

Abstract
The Receptor Interacting serine/threonine Kinase1 and 3 (RIPK1, RIPK3) are regulators of cell death and survival. RIPK1 kinase activity is required for necroptosis and apoptosis, while its scaffolding function is necessary for survival. Although both proteins can mediate apoptosis, RIPK1 and RIPK3 are most well-known for their role in the execution of necroptosis via the pseudokinase mixed lineage domain like (MLKL). Necroptosis is a caspase-independent regulated cell death program which was first described in cultured cells with unknown physiologic relevance in the liver. Many recent reports have suggested that RIPK1 and/or RIPK3 participate in liver disease pathogenesis and cell death. Notably, both proteins have been shown to mediate inflammation independent of cell death. Whether necroptosis occurs in hepatocytes, and how it is executed in the presence of an intact caspase machinery is controversial. In spite of this controversy, it is evident that RIPK1 and RIPK3 participate in many experimental liver disease models. Therefore, in addition to cell death signaling, their necroptosis independent role warrants further examination.
Keywords: RIPK1, RIPK3, cell death, necroptosis, apoptosis
The Receptor Interacting Protein Kinase Family
The Receptor Interacting serine/threonine Protein Kinase (RIPK) family is comprised of seven members, all of which share a homologous kinase domain with varying intermediary and C-terminal domains [Figure1A.]. These unique non-kinase domains differentiate the proteins and largely dictate their function1. RIPK1, the originally identified member of the family, harbors a C-terminal death domain (DD) which engages death receptors (DR) such as tumor necrosis factor (TNF), TNF related apoptosis inducing ligand (TRAIL), CD95 (FAS) directly, or indirectly via adaptors such as TNF-receptor-associated death domain (TRADD) and Fas-associated protein with death domain (FADD). In between RIPK1’s N-terminal kinase domain and its DD there is an intermediary domain with a RIP homotypic interaction motif (RHIM) that enables its interaction with other proteins with similar domains such as RIPK3, TIR-domain-containing adapter-inducing interferon-β (TRIF), and DNA-dependent activator of IRFs/Z-DNA binding protein 1 (DAI). This RHIM interaction is important for Toll-Like Receptor-3 and 4 (TLR3 and TLR4) engagement of RIPK1 via the adaptor protein TRIF2. RIPK1’s kinase activity is generally thought to be necessary for killing by these stimuli3–5. RIPK2 (a.k.a CARDIAK/RICK) has a caspase activation and recruitment (CARD) domain on its C terminus. RIPK2 plays an essential role in modulation of innate and adaptive immune responses and NOD1/NOD2 mediated NFκB activation6. RIPK3 shares the family’s N-terminal kinase domain and on its C-terminus has a RHIM domain which enables its interaction with RIPK17. This RIPK1/RIPK3 interaction serves to activate RIPK3 as the initiation signal for the formation of the necrosome (discussed below). RIPK4 (DIK/PKK) and RIPK5 (SgK288) are characterized by ankyrin repeats in their C-terminus1. RIPK6 and RIPK7, more commonly known as Leucine-Rich Repeat Kinase 1 and 2 (LRRK1 and 2), share the serine/threonine kinase of the RIPKs, however they contain leucine rich repeat motifs, GTPase domains, as well as, Ras of complex proteins/C-terminal of Roc (Roc/COR) domains1. Interestingly variants of RIPK7/LRRK2 have been associated with familial and sporadic Parkinson’s disease and Crohn’s disease8,9. In general, the functions of RIPK4-7 are poorly understood. For the purpose of this review we will focus on the most studied RIP kinases in the liver: RIPK1 and RIPK3.
Figure 1.
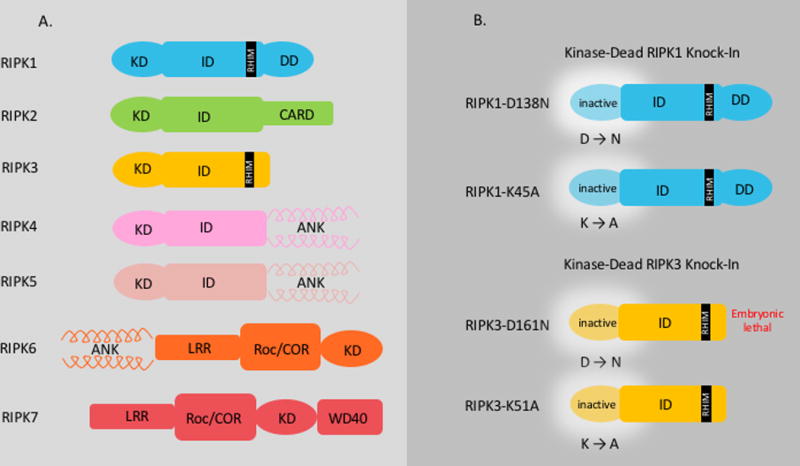
Panel A: The Receptor Interacting Kinase family is comprised of seven diverse members with a homologous Kinase Domain (KD). Each protein contains additional specific domains which are unique to the protein and dictate its function.
Panel B: The mutated Kinase-Dead Knock-In RIPK1 and RIPK3 mice that have been generated to study cell death are depicted. RIPK1-D138N: In order to render the kinase domain inactive, the catalytically important HKD (Asp-Phe-Gly) motif is altered. The conserved aspartate (D) at position 138 is mutated to asparagine (N) resulting in a “kinase-dead” protein. The mice are viable and fertile. RIPK1-K45A: A point mutation in the catalytic lysine (K) to alanine (A) in exon 3 of the RIPK1 gene eliminates all kinase activity resulting in a “kinase-dead” protein. These mice are viable and fertile. RIPK3 D161N: The aspartate (D) at position 161 in the catalytically important DFG (Asp-Phe-Gly) motif of the kinase domain of RIPK3 is mutated to an asparagine (N), rendering the protein catalytically inactive or “kinase-dead”. The mice generated with this mutation are not viable and die at E11.5. RIPK3-K51A: The lysine (K) at position 51 on the ATP-binding pocket is mutated to an alanine (A) resulting in “kinase-dead” RIPK3. These mice are viable and fertile. Abbreviations: ANK: Ankyrin Repeats, CARD: caspase activation and recruitment, KD: Kinase Domain, ID: Intermediary Domain, RHIM: RIP Homeotypic Interaction Motif, LRR: Leucine Rich Repeats, RIPK1: Receptor Interacting Kinase, roc/COR: Ras of complex proteins/C-terminal of Roc, WD40: WD40 Domain.
RIPK1, was first described as a FAS receptor interacting protein and as a mediator of cell death10. The structure of RIPK1 and whether it is in a kinase active conformation or not, dictates its function. When ubiquitinated, RIPK1 is in closed conformation and forms a platform that facilitates congregation of multiprotein complexes resulting in NFκB and MAPK activation11,12. In its kinase-active form, RIPK1 recruits RIPK3 to its RHIM domain initiating cell death pathways. RIPK1 is constitutively expressed in most tissues. However, stress signals, death receptor ligands, or T cell activation can induce the protein1,10. RIPK3 was described subsequent to RIPK1 in 1999 as a novel protein with an N-terminal kinase domain that was closely homologous with RIPK1’s7. RIPK3 was shown to contain the RHIM domain enabling its binding to RIPK1, and RIPK3 over-expression was shown to induce apoptosis7. RIPK3 tissue distribution is not uniform and the protein is difficult to detect in hepatocytes under basal conditions7,13. In fact, the presence of RIPK3 in the liver and whether it is induced and in which models is a matter of intense debate14. Interestingly, unlike RIPK1−/− mice which die within three days of birth11, RIPK3−/− mice showed no phenotype15. However, Kinase-Dead RIPK1 Knock-in mice are viable and fertile, while some Kinase-Dead RIPK3 mice (D161N mutant) die at embryonic day 11.55 [Figure1B.]. Both proteins can mediate apoptosis, through DRs. When caspase-8 is inhibited or absent, TNF can result in an alternative cell death sub-routine termed necroptosis, which is regulated in execution, and necrotic in morphology16. Since the discovery that RIPK1 and RIPK3 mediate the TNF death signal to activate the necroptosis effector mixed lineage domain like (MLKL), these kinases have been largely studied in the context of necroptosis17. These signaling events can occur downstream of various DRs (TNF, FAS, TRAIL, TLR, etc) with some variations. For simplicity we will focus on the extensively studied TNF signaling pathway18–21.
