s to define a useful SAR pattern for this indole scaffold.
Table 1.
Disk diffusion assay zone of inhibition data for the parent Oceanapia sp. C011027 extract, synthetic 2a, various indole analogs of 2, and therapeutic standards compared across five solid tumor cell lines, a leukemia line (CEM), and bone marrow progenitor cells (CFU-GM). Bold numbers indicate selectivity, defined as a differential zone of inhibition greater than 7 mm between solid tumor cell line and at least one normal cell proxy (CEM/CFU-GM).
| nmol/disk | Zone of Inhibition (mm)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| H125 | MCF-7 | OVC-5 | U251N | PANC-1 | CEM | CFU-GM
|
|||
| mouse | human | ||||||||
| gemcitabine | 0.02 | 33 | 13 | 33 | 33 | 13 | 20 | 0 | 33 |
| 5-fluorouracil | 46 | 33 | 33 | 29 | 7 | 18 | 21 | 33 | 33 |
|
| |||||||||
| Oceanapia sp. C011027 crude extract | – | 11 | 8 | 15 | 20 | 23 | 13 | 16 | |
| 6-Br-8-keto-conicamin A (2a) | 112 | 7 | 33 | 10 | 15 | 28 | 7 | 7 | 5 |
| “ | 28 | 16 | 12 | 2.5 | 2 | 0 | |||
| 5-Br-8-keto-conicamin A (3) | 112 | 7 | 33 | 13 | 10 | 20 | 5 | 8 | 5 |
| “ | 28 | 10 | 7 | 20 | 5 | 3 | 0 | ||
| 1H-indole-3-carbonyl cyanide (8) | 352 | 7 | 10 | 7 | 7 | 7 | 10 | ||
| 8-keto-tryptamine (10) | 172 | 7 | 11 | 5 | 5 | 3 | |||
| 6-Br-8-keto-tryptamine (11) | 130 | 2 | 3 | 3 | 2 | 0 | |||
| 5-Br-8-keto-tryptamine (12) | 71 | 0 | 3 | 2 | 2 | 0 | |||
| N,N,N-trimethyl 8-keto-tryptamine (13) | 207 | 2 | 10 | 3 | 2 | 7 | 3 | ||
| 6-Br-N,N-dimethyl-8-keto-tryptamine (14) | 53 | 0 | 3 | 0 | 0 | 2 | |||
| 5-Br-N,N-dimethyl-8-keto-tryptamine (15) | 32 | 0 | 2 | 0 | 0 | 0 | |||
| N,N-dimethyl-8-keto-tryptamine (16) | 59 | 0 | 2 | 0 | 0 | 0 | |||
Cell lines: H-125 = lung adenocarcinoma; MCF-7 = breast adenocarcinoma; OVCAR-5 = ovarian carcinoma; U251N = glioblastoma cells; PANC-1 = prostrate adenocarcinoma; CEM = leukemic lymphoid; CFU-GM = granulocyte-macrophage progenitor cells.
Fig. 1.
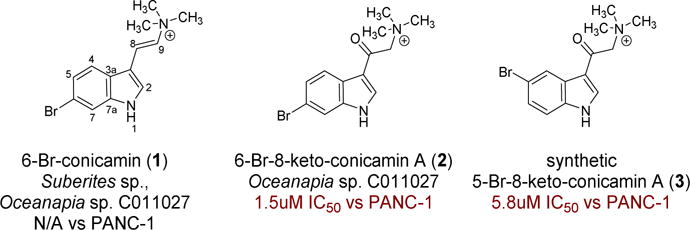
The major products, 1 and 2, isolated from Oceanapia sp. C011027 and the 5-Br synthetic analog 3.
A DDA active fraction (data not shown herein) from the Oceanapia peak library actually contained a mixture of two compounds with m/z of 279 and 295, each having an M+2 peak of equivalent height, indicating the presence of a single bromine atom. The two compounds briefly discussed above, known 6-Br-conicamin (1)8 and new 6-Br-8-keto-conicamin (2), were separated by semi-preparative reversed phase HPLC using 21:89 acetonitrile:H2O. Neither fraction formed adducts in ESI mass spectrometry when spiked with sodium or potassium, consistent with a native positive charge. Accurate mass measurement gave two nearly identical molecular formulas differing by an oxygen: 1: ESI m/z 279.0467 [M]+ (calcd for C13H16N279Br, 279.0491) and 2: ESI m/z 295.0419 [M]+ (calcd for C13H16N2O79Br, 295.0441). The mass data from 1 coupled with a 1H NMR spectrum showing four indole ring protons, two vinyl protons and a singlet for nine protons near δ3 allowed 1 to be identified as 6-bromo-conicamin (1) by dereplication. Despite the structural similarity implied by both MS and NMR data, the structure of 2 had not been reported in the literature. Inspection of the 13C NMR spectrum of 2 revealed that the vinyl carbons in 1 were not present and were replaced by a ketone at δC 185.4 and a CH2 at δC 66.1, respectively (Table 1). The HMBC correlations observed from the N-trimethyl protons at δH 3.33 to the CH2 at δC 66.1 but not the carbonyl at δC 185.4 suggested a ketone-methylene-trimethylamine motif. These data coupled with the remaining long range correlations observed by gHMBCAD enabled provisional assignment of 2. However, placement of the Br at either C-5 or C-6 could not be confidently assigned. This ambiguity was resolved by additional NMR δ analyses discussed next along with NMR data comparison to synthesized products including 6-bromo-8-keto-conicamin A (2a), and 5-bromo-8-keto-conicamin A (3).
