

Abstract
A nonsense mutation in the human cereblon gene (CRBN) causes a mild type of autosomal recessive non-syndromic intellectual disability (ID). Animal studies show that crbn is a cytosolic protein with abundant expression in the hippocampus (HPC) and neocortex (CTX). Its diverse functions include the developmental regulation of ion channels at the neuronal synapse, the mediation of developmental programs by ubiquitination, and a target for herpes simplex type I virus in HPC neurons. To test the hypothesis that anomalous CRBN expression leads to HPC-mediated memory and learning deficits, we generated germ-line crbn knock-out mice (crbn−/−). We also inactivated crbn in forebrain neurons in conditional knock-out mice in which crbn exons 3 and 4 are deleted by cre recombinase under the direction of the Ca2+/calmodulin-dependent protein kinase II alpha promoter (CamKIIcre/+, crbn−/−). crbn mRNA levels were negligible in the HPC, CTX, and cerebellum (CRBM) of the crbn−/− mice. In contrast, crbn mRNA levels were reduced 3- to 4-fold in the HPC, CTX but not in the CRBM in CamKIIcre/+, crbn−/− mice as compared to wild type (CamKIIcre/+, crbn+/+). Contextual fear conditioning showed a significant decrease in the percentage of freezing time in CamKIIcre/+, crbn−/− and crbn−/− mice while motor function, exploratory motivation, and anxiety-related behaviors were normal. These findings suggest that CamKIIcre/+, crbn−/− mice exhibit selective HPC-dependent deficits in associative learning and supports the use of these mice as in vivo models to study the functional consequences of CRBN aberrations on memory and learning in humans.
Keywords: Non-syndromic mental retardation, Cereblon, Conditional knock-out mice, Fear conditioning, Memory, Association learning
1. Introduction
The identification of the genetic causes of rare human disorders that affect critical steps in cognition has greatly facilitated our understanding of the molecular mechanisms involved in developmental disabilities. The effects of mutated human genes in mild, non-syndromic neurogenetic disorders provide the foundation to create optimal animal models to study the fundamental aspects of human memory and learning because the phenotype does not involve other processes or systems besides the brain. To date, however, there are few such models. For this reason, we developed a mouse model that exhibits selective cognitive deficits that mimic the human condition. The human gene, cereblon (CRBN), on chromosome 3p26.2, is associated with a non-syndromic type of mental retardation/intellectual disability (ID). A homozygous nonsense CRBN mutation (p.R419X) causes a recessively inherited phenotype with intelligent quotients (IQs) between 50 and 70 without physical anomalies, or other neuropsychiatric features [1–3]. Besides CRBN’s direct role in the genetics of non-syndromic ID in humans, it is the primary target for the teratogenic effects of thalidomide [4–7]. Thalidomide mainly causes limb anomalies but it is also responsible for isolated ID when fetal exposure occurs at 20–24 days post conception [8]. The cereblon protein (CRBN) forms an E3 ubiquitin ligase complex with damaged DNA-binding protein-1 (DDB1) and cullin 4A (CUL4A). Thalidomide’s binding to the CRBN–DDB1–CUL4A complex inhibits its associated ubiquitin ligase activity and disrupts downstream pathways involved in physical and mental development [4]. The CRBN protein also binds to the UL14 protein of the herpes simplex virus type 1 (HSV1) [9], a virus with a neurotropism for the hippocampus (HPC) [10–12]. CRBN’s abundance in HPC neurons with prominent expression in the hilus and granule cell layer of the dentate gyrus [13], correlates with the topography of HSV1’s selective regional damage to the HPC [10]. Thus, CRBN may be a target for HSV1 mediated HPC injury and participate in the pathogenesis of the memory deficits in patients who survive HSV1 encephalitis [10–12]. The observation that activation of the NF-E2-related factor 2 (Nrf2)/antioxidant response element site in the promoter region of crbn is responsible for the in vitro increase in crbn expression during hypoxic stress, suggests that crbn has a role in the selective vulnerability of HPC neurons to hypoxic ischemic injury [14]. These converging lines of experimental evidence indicate that CRBN is a key intermediary in human nervous system development and that anomalous CRBN gene expression leads to HPC-mediated memory and learning deficits by molecular mechanisms that are currently unknown.