RIPKs and Apoptosis
TNF is a potent inducer of cytokines and inflammation. TNF activation can also result in the activation of caspases, DNA fragmentation, chromatin condensation, plasma membrane blebbing, cell shrinkage, and ultimately formation of apoptotic bodies22. Binding of TNF to TNFR results in the formation of an intracellular signaling protein complex called Complex-I [Figure2.]23,24. RIPK1 and TRADD are recruited to the receptor and bind via their respective DDs. TRADD recruits TNF receptor associated factor-2 (TRAF2) and the cellular inhibitor of apoptosis (cIAPs). The cIAPs ubiquitinate RIPK1 which leads to the recruitment of linear ubiquitin chain assembly complex (LUBAC, composed of HOIL, HOIP, Sharpin), transforming growth factorβ (TGFβ)-activated-kinase-1 (TAK1), TAB1/2 and the IKK complex (IKK1/ 2 and NEMO)23. Phosphorylation of the IKK complex by TAK1 leads to phosphorylation and proteasomal degradation of IkBα which normally sequesters NFκB and this results in NFκB liberation, nuclear translocation, and the transcription of anti-apoptotic NFκB target genes such as the caspase 8 inhibitor cellular FLICE inhibitory protein (cFLIP), and the de-ubiquitinases cylandromatosis (CYLD) and TNF Tumor necrosis factor alpha-induced protein 3 (TNFAIP3 or A20)25. RIPK1 can also mediate TNF-induced activation of the mitogen-activated protein kinases (MAPKs), such as ERK, p38, and JNK12,26. Therefore, the end result of Complex-I activation is inflammation and survival. In contrast transition of this multi-protein cluster from Complex-I to Complex-II results in cell death. RIPK1’s post-translational modifications control cell fate by determining whether signaling pathways stream through Complex-I or Complex-II. When TNF induced transcription is compromised and RIPK1 is de-ubiquitinated by enzymes such as CLYD, it can move from Complex-I to the pro-death Complex-II [Figure2.]. Recently inhibitory phosphorylation of RIPK1 by the TAK1/P38 activated MAPKAP kinase-2 (MK2) has been described as a key regulator of RIPK1 function27–30. Inhibitory phosphorylation of RIPK1 (on S321 and S336) by MK2 suppresses the cytotoxic potential of RIPK1 and prevents TNF induced cell death28–30. In addition, RIPK1 can be inhibited by caspase8/cFLIP mediated cleavage and IKK mediated phosphorylation preventing its incorporation into Complex-II31. How RIPK1 phosphorylation by MK2 prevents Complex-II formation remains unclear, however since the phosphorylated residues are very close to the capsase8/cFLIP cleavage target it has been suggested that this modification can result in a conformational change exposing the cleavage site32. There are multiple variations and compositions of Complex-II. Complex IIa which is comprised of TRADD, FADD, RIPK1 and caspase 8 results in RIPK1 independent apoptosis via capsase-8 [Figure2.]24. In Complex-IIb RIPK1 associates with FADD and caspase-8 resulting in apoptosis which is RIPK1 kinase dependent24. A complex similar to Complex-IIa can form when cIAPs are depleted (independent of TNF signaling) called the Ripoptosome consisting of RIPK1, RIPK3, FADD, Caspase-8 and cFLIP33–35. Complex-II and the Ripoptosome trigger apoptosis by activating caspase 8 to form homodimers which requires the kinase activity of RIPK124,34,35. In type I cells such as lymphocytes caspase-8 activates the executioner caspases (3 and 7) resulting in apoptosis, in type II cells such as hepatocytes, apoptosis requires mitochondrial participation through caspase-8 mediated cleavage of Bid resulting in mitochondrial outer membrane pore opening (MOMP) and cytochrome C release. This results in the formation of the apoptosome activating caspase 9, which in turn activates the effector caspases (caspase 3/7) to induce apoptosis. This has been reviewed in detail22. In addition to DR signaling and TLRs, anticancer drugs and DNA damage can activate RIPK1 dependent apoptosis36. RIPK3 can also result in apoptosis, unlike the RIPK3−/− mice that have no phenotype, kinase-dead RIPK3 knock-ins (RIPK3D161N) die at E11.55. RIPK3 kinase inhibitors cause a conformational change in RIPK3 resulting in RIPK1 recruitment, caspase-8 activation and apoptosis37. Interestingly not all kinase dead RIPK3 mice develop spontaneous apoptosis, for example the K51A mutant is viable and fertile indicating that the lack of kinase activity is not the impetus for apoptosis induction. Rather certain mutations can cause conformational change in the protein resulting in RIPK1 recruitment and ultimately activation of the Ripoptosome38. An intriguing open question especially in liver disease models is whether the RIPK1 inhibitor, necrostatin-1 (nec-1), can similarly affect RIPK1 platform function by affecting protein conformation.
Figure 2.
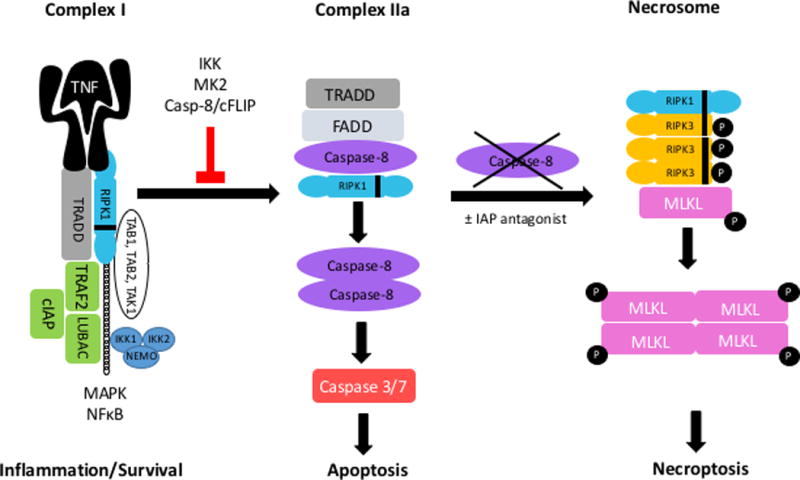
TNF binding to its receptor results in the formation of Complex-1 which includes TRADD, RIPK1, TRAF2, IAP1, IAP2 and LUBAC. Ubiquitination of RIPK1 results in the formation of a platform and subsequent recruitment of IKK1, IKK2, NEMO and TAB/TAK-1 complexes. This results in the activation of NF-κB and MAPK cascade, promoting stress response, inflammation and survival. When RIPK1 is not ubiquitinated, Complex-II forms made up of TRADD, FADD and caspase-8 and RIPK1 (IIa) or RIPK1/RIPK3, FADD, Caspase 8 and FLIPL (IIb, not shown) which leads to caspase-8-mediated activation of caspase 3/7 and apoptosis. In certain cells when caspase-8 is inhibited RIPK1, RIPK3, and MLKL form the necrosome, resulting in MLKL phosphorylation leading to its oligomerization and translocation to cell membrane to induce necroptosis.
RIPKs and Necroptosis
Despite being a critical member of the DR induced Complex-I and participating in apoptosis, RIPK1’s claim to fame is due to its link with the more recently described cell death sub-routine, necroptosis. Necroptosis occurs in certain cell types when TNF binds to TNFR in the presence of a pan-caspase inhibitor (+/− an IAP antagonist)36. It was first described in cells stimulated with TNF or FAS in the presence of a pan caspase inhibitor, carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone (ZVAD-fmk) requiring the kinase activity of RIPK118,39,40. RIPK1 kinase inhibitors, necrostatins (Nec), were later shown to block this form of morphologically necrotic and regulated cell death, confirming the role of the protein in necroptosis execution41,42. When caspase-8 is absent or inhibited, RIPK1 binds to RIPK3 via the RHIM domain, forming a complex called the necrosome which also includes FADD (+/− TRADD) [Figure2.]. RIPK3 then oligomerizes via its RHIM domain and autophosphorylates leading to activation of the pseudokinase, MLKL43. RIPK3 is necessary for necroptosis execution while RIPK1 is most likely dispensable, as recruitment of RIPK3 by other RHIM-containing proteins, such as TRIF and DAI, can promote RIPK3 activation44,45. This has been reported with certain viruses such as herpes simplex and murine cytomegalovirus where RHIM containing proteins can directly engage RIPK3 independent of RIPK146. MLKL, the only known effector of necroptosis, is a pseudokinase with an N-terminal effector domain which is kept inactive by MLKL’s C-terminal pseudokinase domain43,47–49. It is hypothesized that RIPK3 and MLKL exist as pre-assembled non-active complexes and that auto-phosphorylation of RIPK3 results in an open conformational shift exposing the N-terminal four-helical bundle domain and releasing MLKL, leading to oligomerization and translocation to plasma membrane and cell lysis by membrane rupture47–53.RIPK3 is the only known activator of MLKL and therefore it is believed that cells that do not express RIPK3 do not undergo necroptosis36. One intriguing aspect of the relationship between RIPK1 and RIPK3 lies in the observation that in addition to collaboration with RIPK3 to promote cell death, RIPK1 can inhibit RIPK3 under certain conditions. RIPK1 specific knockout in intestinal epithelium or keratinocytes resulted in spontaneous RIPK3 mediated cell death and inflammation54,55. The precise mechanism by which this occurs remains unknown.
RIPKs and Inflammation
While RIPKs were initially described as mediators of cell death and survival, emerging evidence indicates a necroptosis-independent role for the RIPKs in inflammation [Figure3.]56. The most obvious evidence for this lies in the relationship between RIPK1 and NFκB, the main transcription factor for the expression of inflammatory genes. Ubiquitinated RIPK1’s scaffolding function mediates NFκB activation downstream of TNF. Although RIPK1 seems to be dispensable for NFκB activation, some studies have suggested decreased expression of NFκB responsive genes without RIPK157–59. RIPK1 and RIPK3 can participate in inflammatory pathways such as TLR signaling, cytokine production, NOD-like receptor protein 3 (NLRP3) and inflammasome activation independent of MLKL and their role as mediators of necroptosis60–62. The effect of RIPK1 and RIPK3 in inflammation was initially assumed to be due to the inflammatory nature of necroptotic cell death from MLKL mediated plasma membrane rupture and release of intracellular immunogenic molecules. However there is accumulating evidence that the RIPKs directly influence inflammation. An example is in the TNF alpha induced protein 3 (Tnfaip3)/A20 global knockout mouse. A20−/− mice have systemic inflammation resulting in cell death. Deletion of RIPK3 or a catalytic inactive RIPK1 significantly delay mortality in these mice while MLKL deletion does not63. Liver leukocytes isolated from RIPK3−/− mice and stimulated by α-GalCer showed significantly reduced production of cytokines (TNF, IL-4 and IFNγ) compared to WT cells suggesting a role for RIPK3 in NKT cytokine production64. Macrophages lacking cIAPs secret larger quantities of pro-inflammatory TNF and IL-6 which required RIPK1 and RIPK3, but not MLKL65.