Finalizing the Br atom placement in natural product 2 at C-5 vs. C-6 (Fig. 1) should be trivial as it involves assigning the relative position of an indole AB spin system as proximal or distant to the NH. Unfortunately, our attempts to obtain NOE or HMBC correlations to the NH residue for a definitive resolution were unsuccessful. Alternatively, a literature search to provide NMR δH and δC values distinctive for these isomeric frameworks provided diagnostic patterns shown in Figure 1. First, the δH shifts at H-4/H-5 (3JH-H portion), and H-7 for a 6-bromo isomer are different vs. those at H-4, and H-6/H-7 (3JH-H portion) for a 5-bromo isomer as shown in Fig. 2. A key is that the relative ΔδH for the o-proton doublets is small (≅ 0.2 ppm) for the 5-Br isomer but large (> 0.5 ppm) for the 6-Br isomer. Second, the δC at C-3a is deshielded by approximately 3 ppm in the 5-Br relative to that of the 6-Br indole.9, 10 The observed patterns for 2 matched that of the lower example in Figure 2: vicinal proton ΔδH = 0.69 and δC-3a = 124.3. Correspondingly, the synthetic compounds obtained as described in the next section further supported these observations. For example, the spectrum of 3 matched that of the upper example in Figure 2: the vicinal proton ΔδH = 0.20 and δC-3a = 127.7 (see NMR data in Table 1, supplemental Figs. S9 & S10), and the data of the commercially available aldehyde 5 matched that of the lower example in Figure 2: the vicinal proton ΔδH = 0.67 and δC-3a = 123.1.
Fig. 2.
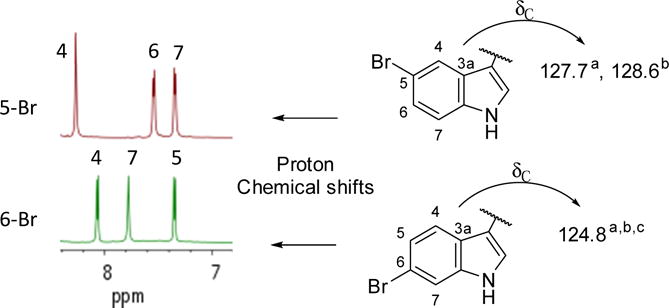
Using NMR data to rapidly distinguish between 5- vs. 6-Br indole frameworks when the diagnostic NOEs to the NH cannot be observed. aThis work; bSeagraves;10 cBartik.9
The process to obtain compound 2 and other analogs by synthesis is shown in Scheme 1. Two indole-3-carboxaldehydes, 4 and 5, were used as separate starting materials to provide the oxotryptamine synthons 10 and 11. While the yield of the cyanohydrin silylether 6 from 4 match that in the literature (60%), the yield of 7 from 5 was poor (20%) vs that in the literature for the 5-Br analog (88%).11 Oxidation of both 6 and 7 via DDQ provided high yields of 8 (83%) and 9 (60%).11 Hydrogenation of 8 afforded 10 in good yield after workup (76%) and reduction of 9 provided the first route to 11.12 Bromination of the keto-tryptamine synthon 10 proceeded in a straightforward fashion, producing the bromo-keto-tryptamines 11 and 12 which were separated by reversed phase HPLC.13 We used 11 prepared from 5 to confidently distinguish between the 5- and 6-bromo isomers (11 and 12) prepared via bromination of 10. NMR spectroscopic experiments on the commercially obtained 5 assured correct assignment (see SI figures S34–38). The final steps involved methylation of 10, 11, or 12 to respectively afford 13, 2a, or 3. Performing these reactions with a reduced molar ratio of methyl iodide provided the dimethyl analogs 14, 15, or 16 from 11, 12, or 10, respectively as minor products. The compounds 2a, 11, or 14 were produced in duplicate from either 5 or 10 as starting material and were equivalent in their spectroscopic and bioassay properties.
Scheme 1.
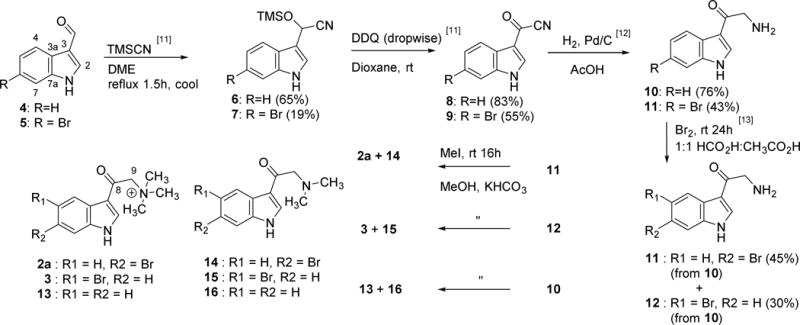
Synthesis of the natural product 6-Br-8-keto-conicamin (2a) and nine key analogs 3, 8, and 10–16 for in vitro cytotoxicity screening against the PANC-1 tumor cell line. Following the synthetic route to 11 from 5 enabled facile differentiation between the 5- and 6-bromo isomers 11 and 12 synthesized from 10. The spectral and biological properties of 2a, 11, or 14 derived from 5 were equivalent to those derived from 10.