CRBN cDNA encodes a 442-amino acid protein with a Lon protease domain, a ‘regulators of G protein-signaling’ (RGS)-like domain, a leucine zipper motif, four putative protein kinase C phosphorylation sites [3,15], and C-terminal binding sites for thalidomide [4] and the HSV1 UL14 protein [9]. In rodents, crbn protein is strongly expressed in the HPC and neocortex (CTX) [13,15] but is distributed in many other tissues [16]. There are at least four transcripts corresponding to 4.4 kb, 4.3 kb, 2.7 kb and 2.6 kb generated by differential splicing in the 5′-coding region and the utilization of two different polyadenylation sites [15]. Although two transcripts (4.4- and 2.7-kb) are found in the brain, the longer transcript is the predominant variant [15]. The crbn protein is localized to the cytosol and plasma membrane and developmentally regulates the neuronal surface expression of ionic channels including the large-conductance calcium-activated potassium (BKCa) and the voltage-gated chloride (ClC-2) channels [15,17,18]. In human lymphoblastoid cell lines, mutant CRBN expressing the nonsense p.R419X mutation causes the continued expression of an immature BKCa channel splice variant [19] and alters Ca2+-mediated signal transduction; a mechanism that is critical for the core processes of learning such as synaptic maturation and connectivity [20].
In order to understand the in vivo role of crbn in learning and memory, we generated both germ-line crbn knock-out mice and conditional crbn knock-out mice under the direction of the Ca2+/calmodulin-dependent protein kinase II alpha (CamKII) promoter [21,22] to inactivate crbn in forebrain neurons. Behavioral testing of these mice in a number of tasks shows selective forebrain impairments. These findings provide validation for the use of this mouse model to study the molecular mechanisms underlying learning and memory associated with human ID.
2. Material and methods
2.1. Construction and validation of the conditional targeting vector for cereblon
The targeting construct was generated using a high fidelity Red/ET recombineering method [23–25] and the mouse C57BL/6 bacterial artificial chromosome (BAC) clone (RPCI-23-24F3) containing the crbn gene. A construct derived from the BAC clone was subcloned into the vector pSP72 (Promega, Madison, WI) containing an ampicillin selection cassette for retransformation of the construct prior to electroporation. The construct was designed such that the short homology arm (SA) extended 3.66 kilobases (kb) 5′ to exon 3. The long homology arm (LA) ended 3′ to exon 4, and is 5.34 kb long (Fig. 1A). The FRT-PGK-EM7-NeobpA-FRT-loxP (Neo) cassette [26] was inserted 227 base pairs (bp) upstream of exon 3 in the antisense direction as shown in Fig. 1A. The polymerase II promoter driven diphtheria toxin gene (DTV840) (Lexicon Genetics, Woodlands, TX) was inserted 5′ to the targeting construct to select against non-homologous recombinants. The single