Figure 3.
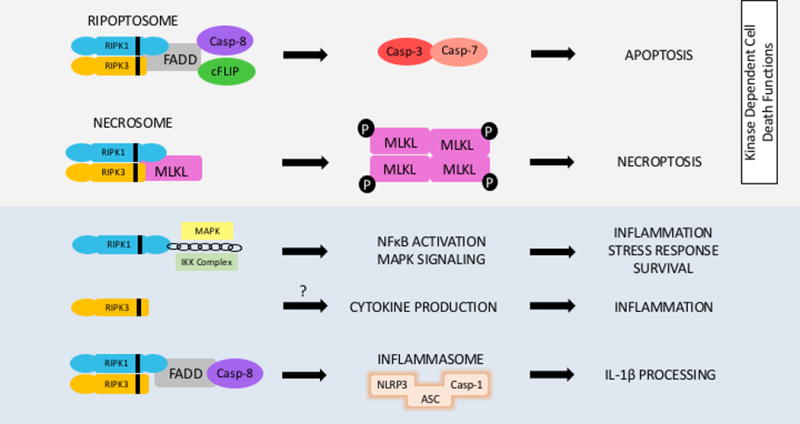
Although the RIPKs are most known in the context of necroptosis they are multifunctional proteins. The kinase activity of RIPK1 mediates apoptosis and necroptosis while its scaffolding activity enables NFκB activation, inflammation and cell survival. RIPK3’s necroptosis independent roles include activation of cytokine secretion in certain models and inflammasome activation via caspase-8. Both RIPK1 and RIPK3 have been implicated in activation the NLRP3 inflammasome pathway.
Inflammasome dependent cell death or pyroptosis, is characterized by the assembly of a multi-protein complex (NLRP3-ASC-Caspase-1) resulting in activation of caspase-1 and release of the pro-inflammatory cytokines IL-1β and IL-1856. RIPK3 can activate caspase-1 and promote IL-1β cleavage thereby contributing to inflammatory cytokine production61,62,66,67. RIPK3 has also been shown to activate the NLRP3 inflammasome pathway in macrophages downstream of TLR3 in response to RNA virus infection68. RIPK1 has been associated with the alternative NLRP3 pathway in human monocytes stimulated by LPS downstream of TLR4 which requires FADD and caspase-869. Interestingly in a mouse model of pyroptosis where NLRP3 was hyperactivated and mutant mice suffered shortened survival, poor growth, and severe liver inflammation; RIPK1 and RIPK3 did not contribute to disease70. Detailed discussion of pyroptosis and inflammasome mediated cell death and inflammation is beyond the scope of this topic and have been recently reviewed extensively56,71,72. The RIPKs role in the inflammasome and in necroptosis may not be mutually exclusive and there may be cross talk between the two pathways56. Therefore, a growing body of evidence suggests that the RIPKs are multi-functional proteins with prominent roles independent of their actions as cell death mediators [Figure3.]. These non-necroptotic properties should be taken into account when interpreting experimental findings14.
RIPKs in the liver
Cell death is an important driver of liver disease and both apoptosis and necrosis occur in the liver with apoptosis being the more common death pathway22,73–75. RIPK1 and RIPK3 have been studied in various liver injury models, mainly in the context of determining the contribution of necroptosis [Figure4.]. Hepatocytes robustly express DRs and many disease states such as NASH, viral hepatitis, alcoholic hepatitis, cholestatic liver disease and auto-immune liver disease have been shown to result in hepatocyte apoptosis76,77. One significant argument against the occurrence of necroptosis in the liver is that under numerous apoptosis-inducing experimental settings pharmacologic or genetic prevention of apoptosis in the liver has been demonstrated to rescue without a switch to necroptosis14. A second important point is that it is not clear that hepatocytes contain all the necessary machinery for necroptosis. RIPK1 and MLKL are clearly expressed in the liver, however RIPK3 expression under basal conditions has been controversial7,13,14,78–80. RIPK3 induction has been suggested in some models and human diseases81–85. While RIPK3 is difficult to detect in freshly isolated hepatocyte it is robustly expressed in the non-parenchymal cell fraction, making it possible that necroptosis occurs in certain cell types in the liver78. A RIPK3 independent model for necroptosis in hepatocytes has been suggested and since MLKL is clearly present in the liver, it is hypothesized that other MLKL activators exist79,80.
Figure 4.
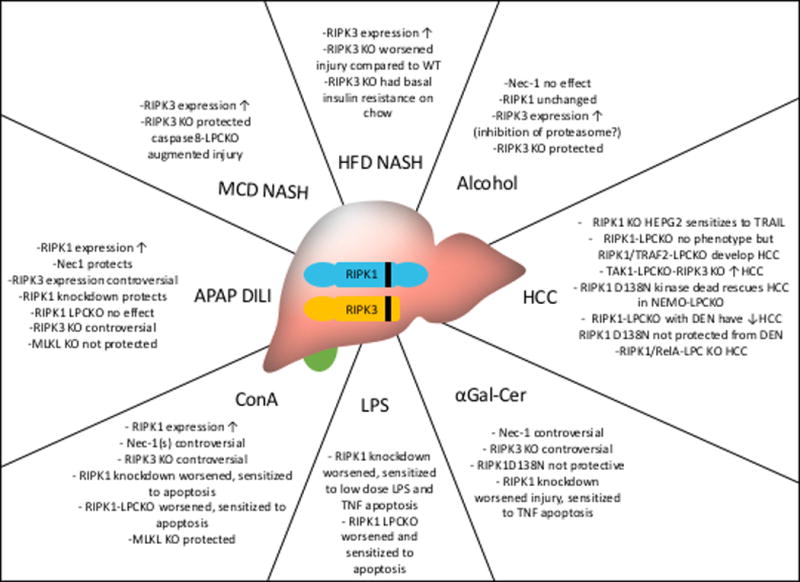
RIPK1 and RIPK3 have been studied in various models of liver disease, generating significant controversy.
RIPKs in APAP toxicity and hepatocyte Death
RIPK1 was the first family member studied in liver disease due to the availability of nec-1, the RIPK1 kinase inhibitor which was shown to protect against necrotic cell death in multiple organ systems41,42. Using nec-1 multiple investigator demonstrated a survival benefit against acetaminophen (APAP) induced liver injury in mice and in hepatocytes in vitro86–90. Unlike the consistent and reproducible results with nec-1, RIPK3’s role in APAP and whether necroptosis occurred in the liver became the source of controversy14,78. Since the cell death mode in APAP is largely necrotic, the abrogation of injury with nec-1 was initially interpreted as inhibition of necroptosis. However nec-1 is neither specific to RIPK1, nor necroptosis38. Nec-1 was discovered to be identical to the indoleamine 2,3-dioxygenase (IDO) inhibitor methyl-thiohydantoin-tryptophan (MTH-Trp)91. IDO is a potent immuno-modulator and is the rate-limiting step of tryptophan catabolism and it has been shown to regulate pathways that lead to pro and anti-inflammatory cytokine production. Therefore additional evidence was needed regarding the role of RIPK1 in APAP induced liver injury91. Using an antisense oligonucleotide knockdown approach, we observed that RIPK1 knockdown in liver protected significantly from APAP induced cell death and provided a survival benefit to mice. In keeping with the controversial reports on this matter, RIPK1’s role in APAP toxicity was soon challenged92. The off target effects of nec-1 combined with fact that necroptosis requires the participation of RIPK3 and MLKL led us to explore APAP toxicity in RIPK3 and MLKL knockout mice to settle the question of APAP-induced necroptosis in the liver78. Previously it was reported that RIPK3−/− mice were protected against APAP at early (6hours) but not late (24hrs) time points88. We were unable to detect any difference in liver injury from RIPK3−/− mice compared with WT animals at either time78. However, knockdown of RIPK1 in RIPK3−/− mice afforded a significant protection, suggesting that RIPK1 was functioning independent in RIPK3 and necroptosis to protect against APAP toxicity78. To confirm this, we used MLKL−/− mice and were unable to detect any attenuation of APAP-induced cell death in MLKL−/− mice78. These results were later independently confirmed by others79. Therefore, the mere participation and even induction of the RIPKs in any disease model does not indicate that the cell death subroutine is necroptosis. In fact, many of the studies in different models and organ systems showing protection using nec-1, RIPK1 kinase dead, and RIPK3−/− mice were not reproducible in MLKL−/− mice, pointing to the multifactorial and non-necroptotic functions of these proteins63.