The synthetic work to prepare 2 and its analogs provided additional information to probe the structure-activity relationship describing the cytotoxicity of Br-indoles. Based on our DDA and IC50 screening of the intermediates and products from the synthesis, only the compounds most similar to 2 (such as 3 and 13) possessed notable cytotoxic activity as shown in Tables 1 and 3. The DDA data (Table 1) indicates similar solid tumor selectivity for 2 and 3 in comparison to the two therapeutic standards. The IC50 data (Table 3) reveals that a quaternary amine functionality is vital for low uM activity against PANC-1 cells, which is on a par with that of the two therapeutic standards, 5-flourouracil and gemcitabine. Also, the presence of the Br atom at C-5 or C-6 on the indole ring and the keto group at C-8 are significant factors in the IC50 activity of 2 and 3. Interestingly, relative to 2, the primary amines (compound 10–12) and tertiary amines (compounds 14 – 16) were inactive in both the DDA and the IC50 determinations. In addition, 13, which lacks a Br, was not selective in the DDA screen and was an order of magnitude less potent vs. 2 against PANC-1 cells based on IC50 data. While the position of the Br (at C-5 or C-6) is less important, it was the 6-bromo-8-keto-conicamin (2) that exhibited the best DDA selectivity for PANC-1 and MCF-7, and greatest potency against PANC-1 (IC50 data against MCF-7 were not measured). Overall, the low micromolar in vitro activity of 2 against the PANC-1 cell line (IC50 1.5 μM for the natural product vs 4.1 μM for the synthetic material) is exciting and is similar to that of the clinical therapeutics (5FU: IC50 = 7.0 μM; gemcitabine: IC50 = 0.02 μM).
Table 3.
In vitro IC50 values against the PANC-1 cell line for natural 2, synthetic 2a, other synthetic analogs, and two therapeutic standards.
| Compound | PANC-1 IC50 (μM) |
|---|---|
| 6-Br-conicamin (1) | NA |
| 6-Br-8-keto-conicamin A (2) [natural product] | 1.5 |
| 6-Br-8-keto-conicamin A (2a) [synthetic product] | 4.1 |
| 5-Br-8-keto-conicamin A (3) | 5.8 |
| 1H-indole-3-carbonyl cyanide (8) | NA |
| 8-keto-tryptamine (10) | NA |
| 6-Br-8-keto-tryptamine (11) | NA |
| 5-Br-8-keto-tryptamine (12) | NA |
| N,N,N-trimethyl 8-keto-tryptamine (13) | 23 |
| 6-Br-N,N-dimethyl-8-keto-tryptamine (14) | NA |
| 5-Br-N,N-dimethyl-8-keto-tryptamine (15) | NA |
| N,N-dimethyl-8-keto-tryptamine (16) | NA |
| 5-fluorouracil | 5.714; 7.0 |
| gemcitabine | 7.215; 0.02* |
NA – compounds inactive in soft-agar disk diffusion assay as shown in Table 1
Note that this IC50 was determined after 5 days compared to 48 hr. We have found in our lab (unpublished data) that there is ~250-fold difference in IC50 values between a 24 hr and a 5-day assay for gemcitabine.
3. Conclusion
The halogenated indole moiety found in 2 has been observed in a number of metabolites from marine macro- and microorganisms, including the topsentins,9 aplysinopsins,10 dragmacidins,16 hypaphorines,17 didemnimides,18 and nakijinamines.8 Thorough reviews of marine-derived indole alkaloids and their activities has been published elsewhere.19,20 The bioactivity of these compounds is diverse: cytotoxicity, antibacterial activity, anti-fungal activity, histamine receptor agonism, and neurotransmitter receptor affinity. The quaternary amine functionality found in 2 is rare, but there are examples of bioactive natural products containing this cationic residue attached to a halogenated indole scaffold (Fig. 3). A prenylated indole from a bryozoan with moderate nAChR activity prompted medicinal chemistry efforts in which some analogs (e.g. 18) attained high nanomolar affinity for a variety of nAChR subtypes.21 The results of this study are modest, yet the in vitro cytotoxicity of 2 against the PANC-1 cell line exceeds that reported for 5-FU and gemcitabine, which are the primary constituents of the current standards of care for pancreatic cancer.14, 15 The structure of 2 was confirmed by synthesis and a number of analogs were made using methodology originally developed for the syntheses of the rhopaladins, dragmacidins, and almazoles.11, 12, 13 The quaternary amine functionality and bromination of the indole ring of 2 was critical for activity against PANC-1. The new small molecule discovered in this work highlights the value of continued pursuit of achiral marine natural products and the effectiveness of continued screening using the NCI Developmental Therapeutics Program extract library. Overall, the lack of small molecule therapies to treat pancreatic cancer makes the results presented in Table 3 reasonably significant.