loxP site, containing an engineered NcoI site, was inserted 225 bp downstream of exon 4. The target region was 1.46 kb and included exons 3 and 4 (Fig. 1A). The targeting vector was confirmed by restriction analysis and sequencing after each modification step. P6 and T7 primers annealed to the vector sequence and read into the 5′ and 3′ ends of the BAC sub clone. The n1 and n2 primer pairs anneal to the 5′ and 3′ ends of the Neo cassette and sequence the SA and LA, respectively. The following PCR primers were used for sequencing: P6 5′ -GAGTGCACCATATGGACATATTGTC-3′; T7 5′-CGATAAGCCAGGTTAACCTGCATTA-3′; n1 5′-TGCGAGGCCAGAGGCCACTTGTGTAGC-3′; n2 5′-TTCCTCGTGCTT-TACGGTATCG-3′; loxP 5′-CACTGCAAACAAGCAAGCATCTTC-3′. A forward primer 5′ to the SA (f 5′-TACACCTGTCATCTGTCCAG-3′) and the n1 primer were used to genotype for the integration of the SA. A reverse primer (r 5′-TCACAGCTTACAGTATGAGC-3′) and the n2 primer were use to link the single loxP site in the LA to the Neo cassette. Two NcoI restriction sites outside the construct permitted the identification of the 16.9 kb wild type (WT) allele by Southern blot analysis using the external pb9 (PCR primers: sense 5′-CTTTCACTGCAACTTAGACGGAGC-3′; antisense 5′-GACCTGTGCAGAGCAGACAAGATT-3′) or internal pb3 (PCR primers: sense 5′-CTCCTGTCAATGCTTGCTTCCTAC-3′; antisense 5′-TGTTGAGGCCTTGTGACAATGAG-3′) probes (Fig. 1A). The additional of NcoI sites at the loxP site and within the Neo cassette allowed the identification of a 10.4 kb fragment using the external probe pb9 or a 5.5 kb fragment using the internal probe pb3 (Fig. 1A). Fifty micrograms of the targeting vector was linearized using NotI prior to electroporation (Bio-Rad Gene Pulser set at 800 V and 3 μF) into embryonic stem cells (ESC) derived from albino C57/BL6 mice. G418 selection was applied 24 h after electroporation, and G418-resistant colonies were isolated on day seven of selection.
Fig. 1.
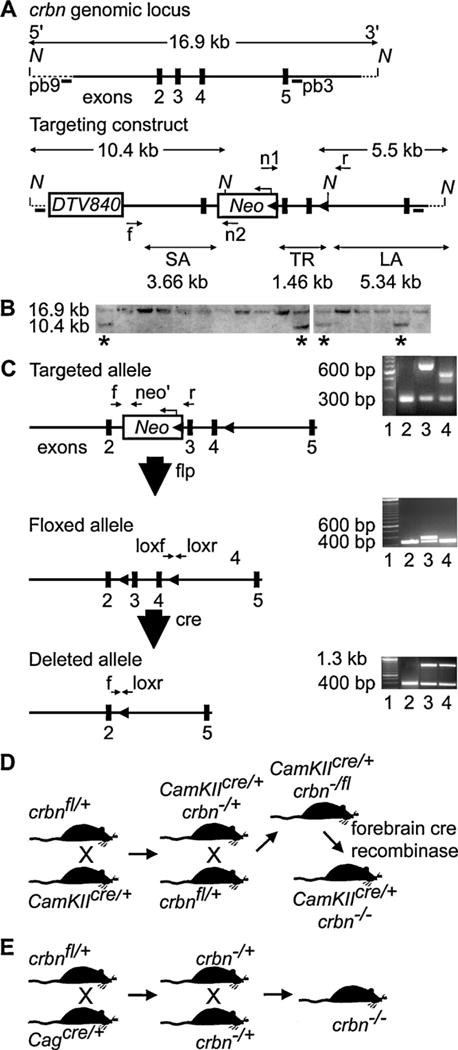
Generation of a crbn conditional null mutation in C57BL/6 mice. (A) Gene targeting strategy. The targeting construct was generated using a high fidelity Red/ET recombineering method to insert the polymerase II promoter driven diphtheria toxin gene (DTV840) and the FRT-PGK-EM7-NeobpA-FRT-loxP (Neo) cassettes, and a single loxP site, containing an engineered NcoI site, 225 bp downstream of exon 4. The short homology arm (SA) extended 3.66 kilobases (kb) 5′ to exon 3 and the 5.34 kb long homology arm (LA) ended 3′ to exon 4. The Neo cassette was inserted 227 base pairs bp upstream of exon 3 in the antisense direction. The DTV840 cassette was inserted 5′ to the targeting construct to select against non-homologous recombinants. The 1.46 target region, flanked by two loxP sites, included exons 3 and 4. The n1 and n2 primer pairs anneal to the 5′ and 3′ ends of the Neo cassette and sequence the SA and LA, respectively. The forward primer (f) 5′ to the SA and the n1 primer were used to genotype