RIPKs in Immune Mediated Liver Injury Models
Compounds that activate the immune system such as Concanavalin A (ConA) and α-galactosylceramide (α-GalCer) are widely used substitute models for human auto-immune hepatitis. Unlike autoimmune hepatitis which is mainly adaptive immune mediated, these compounds generate an innate immune response. ConA, a plant lectin, activates CD4+ T cells, NKT cells, KC and macrophages to induce an immune-mediated hepatitis93. The mode of cell death in ConA has been a source of debate but the preponderance of the evidence suggest it is a necrotic subroutine (no protection by caspase inhibition) although some apoptosis may occur in the early phases of injury94–96. Unlike APAP which is not dependent on external DR signaling, ConA hepatitis is mediated through cytokines and DR. IFN-γ, IL-4, IL-6, FAS and TRAIL have all been reported as mediators of ConA hepatitis and cell death, but the major cytokine contributing to toxicity is known to be TNF96. The necrotic mode of cell death combined with the dependency on DR signaling has led multiple investigators to study necroptosis in ConA induced liver injury. Pre-treatment of nec-1 was suggested by some to markedly protect against ConA97–99. RIPK1 kinase inhibition has been proposed to result in decreased Poly ADP-ribose polymerase-1 (PARP-1) activation, and decreased IL-33 suggesting that RIPK1 is an upstream mediator of these signaling events in ConA97,98. However, in vivo RIPK1 knockdown in the liver (with antisense) resulted in exacerbated injury and increased mortality from ConA which was almost completely prevented by zVAD-fmk57. Using RIPK1 kinase dead knock-in mice (Ripk1-K45A) and nec-1s, Filliol and colleague have reported that RIPK1 kinase activity drives liver injury induced by ConA, as shown by reduced levels of serum transaminases and improved histology100. Interestingly, RIPK1 liver specific knockout RIPK1-LPCKO resulted in more pronounced injury and higher ALT. In agreement with the aforementioned study, the cell death mode in these RIPK1 deficient hepatocytes was caspase-dependent apoptosis100. Furthermore, RIPK1 deletion in parenchymal cells resulted in a more robust inflammatory response. This enhanced sensitivity was TNF mediated as treatment with etanercept, which neutralizes TNF, protected against ConA100. These studies elucidate the complexity with the RIPK1 protein as inhibiting its kinase and platform activities resulted in alternative outcomes. In contrast to the studies reporting benefit with RIPK1 kinase inhibition(genetically or using nec-1), others have observed a worsening of injury and increased mortality (up to 50%) with the more specific nec-1s87. Interestingly these same investigators reported that despite aggravation of liver injury with nec-1s, a modest attenuation was observed in RIPK3 global knockouts87. Kang and colleagues have also reported that ConA injury is attenuated in RIPK3−/− mice64. However, the contribution of RIPK3 to ConA has been challenged by two independent reports indicating no difference in outcomes of liver injury in RIPK3−/− compared to WT mice79,101. Interestingly, Gunther and colleagues were unable to detect any RIPK3 in hepatocytes and failed to detect an induction or increase in transcripts following ConA treatment79. However, despite no role for RIPK3, they reported less liver injury in both MLKL−/− mice and in nec-1s pre-treated mice with ConA suggesting an alternative MLKL activation pathway in hepatocytes79. Further exploration of this alternative, non-canonical necroptotic pathway to MLKL activation is warranted.
The glycolipid α-galactosylceramide (α-GalCer) which is a specific and potent activator for invariant natural killer T cells (NKT) has also been used to study immune-mediated liver injury57. This model resembles the ConA model, but is relatively less severe and restricted to NKT cells, innate immune cells uniquely abundant in the liver. Using both α-GalCer and ConA models, Kang et al have reported attenuation in RIPK3 −\− mice with less cytokine (TNF, IL-4) and IFNγ expression. RIPK3 in bone marrow (BM) cells was essential as ConA-induced liver injury was significantly attenuated in the WT animals that were transplanted with RIPK3−/− BM. This protective effect was suggested to be due to the effect of RIPK3 on cytokine production from NKT cells deficient in RIPK3 and not due to the prevention of hepatocyte necroptosis64. Contrary to the findings by Kang et al, we found no difference in liver injury between global RIPK3−/−, MLKL−/− mice and strain matched controls treated with α-GalCer57. However, knockdown of RIPK1 markedly aggravated α-GalCer-induced hepatitis and resulted in 90% post treatment mortality as early as 6 hours post α-GalCer57. Accompanying the exacerbated liver injury, RIPK1 knockdown significantly increased serum levels of proinflammatory cytokines and chemokines (TNF and IFNγ) 6hrs post injection correlating with increased inflammatory cell infiltration57. The liver cell death was accompanied by caspase 3 activation and inhibited by TNF neutralization and pan-caspase inhibitors without a switch to necroptosis57. This increased sensitivity to TNF was not seen in RIPK1 kinase dead D138N mice or with nec-1 and seems to be due to the scaffolding functions of RIPK157. Increased sensitivity of hepatocytes lacking RIPK1 to TNF mediated apoptosis has been reported using lipopolysaccharide (LPS) and unmethylated CpG oligodeoxynucleotide (CpG-DNA) to induce liver injury as well57,102. In RIPK1 knockdown livers low dose LPS resulted in massive hepatocyte apoptosis57. This injury is thought be mediated by macrophage-produced TNF, as macrophage depletion using clodronate or the use of a TNF neutralizing antibody can rescue from liver injury57,102. While certain aspects of these studies, namely the effect of RIPK3 deletion and RIPK1 inhibitors in immune-mediated hepatitis has been controversial, a common theme that becomes evident is that deletion of RIPK1 aggravates immune-mediated liver injury by sensitizing hepatocytes to TNF mediated apoptosis. The discrepancies in many of these reports could be minimized by using standard dosing regimens and appropriate controls (especially when DMSO is the vehicle). Additionally, interpretation of effects in animal models where littermate controls are not used should be done with caution14.
RIPKs in Fatty Liver Disease and Alcoholic Liver Disease
Alcoholic and non-alcoholic steatohepatitis (ASH and NASH) are major public health problems and leading causes of liver related mortality worldwide and in the U.S.103,104. These distinct disease entities share many common features such as steatosis, Mallory bodies, inflammation, parenchymal fibrosis, and hepatocyte injury leading to cell death. In both entities a major driver of disease progression is cell death with apoptosis as the main subroutine75,105. This is demonstrated by the fact that caspase 8 liver specific KO (casp8-LKO) or caspase inhibition greatly protects against steatohepatitis without a switch to necroptosis in NASH106–108. Studies examining the role of the RIPK proteins in NASH have yielded contradictory results. This may be partly due to the various models employed to induce NASH in rodents. Studies using diets deficient in methionine and choline (MCD) were commonly used to induce steatohepatitis in mice. However, MCD has fallen out of favor as mice on these diets, lose weight and do not exhibit the metabolic syndrome phenotype despite developing steatohepatitis109. Using the MCD model, an increase in RIPK3 protein as early as two weeks has been reported84. In contrast to the earlier studies showing protection in casp8-LKO mice, using the MCD diet Gautheron and colleagues found higher ALTs and worse fibrosis with deletion of caspase-8 which was somewhat attenuated by RIPK3 knockout84 and accompanied by increased pMLKL staining110. Increased RIPK3 expression has also been reported using immuno-staining of human NASH and NAFLD livers84,85. Afonso and colleagues reported increased RIPK3 and pMLKL expression with High Fat Choline Deficient (HFCD) feeding and attenuation of liver injury, inflammatory cytokines, and fibrosis with RIPK3 deletion85. Interestingly they reported increased RIPK3 protein not only in biopsy samples from NASH patients but also in liver biopsies from patients with other chronic liver diseases such as PBC, ASH, Hepatitis B and C (HBV, HCV)85, raising questions about the specificity of RIPK3 staining or perhaps suggesting that induction of RIPK3 is a common feature of chronic liver disease regardless of underlying etiology14. In contrast to reports of amelioration of injury in RIPK3−/− using the MCD diet, using the Western type High Fat Diet (HFD) mice lacking RIPK3 exhibited significantly worse steatosis, fibrosis, and inflammation compared to WT mice82. The WT mice in this study expressed no basal RIPK3 or pMLKL, but after 12 weeks on the HFD both proteins increased in the fatty liver by immunohistochemistry (IHC)82. However, despite the high expression of pMLKL these mice had very little injury and cell death82. Further complicating matters was the observation that RIPK3−/− mice exhibit glucose intolerance at baseline on a chow diet82. The role of RIPK3 in alcoholic liver injury using the Lieber-DeCarli model of alcohol feeding has been explored83,111. In contrast to the HFD study, RIPK3−/− mice on a Lieber-DeCarli alcohol diet had a modest attenuation of injury compared to WT controls83. The authors also reported increased RIPK3 protein immunostaining in human liver biopsy samples with alcoholic liver disease83. Another group recently studied the mode of cell death in Lieber-DeCarli model using caspase8-LPCKO mice and reported increased apoptosis with caspase-8-LPCKO with no significant RIPK1, RIPK3 and MLKL induction111. The Gao-binge model of alcohol feeding has also been reported to result in increased RIPK3 protein81. The authors found lower transaminase elevations and decreased steatosis from global RIPK3 deletion, but no difference in inflammation and hepatitis. They could not detect a transcriptional change in RIPK3, and attributed the increased protein levels of RIPK3 to impaired hepatic proteasome function81. The contribution of necroptosis to cell death in NASH is arguable and apoptosis seems to be the predominant cell death pathway75. Studies using hepatocyte specific MLKL knockout and littermate controls are needed to settle the matter.
However, given recent reports of insulin resistance in RIPK3−/− mice82, it is compelling to imagine a necroptosis independent contribution of RIPK3 to NASH pathogenesis.