Fig. 3.
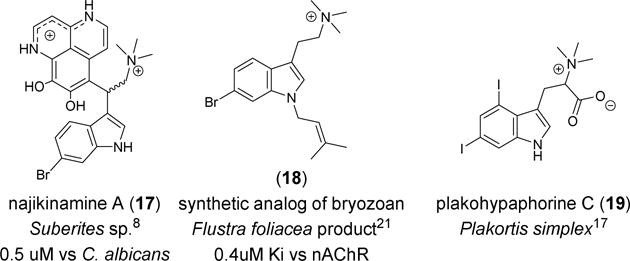
Other natural products featuring halogenated indole with a quaternary amine moiety.
4. Experimental
4.1 General experimental procedures
UV spectra were measured with Thermo Ultimate 3000 DAD. IR spectra were measured with a Perkin-Elmer Spectrum One FTIR spectrometer. Analytical LC-MS analysis was performed on samples at a concentration of 10 mg/mL for crude extracts and 1 mg/mL for pure compounds, using a 150 × 4.60 mm 5 μm C18 Phenomenex Luna column in conjunction with a guard column and 4.0 × 3.0 mm C18 cartridge (Phenomenex, Inc.). 20 uL volumes of sample were injected onto the column using Waters 717 autosampler, with a flow rate of 1 mL/min provided by two Waters 515 HPLC pumps. The eluent was split between an Applied Biosystems Mariner 5200 electrospray ionization time-of-flight (ESI-TOF) mass spectrometer and a Waters 996 photodiode array (PDA) UV detector in line with a SEDERE 75 evaporative light scattering detector (ELSD). Optical rotations were performed on a JASCO P-2000 polarimeter; the average of 10 measurements is reported, each with 10 second digital integration time and 12 second cycle time. All NMR experiments were run on a Varian Unity spectrometer (500 and 125 MHz for 1H and 13C, respectively) or Varian INOVA spectrometer (600 and 150 MHz for 1H and 13C, respectively) equipped with a cryoprobe. High accuracy mass spectrometry measurements were recorded using an Applied Biosystems Mariner 5200 electrospray ionization time of flight (ESI-TOF) mass spectrometer. Synthetic procedures were either performed via Schlenk line with nitrogen or via balloon and septa techniques with argon. Flash chromatography was performed using a Biotage Isolera Prime fraction collector with a 100 g silica gel 60 column.
4.2. Biological material
The sponge Oceanapia sp. C011027 (phylum Porifera, class Demospongia, order Haplosclerida) was collected at 10 m depth off the coast of Sulawesi, Indonesia. Sample collection and identification was done by the Coral Reef Foundation under contract to the National Cancer Institute (National Institutes of Health). A taxonomic voucher specimen (number 0CDN1333) was deposited at the Smithsonian Institution in Suitland, MD (USA).
4.3. Compound isolation
After extract C011027 demonstrated solid tumor selective activity, an HPLC peak library was generated to determine the active constituents. The extract was fractionated using a 250 × 10 mm Phenomonex Luna C18 5μm semi-preparative column equipped with an analytical guard column (4.0 × 3.0 mm C18 cartridge) and two Waters 515 HPLC pumps. The eluent was monitored by a Waters 996 detector before being fractionated by a Gilson 215 Liquid Handler that generated one fraction per minute. A Waters 717 autosampler injected 100 uL of extract at 70 mg/mL onto the column; the first gradient 10:90 acetonitrile:H2O was held for 1 minute, then linearly shifted to 100:0 acetonitrile:H2O over 29 minutes, and finally held at 100:0 acetonitrile:H2O for 10 minutes. Fractions H16 and H17 were the only fractions with significant zones of inhibition when evaluated in the DDA. These contained a mixture of 1 and 2. Compounds 1 and 2 were separated using the following isocratic conditions: 21:79 acetonitrile:H2O with tR = 12.5 and 15.5, respectively. Note that the charge of these compounds leads to significant tailing and inconsistent retention times over longer timescales (i.e. weeks). This can be mitigated to some extent by using TFA in the mobile phase. Fraction collection was done manually using the same column as above with two Waters 510 HPLC pumps, a Waters Model 680 gradient controller, and an ABI Analytical Spectroflow 783 UV detector monitoring at 230nm.
4.3.1. 6-bromo-conicamin (1)
HAESIMS m/z 279.04667 [M]+ (calcd for C13H16N279Br, 279.04914). NMR spectra were in accordance with literature.8
4.3.2. 6-bromo-8-keto-conicamin (2)
Light pink crystalline solid; UV (MeCN:H2O) 209, 247, 271, 308 nm; IR (film) υmax 3438, 3250, 1637, 1520, 1443, 1414, 1384, 1048, 891, 595 cm−1; 1H NMR (DMSO-d6, 600 MHz) δ 8.58 (1H, bs, H-2), 8.08 (1H, d, J = 8.5 Hz, H-4), 7.77 (1H, s, H-7), 7.39 (1H, dd, J = 1.3, 8.5 Hz, H-5), 4.99 (2H, s, H-1′), 3.33 (9H, s, N-(CH3)3); 13C NMR (DMSO-d6, 150 MHz) δ 185.4 (C, C-2′), 138.1 (C, C-7a), 136.9 (CH, C-2), 125.4 (CH, C-5), 124.3 (C, C-3a), 122.7 (CH, C-4), 115.9 (C, C-6), 115.6 (CH, C-7), 114.6 (C, C-3), 66.1 (CH2, C-1′), 53.5 (CH3, N-(CH3)3); HAESIMS m/z 295.04191 [M]+ (calcd for C13H16N2O79Br, 295.04405).