for the integration of the SA. A reverse primer (r) and the n2 primer were use to link the single loxP site in the LA to the Neo cassette. Two NcoI sites at the loxP site and within the Neo cassette allowed the identification of a 10.4 kb fragment using the external probe pb9 or a 5.5 kb fragment using the internal probe pb3. (B) Southern blot confirmation of targeted embryonic stem cell clones derived from albino C57/BL6 mice. After electroporation of the construct, and positive-negative selection, ESC digested with NcoI digest was screened by Southern blot analysis. Four of 48 ESC (starred) showed the presence of the 10.4 kb recombined locus using the external probe (pb9). (C) Strategy for breeding and genotyping crbn conditional transgenic mice. Mouse lines derived from two ESC clones crossed with flp-deleter mice to excise the Neo cassette. Three sets of primers (f, neo’, and r) were used for genotyping for the targeted allele. The PCR assay on the right shows the results of flp-mediated excision of the Neo cassette by flp-deleter mice. Lane 1 is a 100-bp ladder. The wild-type PCR product of 320 bp is amplified by primers f and r (lane 2). Mice heterozygous for the Neo insertion using primers f and neo’ have an allele larger (lane 3) than mice without Neo (lane 4). Mice without Neo mice were genotyped using primers loxf and loxr to analyze the floxed allele. The PCR assay on the right shows a 100-bp ladder (lane 1). Lanes 2 and 4 show the 320 bp PCR product that results from the lack of the downstream loxP site. Lane 3 shows the insertion (400 bp) of the downstream loxP site. Cre-mediated excision of crbn exons 3 and 4 was confirmed by genotyping genomic DNA of the forebrains or tail DNA of weaned mice from the breeding schemes shown in (D) and (E). (D) Floxed mice were bred with cre transgenic mice that contained a cre transgene under the transcriptional control of the CaMKII promoter, termed CaMKIIcre/+, to yield a conditional knock-out of crbn in the forebrain of mice (CaMKIIcre/+, crbn−/−). (E) Floxed mice were bred with transgenic mice where cre recombinase is active in the ovum (Cagcre/+) to generate mice where crbn is deleted in all tissues (crbn−/−). These mice were used as controls in the quantitative RT-PCR experiments to confirm the regional specificity of the forebrain-deletion in CaMKIIcre/+, crbn−/− mice.
Forty-eight resistant colonies (Fig. 1B) were screened for homologous recombination by Southern blot analysis using internal and external probes. Eight percent of the ESC clones were homologous recombinants (n = 4). These recombinants (Fig. 1B) were chosen for injection into blastocysts obtained from C57/BL6 mice and the injected blastocysts were transferred into pseudopregnant mothers. From 42 chimeric mice obtained from three of the four ESC clones injected, five germline chimeras were identified. Of the 52 albino F1 offspring genotyped, 26 showed transmission of the targeted crbn allele.