RIPKs in HCC
Hepatocellular carcinoma (HCC), the most common primary liver tumor which often occurs in the context of chronic inflammation. Cell death is both a consequence and a cause of liver inflammation and therefore a strong link exists between hepatocyte death and HCC development. The role of the RIPKs has been explored in a few models of HCC59,112,113. In the human hepatoma cell line HepG2 cells, deletion of RIPK1 or cFLIP sensitized to TRAIL mediated apoptosis114. Surprisingly, low RIPK1 expression which is seen in some human HCCs has been associated with a worse prognosis and decreased survival112,115. Conditional deletion of the mitogen activated protein 3 (MAP3)-kinase, TAK1, in liver parenchymal cells (TAK1-LPCKO) results in hepatic inflammation, cholestasis, fibrosis which leads to HCC116. These TAK1-LPCKO mice exhibited defective NF-kB activation, increased spontaneous apoptosis and increased RIPK3 protein expression. TAK1-caspase8-LPCKO resulted in less injury and less caspase 3 cleavage without a switch to necroptosis116. The combination of global RIPK3 knockout with TAK1-LPCKO resulted in less necrosis, less cholestasis and decreased ALT. At 38 weeks TAK1-caspase8-LPCKO did not exhibit any tumors while TAK1-LPCKO-RIPK3−/− mice exhibited a massive tumor burden suggesting a role for RIPK3 in modulating hepatocyte apoptosis116. TAK1LPCKO-RIPK3−/− mice had increased JNK activation and the addition of the JNK inhibitor significantly decreased hepatocytes proliferation (Ki67 and cyclin D1 levels)116. NEMO liver parenchymal cell knockout (NEMO-LPCKO) results in hepatitis, hepatocyte apoptosis and HCC, suggesting a tumor suppressor function of NF-kB signaling in the liver117. The development of hepatitis and HCC in NEMOLPC-KO mice is triggered by FADD- and caspase-8-dependent apoptosis. Surprisingly NFKB-LPCKO mice, which lack all subunits in the liver parenchymal cells, have a much milder phenotype compared to NEMO-LPCKO suggesting that NEMO has NF-kB-independent pro-survival functions in hepatocytes113. The role of RIPK1 in mediating cell death from the NEMO-LPKO mice has been explored using a genetic kinase dead RIPK1 knock-in mouse (D138N)113. The addition of the kinase dead RIPK1 to the NEMO-LPCKO strongly inhibited hepatocyte death, prevented liver damage and HCC development. In contrast, the global deletion of RIPK3−/− in the NEMO-LPCKO did not prevent these outcomes, suggesting that RIPK3/MLKL-driven necroptosis does not play an important role in this model113. Further supporting this was the fact that the dying hepatocytes stained positively for cleaved caspase 3, and ablation of caspase-8 or FADD inhibited cell death indicating RIPK1 kinase dependent apoptosis as the cell death subroutine. The expression of kinase inactive RIPK1, fully restored the expression of cFLIP and partly normalized the expression of TRAF2 and cIAP1 proteins in the NEMO-deficient livers. However, RIPK1 ablation in LPC using RIPK1 floxed mice could not prevent the death of NEMO-deficient hepatocytes showing that kinase independent platform properties of RIPK1 are required to prevent hepatocyte apoptosis (this was prevented with the additional deletion of TRADD)113. FADD and caspase-8 co-immunoprecipitated with RIPK1 suggesting that NEMO prevents formation of a RIPK1/FADD/Casp8 complex and that in the absence of NEMO RIPK1 mediates hepatocyte apoptosis via its kinase activity through this complex. In a recent report genetic deletion of RIPK1 in parenchymal cells (RIPK1-LPCKO) had no spontaneous phenotype. However when exposed to LPS/TNF these mice exhibited enhanced apoptosis due to the proteasomal degradation of TRAF2 leading to caspase dependent cell death112. The mode of cell death was caspase-8 dependent apoptosis as the additional knockout of caspase 8 in these mice completely rescued the phenotype112. Unlike the RIPK1-LPCKO, inhibiting the kinase activity of RIPK1 genetically or pharmacologically did not sensitize to TNF mediated cell death suggesting the platform function of RIPK1 is instrumental in cell survival. Interestingly like RIPK1-LPCKO, the TRAF2-LPCKO did not have a spontaneous phenotype but the double KO of RIPK1/TRAF2 resulted in hepatitis, impaired NF-kB activation and caspase 8 dependent apoptosis and HCC112. In this study human liver HCC samples higher expression of RIPK1 and TRAF2 was associated with a significant survival benefit and low expression of these proteins predicted a worse prognosis112. In contrast, another study from China has reported increased RIPK1 expression in HCC samples as a poor prognostic indicator after resection118. Patients expressing more RIPK1 by immunostaining of tumor were more likely to have advanced stage, intra-hepatic metastasis, and portal vein invasion. Interestingly, patients with increased RIPK1 in HCC were more likely to be HBV infected118.
Van and colleagues have used the diethylnitrosamine (DEN) induced HCC model in RIPK1-LPCKO mice and have also observed increase in apoptosis with RIPK1 deletion and reduction in tumor size59. Interestingly while ablation of either RIPK1 or RelA in liver parenchymal cells did not cause spontaneous liver pathology, mice with combined deficiency of RIPK1 and RelA in LPCs showed increased hepatitis, increased hepatocyte apoptosis (cleaved caspase-3) and developed spontaneous chronic liver disease and cancer59. Lack of RIPK1 kinase activity did not inhibit DEN-induced liver tumor formation, confirming that kinase-independent functions of RIPK1 promote carcinogenesis in a TNFR1-dependent manner59. An interesting finding here was that unlike previous studies, RIPK1-LPCKO resulted in impaired NF-κB signaling and reduction in the TNF-induced expression of A20, baculoviral IAP repeat-containing 3 (Birc3), nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor-alpha (Nfkbia), and Tnf59. It has now been shown in multiple models that knockdown and knockout of RIPK1 in vivo sensitizes hepatocytes to TNF mediated apoptotic cell death57,59,100,112. RIPK1 appears an attractive therapeutic target for HCC treatment if targeting cancer cells for RIPK1 ablation or knockdown and sparing healthy hepatocytes becomes possible.
RIPKs in other liver diseases
The contribution of the RIPKs to cholestatic and viral liver disease is less extensively characterized. RIPK1 but not RIPK3 deletion completely rescues the growth retardation, ductopenia and cholestatic phenotype of liver parenchymal cell specific deletion of the IKKα/β subunits (IKKα/β-LPCKO)115. Despite ductopenia and ongoing injury, no HCC developed in the IKKα/β-LPKO livers suggesting a failure of cholangiocyte proliferation. Staining for Ki-67 revealed increased proliferation in the combined IKKα/β-LPKO/RIPK1 KO compared to IKKα/β-LPKO115. The authors proposed that RIPK1 is the direct phosphorylation target of the IKK subunits and functions as an anti-proliferative protein in cholangiocyte homeostasis115. The mode of cell death in this model was proposed to be a combination of apoptosis and necrosis without a significant contribution from necroptosis as RIPK3−/− did not affect the phenotype115. Afonso and colleagues have examined the effect of RIPK3 deletion in a bile duct ligation (BDL) model of liver injury119. No difference was detected after three days however, two weeks post BDL, RIPK3−/− mice displayed less liver injury compared to strain matched controls119. RIPK3 deficiency attenuated inflammatory cell infiltration but did not affect BDL induced fibrosis119. How necroptosis is activated in this model with intact caspase-8 is unclear. High levels of RIPK3 and MLKL expression in the liver of PBC patients (hepatocytes and cholangiocytes) using immunostaining was also reported119. Cell death in viral hepatitis is due to a cytotoxic CD8+ T cell immune response120. Viral hepatitis is associated with increased expression of DR ligands such as soluble FAS, TRAIL and TNF resulting in apoptotic hepatocyte death121–125. There is not much data on the RIPKs in viral hepatitis. In an in vitro HCV culture system it was reported that the addition of nec-1 to a pancaspase inhibitor abated HCV related cell death and resulted in improved survival compared to caspase inhibition alone126.
Concluding Remarks
RIPK1 and RIPK3 are the two most studied proteins in the RIPK family. Both proteins are multi-functional and can mediate apoptosis, necroptosis or activate inflammatory pathways independent of cell death. It is well established that RIPK1 kinase activity is necessary for necroptosis, but how RIPK1 is activated and what target protein it phosphorylates remain unclear38. RIPK1 kinase inhibition does not only block necroptosis; it also interferes with TNF induced apoptosis in certain contexts24,35. Therefore, it is important to consider that the effect of necrostatins on hepatocytes and in animal models can be due to interference with apoptosis or necroptosis38. Nec-1 has also been known to have off target effects and studies using nec-1 should be interpreted with caution91. Necroptosis is thought to have evolved as an alternative cell death pathway when apoptosis is not possible. Recently the positive effects of catalytic inactive RIPK1 kinase dead and RIPK3 ablation in different pathologies was challenged by Newton et al. as MLKL loss was not as effective as RIPK3 knockout or catalytic inactive RIPK1 in these models, re-iterating the MLKL independent roles of the RIPKs63. In addition to its kinase activity, ubiquitinated RIPK1 can form a platform and participate in the activation of signaling cascades by approximating multiprotein complexes. For example, RIPK1 serves to facilitate NFκB and MAPK activation. This platform function of RIPK1 is important for the integrity of hepatocyte function as hepatocyte RIPK1KO or knockdown sensitizes to TNF mediated apoptosis (without affecting NFκB activation in most reports)57–59,100,112. It is therefore not surprising that in many animal models of liver disease, RIPK1 kinase inhibition genetically or pharmacologically had a different effect than RIPK1 ablation in liver parenchymal cells. Furthermore, the effect of RIPK1 in the immune cells and non-parenchymal liver cells in these injury models has not been fully established14.