4.4.1. 2-(1H-indole-3-yl)-2-(trimethylsiloxy)acetonitrile (6)
Procedure adapted from Janosik.11 To a solution of 1.16g 1H-indol-3-yl-carboxaldehyde (Combi-Blocks, San Diego, CA, USA) in 10mL acetonitrile, 1.3 mL TMSCN was added. The reaction solution was heated and maintained at reflux for 2 h before cooling. After evaporation of the solvent, reaction products were purified from the resulting oil using flash chromatography (isocratic 80:20 hexane-ethyl acetate) to afford 6 (1.2g, 65%) as a lime-green oil. MS (ESI) 243 m/z (M-H)−. NMR data were in accordance with literature.
4.4.2. 2-(6-bromo-1H-indol-3-yl)-2-(trimethylsiloxy)acetonitrile (7)
Adapted from procedure for 5-bromo isomer from Janosik.11 To a solution of 1.08g 6-bromo-1H-indol-3-yl-carboxaldehyde (Tokyo Chemical Industry, Tokyo, Japan) in 10mL DME, 0.73 mL TMSCN was added. The reaction solution was heated and maintained at reflux for 2.5 h before cooling. After evaporation of the solvent, reaction products were purified by flash chromatography (hexane-ethyl acetate gradient) to afford 7 (0.35g, 19%) as a light yellow oil. 1H NMR (DMSO-d6, 600 MHz) δ11.45 (1H, S), 7.63 (1H, d, J = 1.9), 7.60 (1H, d, J = 8.4), 7.56 (1H, d, J = 2.6), 7.24 (1H, dd, J = 8.5, 1.8), 6.18 (1H, s), −0.11 (9H, s); 13C NMR (DMSO-d6, 150 MHz) δ 137.4, 126.0, 123.8, 122.5, 120.4, 119.9, 114.7, 114.6, 111.0, 56.8, −0.23. MS (ESI) 321 and 323 m/z (M-H)−.
4.4.3. 1H-indol-3-carbonyl cyanide (8)
A solution of DDQ (1.21 g, 5.3 mmol) in 60 mL dioxane was added dropwise by addition funnel to a stirring solution of 6 (1.18 g, 4.8 mmol) in 10 mL dioxane. After 2.5 h the reaction mixture was filtered and the filtrate dried to a purple-black solid. Flash chromatography (DCM-EtOAc gradient) afforded 8 (0.68g, 83%) as a yellow oil. MS (ESI) 171 m/z [M+H]+. NMR data were in accordance with literature.
4.4.4. 6-bromo-1H-indol-3-carbonyl cyanide (9)
The procedure above was used with 6-bromo-indole-TMS (7) (0.35g, 1.1 mmol) and DDQ (0.27g). Flash chromatography (hexanes-EtOAc gradient) afforded 9 (0.15g, 55%) as a fluffy yellow solid. MS (ESI) 247/249 [M-H]−.
4.4.5. 8-keto-tryptamine (10)
To 150 mg of 10% Pd/C, a solution of 1 mmol of 1H-indol-3-carbonyl cyanide (8) in 10 mL acetic acid was added. Hydrogen gas was added via balloon and septum. After 15 h, the reaction mixture was filtered using diatomaceous earth and the filtrate was dried, resuspended in water, basified, then extracted with ethyl acetate to give 0.76 mmol of the free base 10. NMR data were in accordance with literature.
4.4.6. 6-bromo-8-keto-tryptamine (11)
The first route to 11 employed the procedure above (10) using 9 as starting material. The second route was performed as follows. To a stirring solution of β-keto-tryptamine (10) in 1:1 formic acid:acetic acid, 1.2 equivalents of Br2 was added dropwise. After 24 h, the reaction mixture was dried and subjected to reversed phase HPLC to afford 11 and 12. 1H NMR (DMSO, 500 MHz) δ 12.47 (1H, s, NH), 8.52 (1H, d, J = 3 Hz, H-2), 8.1 (1H, d J = 8.4 Hz, H-4), 7.74 (1H, s, H-7), 7.4 (1H, d J = 8.5 Hz, H-5), 4.38 (2H, s, H-9); 13C NMR (DMSO, 125 MHz) δ 187 (CO), 137.5, 136, 123.3, 124.1, 122.6, 115.6, 115.8, 115.2, 113.1, 44.1. MS (ESI) 252/254 [M+H]+.