2.2. Generation of mice with a germ-line and conditional deletion of cereblon
Mouse lines derived from two ESC clones (Fig. 1B) were expanded and crossed to excise the Neo cassette. Fig. 1C shows the PCR strategy used to genotype the mice. The PCR assay shows the results of flp-mediated excision of Neo by deleter mice using three sets of primers (F 5′-TTGTTTCAGAACTGCTGGGATGTG-3′, Neo 5′-GTACTCGGATGGAAGCCGGTCTT-3′, R 5′-AGGTACTACTCAAGAGCACAGAGT-3′). Primers that amplify the WT allele (320 bp) were in intron 2 (F) and bridge the junction of intron 2 and exon 3 (R). Mice heterozygous for the Neo insertion (HET) using primers F and Neo have an allele larger than mice without Neo (ΔNeo). ΔNeo mice were genotyped using a PCR assay that detects the insertion (400 bp) or lack (320 bp) of the downstream loxP site (loxF 5′-AGGAGCACTGAACGGCTTACAG-3′; loxR 5′-CGCATGCTGACTGATCACAGC-3′). Cre-mediated excision of crbn exons 3 and 4 was confirmed by genotyping genomic DNA of the forebrains of weaned mice from the breeding scheme shown in Fig. 1D and tail DNA shown in Fig. 1E. Fig. 1D shows the use of cre transgenic mice that contained a cre transgene under the transcriptional control of the CaMKII α promoter, termed CamKIIcre/+ [21,22], to yield a conditional knock-out of crbn in the forebrain of mice beginning at postnatal day 7. Fig. 1E shows the use of Cagcre/+ deleter mice to generate mice where cre recombinase is active in the ovum [27] and deletes crbn in all tissues (i.e. germ-line).
2.3. Quantitative RT-PCR
Mouse brain tissue was sectioned in a cryostat in the coronal plane according to the stereotactic coordinates relative to the bregma established by Franklin and Paxinos [28]. Quantitative RT-PCR was performed by methods previously published [13]. The differences in the mean mRNA levels for each brain region were compared by a one-way analysis of variance (ANOVA) with post hoc comparisons using the Fisher’s protected least significant difference (PLSD) test.
2.4. Behavioral tests
Behavioral experiments were conducted in adult male mice (P60-P70). Mice were housed under a 12 h light/12 h dark cycle with ad libitum access to food and water. All of the investigations involving the mice were in compliance with the Weill Cornell Medical College Institutional Animal Care and Use Committee. Each animal had baseline assessments including home cage observations and general health assessments [29,30]. Behavioral assays compared the conditional knock out line, CamKIIcre/+, crbn−/−, to their wild type counterpart, CamKIIcre/+, crbn+/+ mice, at P60. The germ-line knockout line, crbn−/− was compared to their wild type counterpart, crbn+/+ at P60 to validate the results of the contextual fear conditioning test. Heterozygous and wild type mice were used as controls for the contextual fear conditioning behavioral tests. The data from each mouse line was analyzed separately, using control comparisons relevant to the behavioral parameter(s) of the specific task. The tests were performed blind to the mouse genotypes.
2.4.1. Basal locomotor activity
Horizontal locomotor activity was assessed by computer-based activity monitoring software (Med Associates, St. Albans, VT) in a polycarbonate/polypropylene test chamber (27.3 cm × 27.3 cm) equipped with three infrared beam arrays. Locomotor activity was measured as sequential adjacent beam breaks and reported as a measure of distance (cumulative total of cm. covered). For each test session, animals were habituated for 1 h prior to testing and subsequently placed in the chamber where locomotor activity was recorded for 2 h without interruption.
2.4.2. Open field test
Mice were placed in a Plexiglas open field arena (38 cm × 54 cm) and their activity was monitored for 10 min with a video tracking system using EthoVision software (Noldus Information Technology, Leesburg, VA). The duration of time spent in the center of the open field (13 cm × 28 cm) and the frequency to enter the center of the open field were analyzed.
2.4.3. Contextual fear conditioning
Fear conditioning tests was performed using a Coulbourn Habitest Modular Test System (Coulbourn Instruments, Whitehall, PA, USA) with a stainless-steel grid floor for administration of the foot shock as published previously [31–33]. Mice were habituated to the behavior room for an hour before trials. Before and after each trial the apparatus was cleansed with 70% ethanol and 0.1% peppermint as an odorant. On the first day, the mice were habituated in the apparatus for 90 s and lights were flashed once at the beginning and at the end of a 30-s 85 dB tone. The second flash directly coincided with a 1-s 0.7-mA shock. The mice were allowed to rest for 40 s between four consecutive shocks and for 1 min after the fifth shock. Twenty-four hours post-conditioning, the mice were placed back into the Habitest apparatus, and habituated for 1 min. Lights were flashed once at the beginning of the testing period and freezing time was monitoring for 4.5 min.