RIPK3’s role in the liver has been quite controversial. Although RIPK3 is most known as the kinase activating MLKL for necroptosis execution, RIPK3 can also mediate apoptosis. Mutant kinase inactive RIPK3D161N protein and RIPK3 small molecule inhibitors promote apoptosis, due to the induction of a conformational change in RIPK3 resulting in RIPK1 mediated caspase-8 recruitment5,37. Whether necroptosis occurs in liver disease and in hepatocytes in particular is controversial14,106. It is generally accepted that cells that do not express RIPK3 do not undergo necroptosis36. Therein lies one of the controversies regarding the RIPKs and necroptosis in liver disease, as RIPK3 is not expressed in hepatocytes under basal conditions7,13,78,79. However, increased expression of RIPK3 in both animal models of acute and chronic liver injury and human liver diseases has been suggested by some investigators, but not all81–85,87,119. In one model, due to a lack of increase in RIPK3 mRNA, impaired proteasome function was implicated as the cause of increased RIPK381. Much of the work on the RIPKs was done by immunostaining of liver tissue and most investigators have interpreted increased RIPK3 staining as the “induction of necroptosis” bringing up the issues of specificity of these stains on injured liver, and the quality of the reagents used14. In addition, most genetic knockout studies use complex embryonic multiprotein knockout models which could result in unforeseen compensatory biologic consequences and complicate interpretation of data and extrapolation to human physiology. Since these critiques are universal limitations of experimental models multiple scientific approaches and more consistent findings by independent laboratories are necessary to draw firm conclusions regarding RIPK3 and necroptosis in the liver. We have been unable to detect RIPK3 in diseased human livers (unpublished data). In mice we were only able to detect RIPK3 in the NPC fraction of the liver including immune cells and endothelial cells and others have confirmed this data78,79. However, MLKL is robustly expressed. A few key points and open questions remain that will guide future research in the field. Why is MLKL present in hepatocytes in the absence of RIPK3 and what other protein(s) can activate it? What are the non-necroptosis functions of RIPK1 and RIPK3 in the liver? What is the role of the RIPKs in the non-parenchymal liver cells? Given the heterogeneity of the liver and its diverse cell population, a more detailed examination of the effect of the RIPKs in the liver in a cell specific manner should be undertaken. It is evident more than ever that non-necroptotic functions of the RIPKs in the liver (including apoptosis) may contribute to some of the findings in liver disease models. It is also important to consider that the cell death functions and non-cell death/ inflammatory functions of the RIPKs may not be mutually exclusive. Finally, the most pressing clinical issue remains, which is to understand if necroptosis occurs with an intact apoptotic machinery and in the presence of caspase-8 in the physiologic setting or if activation of necroptosis is a potential complication of the use of caspase inhibitors therapeutically in human liver disease. There is still much to be learned about the functions of the RIPKs and future studies will shed some light on the complex biology of these multifunctional proteins.
Main Concepts and Learning Points.
-
-
RIPK1 and RIPK3 participate in both apoptotic and necroptotic cell death and this is largely context dependent
-
-
RIPK1 and RIPK3 have cell death independent functions
-
-
RIPK1 knockdown and knockout sensitizes hepatocytes to apoptotic cell death in TNF/DR mediated cell death models, indicating its scaffolding function mediates hepatocyte survival
-
-
RIPK3 is undetectable in hepatocytes under basal conditions; however, it may be induced in certain diseases
-
-
Whether necroptosis occurs in hepatocytes is unclear
-
-
The contribution of RIPK1 and RIPK3 in various liver disease models has been examined and is discussed below
Acknowledgments
The author would like to thank Dr. Neil Kaplowitz, for his critical insight, discussions and editorial comments on the topic of this review.
Financial Support: This work was supported by NIH grant K08DK109141(LD)
Abbreviations
- ALT
Alanine Aminotransferase
- APAP
Acetaminophen
- α-GalCer
α-galactosylceramide
- ASC
Apoptosis-associated Speck-like protein containing a Carboxy-terminal CARD
- ASH
Alcoholic Steatohepatitis
- Birc3
Baculoviral IAP repeat-containing 3
- cIAP
cellular Inhibitor of Apoptosis
- cFLIP
cellular FLICE inhibitory protein
- ConA
Concanavalin A
- CYLD
cylandromatosis
- DAI
DNA-dependent Activator of IRFs/Z-DNA binding protein 1
- DD
Death Domain
- DR
Death Receptor
- DEN
Diethylnitrosamine
- ERK
Extracellular signal-regulated kinase
- FADD
Fas-Associated protein with Death Domain
- DMSO
Dimethyl Sulfoxide
- IHC
Immunohistochemistry
- IDO
Indoleamine 2,3-Dioxygenase
- KO
Knockout
- KC
Kupffer cell
- LRRK1 and 2
Leucine-Rich Repeat Kinase 1 and 2
- LPCKO
Liver Parenchymal Cell Knockout
- LUBAC
linear ubiquitin chain assembly complex
- MCD
Metheionine Choline Deficient Diet
- MTH-Trp
methyl-thiohydantoin-tryptophan
- MLKL
Mixed lineage kinase domain-like
- MOMP
mitochondrial outer membrane pore opening
- MKK2
MAP kinase-activated protein kinase 2
- NAPQI
N-Acetyl-p-benzoquinone imine
- NF-kB
nuclear factor kB
- NEMO
NF-kappa-B Essential Modulator
- Nfkbia
Nuclear factor of kappa light polypeptide gene enhancer in B cells inhibitor, alpha
- NAFLD
non alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NK
natural killer cell
- NLRP3
NOD-like receptor protein 3
- NPC
non-parenchymal cells
- Roc/COR
Ras of complex proteins/C-terminal of Roc
- RIPK1
Receptor Interacting serine/threonine Protein Kinase 1
- RIPK3
Receptor Interacting serine/threonine Protein Kinase 3
- RHIM
RIP Homology Interaction Motif
- TNF
Tumor Necrosis Factor
- Tnfaip3
TNF alpha induced protein 3
- TGFβ
transforming growth factorβ
- TAK1
-activated-kinase-1
- TAB2
TAK1 Binding protein-2
- TAB3
TAK1 Binding protein-3
- TLR3 and TLR4
Toll-Like Receptor-3 and 4
- TRAIL
TNF Related Apoptosis Inducing Ligand
- TRADD
TNF Receptor Associated Death Domain
- TRAF
TNF Receptor Associated Factor
- TRIF
TIR-domain-containing adapter Inducing Interferon-β
- WT
wild type
- ZVAD-fmk
carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]- fluoromethylketone
Footnotes
Conflict of interest statement: The author has no conflict of interest to state.