4.4.7. 5-bromo-8-keto-tryptamine (12)
The second procedure above for 11 also afforded 12 by reversed phase HPLC. 1H NMR (DMSO, 500 MHz) δ 8.54 (1H, d J=3.0 Hz, H-2), 8.29 (1H, d J = 1.8 Hz, H-4), 7.53 (1H, d J = 8.6 Hz, H-7), 7.41 (1H, dd J = 8.6, 1.7 Hz, H-6), 4.39 (2H, s, H-9); 13C NMR (DMSO, 125 MHz) δ 187, 136.3, 135.4, 126.8, 125.9, 123.0, 115.1, 114.7, 112.6, 44.1. MS (ESI) 252/254 [M+H]+.
4.4.8. 8-keto-N,N,N-trimethyltryptamine (13)
To a solution of 8-keto-tryptamine (10) in KHCO3-saturated MeOH, 3 equivalents of methyl iodide were added and stirred overnight. The product mixture under these conditions contained unreacted starting material (10), 13, 14, and a minor amount of the single methyl analog which were separated by reverse phase HPLC. Using excess MeI (> 5 eq) yields exclusively the quaternary amine product. Yellow solid; 1H NMR (DMSO 600 MHz) δ 8.51 (1H, s), 8.15 (1H, d J = 7.3 Hz), 7.57 (1H, d J = 7.3 Hz), 7.24 (2H, m), 4.93 (2H, s), 3.32 (9H, s). 13C NMR (DMSO, 150 MHz) δ 184.9, 137.4, 136.5, 125.4, 123.3, 122.4, 121.0, 114.6, 113.1, 66.05, 53.5. HAESIMS m/z 217.1327 [M+H]+ (cald for C13H17ON2, 217.1341)
4.4.9. 6-bromo-8-keto-conicamin (2a)
See above procedure for 13, but using compound 11 as starting material. The product mixture primarily contained 2a and 14, which were seprable by reverse phase HPLC. Off-white crystalline solid; 1H NMR (DMSO 600 MHz) δ 8.51 (1H, s), 8.07 (1H, d J = 8.41 Hz), 7.78 (1H, s), 7.35 (1H, d J = 8.46 Hz), 4.90 (2H s), 3.31 (9H, s). 13C NMR (DMSO, 150 MHz) δ 184.6, 139.5, 138.2, 125.0, 124.8, 122.5, 116.1, 115.5, 114.5, 66.1, 53.5. HAESIMS m/z 295.0443 [M]+ (calcd for C13H16N2O79Br, 295.0441).
4.4.10. 5-bromo-8-keto-conicamin (3)
See above procedure for 13, but using compound 12 as starting material. The product mixture primarily contained 3 and 15. Off-white crystalline solid; 1H NMR (DMSO 600 MHz) δ8.52 (1H, s), 8.27 (1H, s), 7.54 (1H, d, J = 8.3 Hz), 7.34 (1H, d, J = 8.0), 4.91 (2H, s), 3.32 (9H, s). 13C NMR (DMSO, 150 MHz) δ184.4, 138.8, 137.6, 127.7, 125.4, 123.0, 115.6, 114.8, 114.0, 66.0, 53.5. HAESIMS m/z 295.0442 [M]+ (calcd for C13H16N2O79Br, 295.0441)
4.4.11. 6-bromo-8-keto-N,N-dimethyltryptamine (14)
See above procedure for 13, but using compound 11 as starting material. 1H NMR (DMSO 600 MHz) δ12.26 (1H, s), 8.45 (1H, s), 8.10 (1H, d, J = 8.5 Hz), 7.68 (1H, s), 7.31 (1H, dd, J = 8.4, 1.4 Hz), 3.52 (2H, s), 2.26 (6H, s) 13C NMR (DMSO, 150 MHz) see supplemental Figs. S29 and S30. HAESIMS m/z 281.0285 [M+H]+ (calcd for C12H14N2O79Br, 281.0290)
4.4.12. 5-bromo-8-keto-N,N-dimethyltryptamine (15)
See above procedure for 13, but using compound 12 as starting material. 1H NMR (DMSO 600 MHz) δ8.46 (1H, s), 8.30 (1H, bs), 7.46 (1H, d, J = 8.6 Hz), 7.32 (1H, broad d, J = 8.6 Hz), 3.51 (2H, s), 2.26 (6H, s) 13C NMR (DMSO, 150 MHz) see supplemental Figs. S32 and S33. HAESIMS m/z 281.0287 [M+H]+ (calcd for C12H14N2O79Br, 281.0290)
4.4.13. 8-oxo-N,N-dimethyltryptamine (16)
See above procedure for 13. 1H NMR (DMSO 600 MHz) δ 11.98 (1H, s), 8.42 (1H, d, J = 2.7), 8.17 (1H, d, J = 7.9 Hz), 8.15 (1H, d J =7.29 Hz), 7.47 (1H, d, J = 7.7 Hz), 7.20 (1H, td, J = 7.7, 7.1, 1.5 Hz), 7.17 (1H, td, J = 7.9, 7.0, 1.3 Hz), 3.52 (2H, s), 2.27 (6H, s). 13C NMR via HMBC (DMSO, 150 MHz) δ 192.9, 136.2, 135.9, 125.6, 122.6, 121.6, 121.3, 115.0, 112.1, 66.4, 45.4. HAESIMS m/z 203.1179 [M+H]+ (cald for C12H15ON2, 203.1184)
4.5. Cytotoxic assays