2.4.4. Elevated plus maze
The elevated plus maze was constructed based on that previously described [34,35] and was composed of a cross-shaped maze with two open and two closed arms (50 cm) elevated to a height of 38 cm above the floor. Mice were placed in the intersection of the four arms of the elevated plus maze and the time spent and entries made on the open and closed arms were recorded for 5 min. Digitized video recordings (30 frames/s) using EthoVision software (Noldus Information Technology, Leesburg, VA) were used to score the number of entries (all four paws into the arm) into open and closed arms, and the time spent in the open and closed arms.
2.5. Data analysis
Data was analyzed using JMP version 5 software (SAS Institute, Cary, NC). The differences in the mean mRNA levels for each and brain region were compared by a one-way analysis of variance with post hoc comparisons using the Fisher’s protected least significant difference test. Behavioral data was analyzed using the two-way ANOVA. All values in the text and figures are expressed as means ± SEM. For all comparisons, the level of statistical significance was set at P = 0.05.
3. Results
3.1. Generation of mice with a conditional deletion of cereblon
We generated transgenic mice carrying two loxP sites flanking crbn exons 3 and 4 (Fig. 1A–C, crbnfl/+) to investigate whether a homozygous deletion of crbn in the forebrain influences memory and learning in postnatal mice. Two transgenic lines were established by breeding the crbnfl/+ mice with two types of deleter mice (Cagcre/+ and CamKIIcre/+) expressing different temporal and spatial-specific cre recombinase tissue expression (Fig. 1D and E). Mice produced by crossing crbnfl/+ with CamKIIcre/+ mice activate the deletion of crbn in the neurons of forebrain regions such as the HPC and CTX by postnatal day 21 (P21) (CamKIIcre/+, crbn−/− mice) [22,36] (Fig. 1D). Breeding crbnfl/+ mice with Cagcre/+ mice deletes crbn at fertilization [27] to generate crbn−/− mice with a germ-line crbn deletion in all tissues (Fig. 1E). Quantitative RT-PCR analysis showed that crbn mRNA levels were negligible at P60 in crbn−/− mice (n = 4) in the HPC (0.0008 ± 0.003, mean ± SEM; P = 0.000004), CTX (0.03 ± 0.02; P = 0.0005), and cerebellum (CRBM; 0.02 ± 0.0009; P = 0.0006) as compared to wild type (crbn+/+) mice (n = 6, HPC = 1.0 ± 0.05; CTX = 1.0 ± 0.15; CRBM = 1.0 ± 0.001) (Fig. 2A). The crbn mRNA levels were significantly reduced in the forebrain regions, HPC (0.33 ± 0.06, P = 0.002) and CTX (0.27 ± 0.06; P = 0.0006), at P60 in the CamKIIcre/+, crbn−/− mice (n = 4) but not in the CRBM as compared to their wild type counterparts (CamKIIcre/+, crbn+/+; n = 6, HPC = 1.0 ± 0.12; CTX = 1.0 ± 0.10; CRBM = 1.0 ± 0.01) (Fig. 2B). Post hoc comparisons using the Fisher’s protected least significant difference (PLSD) test showed that crbn mRNA levels were decreased 3-fold in the HPC (P = 0.002), 4-fold in the CTX (P = 0.0006), and were the same in the CRBM of CamKIIcre/+, crbn−/− as compared to wild type CamKIIcre/+, crbn+/+ mice.
Fig. 2.