References
- 1.Zhang D, Lin J, Han J. Receptor-interacting protein (RIP) kinase family. Cell Mol Immunol. 2010;7(4):243–249. doi: 10.1038/cmi.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meylan E, Burns K, Hofmann K, et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat Immunol. 2004;5(5):503–507. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- 3.Kaiser WJ, Daley-Bauer LP, Thapa RJ, et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U S A. 2014;111(21):7753–7758. doi: 10.1073/pnas.1401857111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polykratis A, Hermance N, Zelic M, et al. Cutting edge: RIPK1 Kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193(4):1539–1543. doi: 10.4049/jimmunol.1400590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newton K, Dugger DL, Wickliffe KE, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343(6177):1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- 6.Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. 2008;27(2):373–383. doi: 10.1038/sj.emboj.7601962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun X, Lee J, Navas T, Baldwin DT, Stewart TA, Dixit VM. RIP3, a novel apoptosis-inducing kinase. J Biol Chem. 1999;274(24):16871–16875. doi: 10.1074/jbc.274.24.16871. [DOI] [PubMed] [Google Scholar]
- 8.Rosenbusch KE, Kortholt A. Activation Mechanism of LRRK2 and Its Cellular Functions in Parkinson’s Disease. Parkinsons Dis. 2016;2016:7351985. doi: 10.1155/2016/7351985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bae JR, Lee BD. Function and dysfunction of leucine-rich repeat kinase 2 (LRRK2): Parkinson’s disease and beyond. BMB Rep. 2015;48(5):243–248. doi: 10.5483/BMBRep.2015.48.5.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81(4):513–523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 11.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8(3):297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 12.Lee TH, Shank J, Cusson N, Kelliher MA. The kinase activity of Rip1 is not required for tumor necrosis factor-alpha-induced IkappaB kinase or p38 MAP kinase activation or for the ubiquitination of Rip1 by Traf2. J Biol Chem. 2004;279(32):33185–33191. doi: 10.1074/jbc.M404206200. [DOI] [PubMed] [Google Scholar]
- 13.Kasof GM, Prosser JC, Liu D, Lorenzi MV, Gomes BC. The RIP-like kinase, RIP3, induces apoptosis and NF-kappaB nuclear translocation and localizes to mitochondria. FEBS Lett. 2000;473(3):285–291. doi: 10.1016/s0014-5793(00)01473-3. [DOI] [PubMed] [Google Scholar]
- 14.Dara L, Liu ZX, Kaplowitz N. Questions and controversies: the role of necroptosis in liver disease. Cell Death Discov. 2016;2:16089. doi: 10.1038/cddiscovery.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24(4):1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137(6):1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 17.Newton K. RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol. 2015 doi: 10.1016/j.tcb.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 18.Vercammen D, Beyaert R, Denecker G, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187(9):1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A. 2011;108(50):20054–20059. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thapa RJ, Nogusa S, Chen P, et al. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc Natl Acad Sci U S A. 2013;110(33):E3109–3118. doi: 10.1073/pnas.1301218110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saveljeva S, Mc Laughlin SL, Vandenabeele P, Samali A, Bertrand MJ. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015;6:e1587. doi: 10.1038/cddis.2014.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guicciardi ME, Malhi H, Mott JL, Gores GJ. Apoptosis and necrosis in the liver. Comprehensive Physiology. 2013;3(2):977–1010. doi: 10.1002/cphy.c120020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133(4):693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 25.Silke J, Rickard JA, Gerlic M. The diverse role of RIP kinases in necroptosis and inflammation. Nat Immunol. 2015;16(7):689–697. doi: 10.1038/ni.3206. [DOI] [PubMed] [Google Scholar]
- 26.Yuasa T, Ohno S, Kehrl JH, Kyriakis JM. Tumor necrosis factor signaling to stress-activated protein kinase (SAPK)/Jun NH2-terminal kinase (JNK) and p38. Germinal center kinase couples TRAF2 to mitogen-activated protein kinase/ERK kinase kinase 1 and SAPK while receptor interacting protein associates with a mitogen-activated protein kinase kinase kinase upstream of MKK6 and p38. J Biol Chem. 1998;273(35):22681–22692. doi: 10.1074/jbc.273.35.22681. [DOI] [PubMed] [Google Scholar]
- 27.Geng J, Ito Y, Shi L, et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat Commun. 2017;8(1):359. doi: 10.1038/s41467-017-00406-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menon MB, Gropengiesser J, Fischer J, et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat Cell Biol. 2017;19(10):1248–1259. doi: 10.1038/ncb3614. [DOI] [PubMed] [Google Scholar]
- 29.Jaco I, Annibaldi A, Lalaoui N, et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol Cell. 2017;66(5):698–710.e695. doi: 10.1016/j.molcel.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dondelinger Y, Delanghe T, Rojas-Rivera D, et al. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat Cell Biol. 2017;19(10):1237–1247. doi: 10.1038/ncb3608. [DOI] [PubMed] [Google Scholar]
- 31.Dondelinger Y, Jouan-Lanhouet S, Divert T, et al. NF-kappaB-Independent Role of IKKalpha/IKKbeta in Preventing RIPK1 Kinase-Dependent Apoptotic and Necroptotic Cell Death during TNF Signaling. Mol Cell. 2015;60(1):63–76. doi: 10.1016/j.molcel.2015.07.032. [DOI] [PubMed] [Google Scholar]
- 32.Oberst A. MK2 balances inflammation and cell death. Nat Cell Biol. 2017;19(10):1150–1152. doi: 10.1038/ncb3619. [DOI] [PubMed] [Google Scholar]
- 33.Feoktistova M, Geserick P, Kellert B, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43(3):449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tenev T, Bianchi K, Darding M, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43(3):432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Dondelinger Y, Aguileta MA, Goossens V, et al. RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 2013;20(10):1381–1392. doi: 10.1038/cdd.2013.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Degterev A, Zhou W, Maki JL, Yuan J. Assays for necroptosis and activity of RIP kinases. Methods in enzymology. 2014;545:1–33. doi: 10.1016/B978-0-12-801430-1.00001-9. [DOI] [PubMed] [Google Scholar]
- 37.Mandal P, Berger SB, Pillay S, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56(4):481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newton K, Manning G. Necroptosis and Inflammation. Annu Rev Biochem. 2016 doi: 10.1146/annurev-biochem-060815-014830. [DOI] [PubMed] [Google Scholar]
- 39.Holler N, Zaru R, Micheau O, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1(6):489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 40.Vercammen D, Brouckaert G, Denecker G, et al. Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J Exp Med. 1998;188(5):919–930. doi: 10.1084/jem.188.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 43.Sun L, Wang H, Wang Z, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1-2):213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 44.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe. 2012;11(3):290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nature reviews Immunology. 2011;12(2):79–88. doi: 10.1038/nri3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang X, Li Y, Liu S, et al. Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc Natl Acad Sci U S A. 2014;111(43):15438–15443. doi: 10.1073/pnas.1412767111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hildebrand JM, Tanzer MC, Lucet IS, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111(42):15072–15077. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dondelinger Y, Declercq W, Montessuit S, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell reports. 2014;7(4):971–981. doi: 10.1016/j.celrep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 49.Wang H, Sun L, Su L, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014;54(1):133–146. doi: 10.1016/j.molcel.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 50.Cai Z, Jitkaew S, Zhao J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16(1):55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao J, Jitkaew S, Cai Z, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109(14):5322–5327. doi: 10.1073/pnas.1200012109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy JM, Czabotar PE, Hildebrand JM, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39(3):443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 53.Grootjans S, Vanden Berghe T, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017;24(7):1184–1195. doi: 10.1038/cdd.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takahashi N, Vereecke L, Bertrand MJ, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513(7516):95–99. doi: 10.1038/nature13706. [DOI] [PubMed] [Google Scholar]
- 55.Dannappel M, Vlantis K, Kumari S, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513(7516):90–94. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moriwaki K, Chan FK. Necroptosis-independent signaling by the RIP kinases in inflammation. Cell Mol Life Sci. 2016;73(11-12):2325–2334. doi: 10.1007/s00018-016-2203-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suda J, Dara L, Yang L, et al. Knockdown of RIPK1 Markedly Exacerbates Murine Immune-Mediated Liver Injury through Massive Apoptosis of Hepatocytes, Independent of Necroptosis and Inhibition of NF-kappaB. J Immunol. 2016;197(8):3120–3129. doi: 10.4049/jimmunol.1600690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wong WW, Gentle IE, Nachbur U, Anderton H, Vaux DL, Silke J. RIPK1 is not essential for TNFR1-induced activation of NF-kappaB. Cell Death Differ. 2010;17(3):482–487. doi: 10.1038/cdd.2009.178. [DOI] [PubMed] [Google Scholar]
- 59.Van TM, Polykratis A, Straub BK, Kondylis V, Papadopoulou N, Pasparakis M. Kinase-independent functions of RIPK1 regulate hepatocyte survival and liver carcinogenesis. J Clin Invest. 2017;127(7):2662–2677. doi: 10.1172/JCI92508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rajput A, Kovalenko A, Bogdanov K, et al. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity. 2011;34(3):340–351. doi: 10.1016/j.immuni.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 61.Moriwaki K, Bertin J, Gough PJ, Chan FK. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J Immunol. 2015;194(4):1938–1944. doi: 10.4049/jimmunol.1402167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawlor KE, Khan N, Mildenhall A, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Newton K, Dugger DL, Maltzman A, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23(9):1565–1576. doi: 10.1038/cdd.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang YJ, Bang BR, Han KH, et al. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat Commun. 2015;6:8371. doi: 10.1038/ncomms9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wong WW, Vince JE, Lalaoui N, et al. cIAPs and XIAP regulate myelopoiesis through cytokine production in an RIPK1- and RIPK3-dependent manner. Blood. 2014;123(16):2562–2572. doi: 10.1182/blood-2013-06-510743. [DOI] [PubMed] [Google Scholar]