Extract C011027 was evaluated in the in vitro disk diffusion soft agar colony formation assay which defines differential cell killing among 13 cell types. The screen includes CCRF-CEM (leukemia), H125 (lung cancer), OVCAR-5 (ovarian cancer), MCF-7 (breast cancer), PANC-1 (pancreatic cancer), U251N (brain cancer), as well as a normal murine hematopoietic progenitor cell, CFU-GM. The human cancer cell lines are maintained in cell culture in either Dulbecco’s modified MEM, Eagles MEM or RPMI-1640 depending upon the cell lines. The media is supplemented with either 10% or 15% BCS plus Pen-Strept (100,000 I.U. Pen/L; 100,000 μg/L Strept) and L-glutamine (292 mg/L). Cells are removed from cultures by a trypsin solution (0.05%). Plating efficiencies are sufficiently high that 30,000 to 60,000 cells in 3 mL produce the desired number of colonies (over 10,000 per plate) in 60 mm plates. This soft agar top layer (0.3% with the serum and media as above) plus the titrated tumor cells are poured into plates and allowed to solidify. A volume of 15 μL of each sample is dropped onto a 6.5 mm disk (Whatman filter disks). The disk is allowed to dry overnight and then placed close to the edge of the petri dish. The plates are incubated for 7–10 days (depending upon cell type) and examined by an inverted stereo-microscope (10×) for measurement of the zone of inhibition measured from the edge of the filter disk to the beginning of normal-sized colony formation. The diameter of the filter disk, 6.5 mm, and a difference in zones between solid tumor cells and either normal or leukemia cells of 6.5 mm or greater defines solid tumor selective compounds. If the test material is excessively toxic at the first dosage, a range of dilutions of the agents (at 1:4 decrements) are tested against the same tumors. At some dilution, quantifiable cytotoxicity is invariably obtained.
IC50 assays were performed as follows. PANC-1 human pancreatic cancer (epithelioid carcinoma) cells are plated at 5×104 cells in T25 tissue culture flasks (Falcon Plastics, New Jersey) with 5 mL media RPMI 1640 (Cellgro, Virginia) supplemented with 15% BCS (Hyclone, Utah), 1% Penicillin-Streptomycin, and 1% Glutamine (Cellgro). Three days later (cells in logarithmic growth phase), test compounds are added to the flasks to achieve concentrations ranging from 100 to 10−5 μg/mL (10-fold dilutions). Seventy-two hours later, the cells in the flasks are washed, trypsinized, spun down and counted for both viable and dead cells using 0.04% trypan blue (Gibco, Maryland). Viable cell number as a function of concentration is plotted and the IC50 value determined by interpolation. Each point is done in duplicate and a standard deviation determined. A second, definitive run is then carried out with four two-fold drug dilutions around the initial IC50 value and these are the IC50 values reported in Table 2.
Table 2.
NMR Dataa (DMSO-d6) for natural 6-Br-8-keto-conicamin A (2), synthetic 6-Br-8-keto-conicamin (2a), and synthetic 5-Br-8-keto-conicamin A (3).
| position | 2
|
2a
|
3
|
||||
|---|---|---|---|---|---|---|---|
| δC, Typeb | δH, mult (J, Hz) | HMBCc | δC | δH, mult (J, Hz) | δC | δH, mult (J, Hz) | |
| 1 | 8.47, bs | 8.51, s | 8.54, s | ||||
| 2 | 136.9, CH | 8.58, bs | 3, 3a, 7a | 138.2 | 8.51, s | 138.8 | 8.52, s |
| 3 | 114.6, C | – | 114.5 | – | 114.1 | – | |
| 3a | 124.3, C | – | 124.8 | – | 127.7 | – | |
| 4 | 122.7, CH | 8.08, d (8.5) | 3, 3a, 5, 7, 7a | 122.5 | 8.07, d (8.5) | 123.0 | 8.27, s |
| 5 | 125.4, CH | 7.39, dd (1.3, 8.5) | 3a, 7 | 125.0 | 7.35, bd (8.5) | 115.6 | – |
| 6 | 115.9, C | – | 115.5 | – | 125.4 | 7.34, d (8.4) | |
| 7 | 115.6, CH | 7.77, bs | 3a, 5, 6, 7a | 116.1 | 7.78, bs | 114.8 | 7.54, d (8.6) |
| 7a | 138.1, C | – | 139.5 | – | 137.6 | – | |
| 8 | 185.4, C | – | 184.6 | – | 184.4 | – | |
| 9 | 66.1, CH2 | 4.99, s | 8, N-(CH3)3 | 66.1 | 4.90, s | 66.0 | 4.91, s |
| N-(CH3)3 | 53.5, CH3 | 3.33, s | 9 | 53.5 | 3.31, s | 53.5 | 3.32, s |
Measured at 600 MHz (1H) and 150 MHz (13C);
Carbon type determined by gHSQC;
gHMBC(AD) correlations to indicated carbon with JnCH optimized for 6 Hz couplings and using a two second relaxation delay.