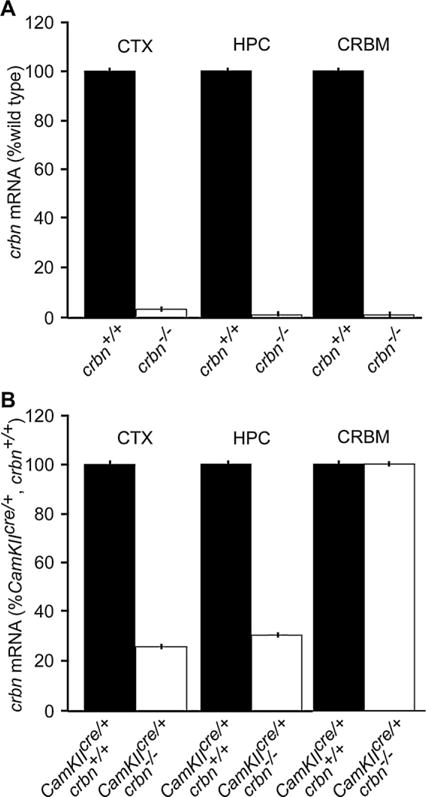
Quantitative RT-PCR analysis of crbn mRNA levels in the hippocampus (HPC), neocortex (CTX), and cerebellum (CRBM) of crbn−/− and CaMKIIcre/+, crbn−/− mice at P60. (A) A complete crbn knock-out (crbn−/−) does not show regional specificity of crbn mRNA expression in the brain. The crbn mRNA levels were negligible in the HPC, CTX, and CRBM (P < 0.001) of crbn−/− mice (n = 4) as compared to wild type (crbn+/+) mice (n = 6). (B) The specificity of the crbn conditional knock-out in the forebrain of CaMKIIcre/+, crbn−/− mice. The crbn mRNA levels were significantly reduced in the HPC (P < 0.01) and CTX (P < 0.001) but not in the CRBM in CamKIIcre/+, crbn−/− mice (n = 4) as compared to their wild type counterparts, CamKIIcre/+, crbn+/+ (n = 6). The vertical error bars represent a SEM of less than 1.0%.
3.2. CamKIIcre/+, crbn−/− mice exhibit abnormal contextual fear conditioning
Home cage observations and general health were normal in all mouse lines at P60 to P80. Several types of neurobehavioral tests were performed to investigate the involvement of crbn in forebrain-dependent psychomotor behaviors attributed to memory and learning in mice. The results for all behavioral tests in heterozygous CamKIIcre/+, crbn+/− (n = 9) mice, were the same as the wild type CamKIIcre/+, crbn+/+ (n = 18). The freezing time on the contextual fear conditioning task were the same when heterozygous CamKIIcre/+, crbn+/− (n = 18) were compared to heterozygous crbn+/− (n = 7) mice.
3.2.1. Basal locomotor activity
The cumulative distance traveled in centimeters (cm) over 2 h between CamKIIcre/+, crbn−/− (n = 10; 12,910 ± 1156) and CamKIIcre/+, crbn+/+ (n = 18; 14,360 ± 873) mice were not significantly different [F(1,26) = 1.00, P = 0.3258] (Fig. 3A).
Fig. 3.
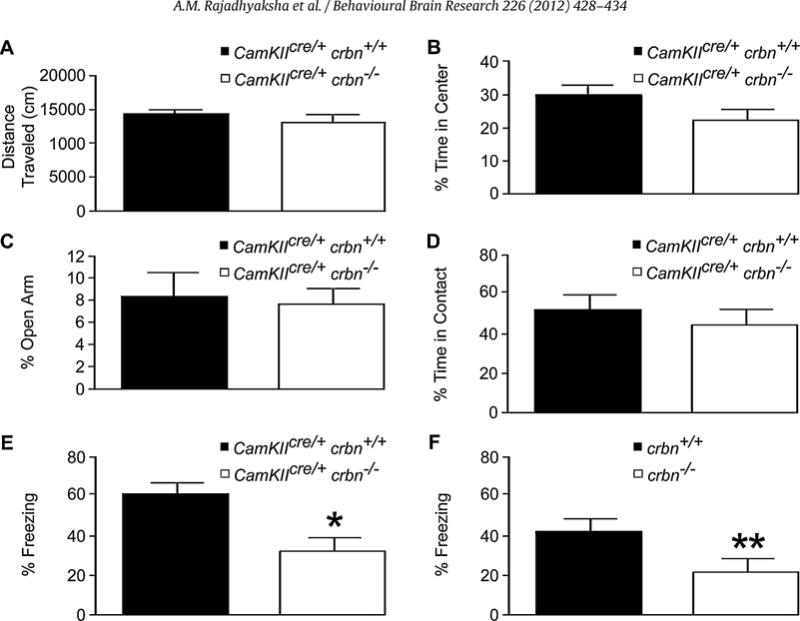
Performance of wild type and crbn knock-out mice on behavioral tests at P60. There were no significant differences between wild type, CamKIIcre/+, crbn+/+ and CamKIIcre/+, crbn−/− mice on the (A) 2 h basal locomotor activity, (B) motor activity on the open field box, (C) elevated plus maze and (D) social approach tasks. (E) Contextual fear conditioning task was assessed in wild type, CamKIIcre/+, crbn+/+ versus the forebrain-specific conditional knock-out, CamKIIcre/+, crbn−/− mice, and (F) wild type, crbn+/+ and germ-line knock-out, crbn−/− mice. The mean percentage freezing per minute was significantly different between the CamKIIcre/+, crbn+/+ (n = 10; 62.3 ± 5.1) versus the CamKIIcre/+, crbn−/− [n = 10; mean ± SEM, 33.3 ± 5.8] mice, and between the crbn+/+ (n = 9; 41.5 ± 6.1) versus the crbn−/− [n = 12; 22.4 ± 5.3] mice. The error bars represent the SEM. *P = 0.0015, **P = 0.0296.