- 66.Vince JE, Wong WW, Gentle I, et al. Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity. 2012;36(2):215–227. doi: 10.1016/j.immuni.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 67.Yabal M, Muller N, Adler H, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell reports. 2014;7(6):1796–1808. doi: 10.1016/j.celrep.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 68.Kang S, Fernandes-Alnemri T, Rogers C, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun. 2015;6:7515. doi: 10.1038/ncomms8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gaidt MM, Ebert TS, Chauhan D, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. 2016;44(4):833–846. doi: 10.1016/j.immuni.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 70.Wree A, Eguchi A, McGeough MD, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898–910. doi: 10.1002/hep.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.He Y, Hara H, Nunez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci. 2016;41(12):1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol. 2015;12(7):387–400. doi: 10.1038/nrgastro.2015.94. [DOI] [PubMed] [Google Scholar]
- 73.Guicciardi ME, Gores GJ. Life and death by death receptors. FASEB J. 2009;23(6):1625–1637. doi: 10.1096/fj.08-111005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iorga A, Dara L, Kaplowitz N. Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. Int J Mol Sci. 2017;18(5) doi: 10.3390/ijms18051018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hirsova P, Gores GJ. Death Receptor-Mediated Cell Death and Proinflammatory Signaling in Nonalcoholic Steatohepatitis. Cell Mol Gastroenterol Hepatol. 2015;1(1):17–27. doi: 10.1016/j.jcmgh.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med. 2004;8(4):445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. 2014;147(4):765–783.e764. doi: 10.1053/j.gastro.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dara L, Johnson H, Suda J, et al. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology. 2015 Dec;62(6):1847–1857. doi: 10.1002/hep.27939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gunther C, He GW, Kremer AE, et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independently of RIPK3 during hepatitis. J Clin Invest. 2016;126(11):4346–4360. doi: 10.1172/JCI87545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dara L, Liu ZX, Kaplowitz N. A murder mystery in the liver: who done it and how? J Clin Invest. 2016;126(11):4068–4071. doi: 10.1172/JCI90830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang S, Ni HM, Dorko K, et al. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget. 2016;7(14):17681–17698. doi: 10.18632/oncotarget.6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roychowdhury S, McCullough RL, Sanz-Garcia C, et al. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology. 2016;64(5):1518–1533. doi: 10.1002/hep.28676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57(5):1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gautheron J, Vucur M, Reisinger F, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO molecular medicine. 2014;6(8):1062–1074. doi: 10.15252/emmm.201403856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Afonso MB, Rodrigues PM, Carvalho T, et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci (Lond) 2015;129(8):721–739. doi: 10.1042/CS20140732. [DOI] [PubMed] [Google Scholar]
- 86.An J, Mehrhof F, Harms C, et al. ARC is a novel therapeutic approach against acetaminophen-induced hepatocellular necrosis. J Hepatol. 2013;58(2):297–305. doi: 10.1016/j.jhep.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 87.Deutsch M, Graffeo CS, Rokosh R, et al. Divergent effects of RIP1 or RIP3 blockade in murine models of acute liver injury. Cell Death Dis. 2015;6:e1759. doi: 10.1038/cddis.2015.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. The receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology. 2013 doi: 10.1002/hep.26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Takemoto K, Hatano E, Iwaisako K, et al. Necrostatin-1 protects against reactive oxygen species (ROS)-induced hepatotoxicity in acetaminophen-induced acute liver failure. FEBS Open Bio. 2014;4:777–787. doi: 10.1016/j.fob.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang YF, He W, Zhang C, et al. Role of receptor interacting protein (RIP)1 on apoptosis-inducing factor-mediated necroptosis during acetaminophen-evoked acute liver failure in mice. Toxicol Lett. 2014;225(3):445–453. doi: 10.1016/j.toxlet.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 91.Takahashi N, Duprez L, Grootjans S, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schneider AT, Gautheron J, Tacke F, Vucur M, Luedde T. Receptor Interacting Protein Kinase-1 (RIPK1) in hepatocytes does not mediate murine acetaminophen toxicity. Hepatology. 2015 doi: 10.1002/hep.28225. [DOI] [PubMed] [Google Scholar]
- 93.Heymann F, Hamesch K, Weiskirchen R, Tacke F. The concanavalin A model of acute hepatitis in mice. Lab Anim. 2015;49(1 Suppl):12–20. doi: 10.1177/0023677215572841. [DOI] [PubMed] [Google Scholar]
- 94.Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A. Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am J Pathol. 1995;146(5):1220–1234. [PMC free article] [PubMed] [Google Scholar]
- 95.Mizuhara H, O’Neill E, Seki N, et al. T cell activation-associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin 6. J Exp Med. 1994;179(5):1529–1537. doi: 10.1084/jem.179.5.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trautwein C, Rakemann T, Brenner DA, et al. Concanavalin A-induced liver cell damage: activation of intracellular pathways triggered by tumor necrosis factor in mice. Gastroenterology. 1998;114(5):1035–1045. doi: 10.1016/s0016-5085(98)70324-5. [DOI] [PubMed] [Google Scholar]
- 97.Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012;19(12):2003–2014. doi: 10.1038/cdd.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Arshad MI, Piquet-Pellorce C, Filliol A, et al. The chemical inhibitors of cellular death, PJ34 and Necrostatin-1, down-regulate IL-33 expression in liver. J Mol Med (Berl) 2015;93(8):867–878. doi: 10.1007/s00109-015-1270-6. [DOI] [PubMed] [Google Scholar]
- 99.Zhou Y, Dai W, Lin C, et al. Protective effects of necrostatin-1 against concanavalin A-induced acute hepatic injury in mice. Mediators Inflamm. 2013;2013:706156. doi: 10.1155/2013/706156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Filliol A, Piquet-Pellorce C, Le Seyec J, et al. RIPK1 protects from TNF-alpha-mediated liver damage during hepatitis. Cell Death Dis. 2016;7(11):e2462. doi: 10.1038/cddis.2016.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Weinlich R, Oberst A, Dillon CP, et al. Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell reports. 2013;5(2):340–348. doi: 10.1016/j.celrep.2013.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Filliol A, Piquet-Pellorce C, Raguenes-Nicol C, et al. RIPK1 protects hepatocytes from Kupffer cells-mediated TNF-induced apoptosis in mouse models of PAMP-induced hepatitis. J Hepatol. 2017;66(6):1205–1213. doi: 10.1016/j.jhep.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 103.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332(6037):1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol. 2015;12(4):231–242. doi: 10.1038/nrgastro.2015.35. [DOI] [PubMed] [Google Scholar]
- 105.Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125(2):437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 106.Hirsova P, Gores GJ. Reply. Cellular and Molecular Gastroenterology and Hepatology. 1(3):265–266. doi: 10.1016/j.jcmgh.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hatting M, Zhao G, Schumacher F, et al. Hepatocyte caspase-8 is an essential modulator of steatohepatitis in rodents. Hepatology. 2013;57(6):2189–2201. doi: 10.1002/hep.26271. [DOI] [PubMed] [Google Scholar]
- 108.Witek RP, Stone WC, Karaca FG, et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology. 2009;50(5):1421–1430. doi: 10.1002/hep.23167. [DOI] [PubMed] [Google Scholar]
- 109.Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical Models of Nonalcoholic Fatty Liver Disease. J Hepatol. 2017 doi: 10.1016/j.jhep.2017.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gautheron J, Vucur M, Luedde T. Necroptosis in Nonalcoholic Steatohepatitis. Cellular and Molecular Gastroenterology and Hepatology. 1(3):264–265. doi: 10.1016/j.jcmgh.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hao F, Cubero FJ, Ramadori P, et al. Inhibition of Caspase-8 does not protect from alcohol-induced liver apoptosis but alleviates alcoholic hepatic steatosis in mice. Cell Death Dis. 2017;8(10):e3152. doi: 10.1038/cddis.2017.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schneider AT, Gautheron J, Feoktistova M, et al. RIPK1 Suppresses a TRAF2-Dependent Pathway to Liver Cancer. Cancer Cell. 2017;31(1):94–109. doi: 10.1016/j.ccell.2016.11.009. [DOI] [PubMed] [Google Scholar]
- 113.Kondylis V, Polykratis A, Ehlken H, et al. NEMO Prevents Steatohepatitis and Hepatocellular Carcinoma by Inhibiting RIPK1 Kinase Activity-Mediated Hepatocyte Apoptosis. Cancer Cell. 2015;28(5):582–598. doi: 10.1016/j.ccell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sun J, Yu X, Wang C, et al. RIP-1/c-FLIPL Induce Hepatic Cancer Cell Apoptosis Through Regulating Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Med Sci Monit. 2017;23:1190–1199. doi: 10.12659/MSM.899727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Koppe C, Verheugd P, Gautheron J, et al. IkappaB kinasealpha/beta control biliary homeostasis and hepatocarcinogenesis in mice by phosphorylating the cell-death mediator receptor-interacting protein kinase 1. Hepatology. 2016;64(4):1217–1231. doi: 10.1002/hep.28723. [DOI] [PubMed] [Google Scholar]
- 116.Vucur M, Reisinger F, Gautheron J, et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell reports. 2013;4(4):776–790. doi: 10.1016/j.celrep.2013.07.035. [DOI] [PubMed] [Google Scholar]
- 117.Luedde T, Beraza N, Kotsikoris V, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell. 2007;11(2):119–132. doi: 10.1016/j.ccr.2006.12.016. [DOI] [PubMed] [Google Scholar]
- 118.Wang C, Yao B, Xu M, Zheng X. RIP1 upregulation promoted tumor progression by activating AKT/Bcl-2/BAX signaling and predicted poor postsurgical prognosis in HCC. Tumour Biol. 2016;37(11):15305–15313. doi: 10.1007/s13277-016-5342-1. [DOI] [PubMed] [Google Scholar]
- 119.Afonso MB, Rodrigues PM, Simao AL, et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis. 2016;7(9):e2390. doi: 10.1038/cddis.2016.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med. 2013;19(7):859–868. doi: 10.1038/nm.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mundt B, Kuhnel F, Zender L, et al. Involvement of TRAIL and its receptors in viral hepatitis. FASEB J. 2003;17(1):94–96. doi: 10.1096/fj.02-0537fje. [DOI] [PubMed] [Google Scholar]
- 122.Song le H, Binh VQ, Duy DN, et al. Variations in the serum concentrations of soluble Fas and soluble Fas ligand in Vietnamese patients infected with hepatitis B virus. J Med Virol. 2004;73(2):244–249. doi: 10.1002/jmv.20082. [DOI] [PubMed] [Google Scholar]
- 123.Streetz K, Leifeld L, Grundmann D, et al. Tumor necrosis factor alpha in the pathogenesis of human and murine fulminant hepatic failure. Gastroenterology. 2000;119(2):446–460. doi: 10.1053/gast.2000.9364. [DOI] [PubMed] [Google Scholar]
- 124.Fang JW, Shen WW, Meager A, Lau JY. Activation of the tumor necrosis factor-alpha system in the liver in chronic hepatitis B virus infection. The American journal of gastroenterology. 1996;91(4):748–753. [PubMed] [Google Scholar]
- 125.Yoon JH, Gores GJ. Death receptor-mediated apoptosis and the liver. J Hepatol. 2002;37(3):400–410. doi: 10.1016/s0168-8278(02)00209-x. [DOI] [PubMed] [Google Scholar]
- 126.Lim EJ, El Khobar K, Chin R, et al. Hepatitis C virus-induced hepatocyte cell death and protection by inhibition of apoptosis. J Gen Virol. 2014;95(Pt 10):2204–2215. doi: 10.1099/vir.0.065862-0. [DOI] [PubMed] [Google Scholar]