Supplementary Material
Acknowledgments
We thank the Natural Products Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute for collection and extraction of Oceanapia sp.27. This work was supported by a grant from the NIH R01 CA47135 (P.C. and F.A.V.). We also acknowledge funding from NSF CHE1427922 for the Thermo Velos Pro electrospray ionization hybrid ion trap-orbitrap mass spectrometer and the NIH 1S10OD018455-01 for the 800 MHz NMR spectrometer and helium cryoprobe. Finally, we thank the members of the Lokey research group at UCSC, especially M. Naylor and J. Schwochert, for their kind advice during the synthetic portion of this work.
Appendix A. Supplementary data
Copies of the NMR spectra for all products are available in the supplemental information and can be found at http://########.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sakai R, Higa T, Jefford CW, Bernardinelli G. J Am Chem Soc. 1986;108:6404–6405. [Google Scholar]
- 2.Waters AL, Peraud O, Kasanah N, Sims JW, Kothalawala N, Anderson MA, Abbas SH, Rao KV, Jupally VR, Kelly M, Dass A, Hill RT, Hamann MT. Front Mar Sci. 2014;1 doi: 10.3389/fmars.2014.00054. Article 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim C-K, Riswanto R, Won TH, Kim H, Elya B, Sim CJ, Oh D-C, Oh K-B, Shin J. J Nat Prod. 2017;80:1575–1583. doi: 10.1021/acs.jnatprod.7b00121. [DOI] [PubMed] [Google Scholar]
- 4.McCloud TG. Molecules. 2010;15:4526–4563. doi: 10.3390/molecules15074526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valeriote FA, Tenney K, Media J, Pietraszkiewicz H, Edelstein M, Johnson TA, Amagata T, Crews P. J Exp Ther Oncol. 2012;10:119–134. [PubMed] [Google Scholar]
- 6.Howlader N, Noone AM, Krapcho M, Miller D, Bishop K, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA, editors. SEER Cancer Statistics Review, 1975–2013. National Cancer Institute; Bethesda, MD: http://seer.cancer.gov/csr/1975_2013/, based on November 2015 SEER data submission, posted to the SEER web site, April 2016. [Google Scholar]
- 7.Lin S, McCauley E, Lorig-Roach N, Tenney K, Naphen C, Yang A-M, Johnson T, Hernadez T, Rattan R, Valeriote F, Crews P. Marine Drugs. 2017;15:98. doi: 10.3390/md15040098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takahashi Y, Tanaka N, Kubota T, Ishiyama H, Shibazaki A, Gonoi T, Fromont J, Kobayashi J. Tetrahedron. 2012;68:8545–8550. [Google Scholar]
- 9.Bartik K, Braekman J-C, Daloze D, Stoller C, Huysecom J, Vandevyver G, Ottinger R. Can J Chem. 1987;65:2118–2121. [Google Scholar]
- 10.Seagraves NL, Crews P. J Nat Prod. 2005;68:1484–1488. doi: 10.1021/np0501334. [DOI] [PubMed] [Google Scholar]
- 11.Janosik T, Johnson AL, Bergman J. Tetrahedron. 2002;58:2813–2819. [Google Scholar]
- 12.Miyake F, Hashimoto M, Tonsiengsom S, Yakushijin K, Horne DA. Tetrahedron. 2010;66:4888–4893. [Google Scholar]
- 13.Miyake FY, Yakushijin K, Horne DA. Org Lett. 2000;2:2121–2123. doi: 10.1021/ol000124g. [DOI] [PubMed] [Google Scholar]
- 14.Guzmán EA, Johnson JD, Carrier MK, Meyer CI, Pitts TP, Gunasekera SP, Wright AE. Anticancer Drugs. 2009;20:149–155. doi: 10.1097/CAD.0b013e32831fa39e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Zhu J, Melvin WS, Bekaii-Saab TS, Chen C-S, Muscarella P. J Gastrointestinal Surg. 2006;10:207–214. doi: 10.1016/j.gassur.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 16.Kohmoto S, Kashman Y, McConnell OJ, Rinehart KL, Wright A, Koehn F. J Org Chem. 1988;53:3116–3118. [Google Scholar]
- 17.Campagnuolo C, Fattorusso E, Taglialatela-Scafati O. Eur J Org Chem. 2003;2:284–287. doi: 10.1021/ol027437r. [DOI] [PubMed] [Google Scholar]
- 18.Vervoort HC, Richards-Gross SE, Fenical W, Lee AY, Clardy J. J Org Chem. 1997;62:1486–1490. [Google Scholar]
- 19.Gul W, Hamann MT. Life Sciences. 2005;78:442–453. doi: 10.1016/j.lfs.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gribble GW. The Alkaloids: Chemistry and Biology. Vol. 71. Elsevier; 2012. Occurrence of Halogenated Alkaloids; pp. 1–165. [DOI] [PubMed] [Google Scholar]
- 21.Pérez EG, Cassels BK, Eibl C, Gündisch D. Bioorg Med Chem. 2012;20:3719–3727. doi: 10.1016/j.bmc.2012.04.050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.