3.2.2. Anxiety-like behaviors
Both the CamKIIcre/+, crbn−/− and CamKIIcre/+, crbn+/+ mice showed similar levels of activity and thigmotaxis in the open field test. There was no difference [F(1,26) = 2.36, P = 0.1368] in the percentage of time spent in the center of the open field between CamKIIcre/+, crbn−/− (n = 10; 22.4 ± 3.3) and CamKIIcre/+, crbn+/+ (n = 18; 29.9 ± 3.1) mice (Fig. 3B). In the elevated plus maze the percentage of time spent exploring the open arms of the maze was no different [F(1,14) = 0.0917, P = 0.7665] in the CamKIIcre/+, crbn−/− (n = 10; 7.7 ± 1.4) compared to the CamKIIcre/+, crbn+/+ (n = 6; 8.4 ± 2.2) mice (Fig. 3D).
3.2.3. Contextual fear conditioning
To investigate the effect of the loss of crbn on associative memory in the forebrain, we conducted a contextual fear conditioning task, in which robust HPC-dependent associative memory can be acquired in a single trial [37]. In this task, mice were trained to associate the conditioned stimulus, the chamber, with an unconditioned stimulus, an electric foot shock of 0.7 mA. When mice were exposed to the same context 24 h later and freezing behavior without the foot shock was measured, both CamKIIcre/+, crbn−/− and crbn−/− mice froze significantly less than CamKIIcre/+, crbn+/+mice and wild type crbn+/+ mice. The mean percentage freezing per min showed significant differences [F(1,18) = 13.95, P = 0.0015] between the CamKIIcre/+, crbn−/− (n = 10; 33.3 ± 5.8) versus the CamKIIcre/+, crbn+/+ mice (n = 10; 62.3 ± 5.1). The mean percentage freezing per min also showed significant differences [F(1,19) = 5.53, P = 0.0296] between, and the crbn−/− (n = 12; 22.4 ± 5.3) versus the crbn+/+ mice (n = 9; 41.5 ± 6.1). These results show that CamKIIcre/+, crbn−/− and crbn−/− mice have deficits in HPC-dependent associative learning (Fig. 3C).
4. Discussion
The results of our study demonstrate that a specific deletion of crbn in forebrain neurons of postnatal mice (CamKIIcre/+, crbn−/−) selectively affects HPC-dependent associative or explicit memory without impairment of motor function, exploratory motivation, social interaction, or anxiety-related behaviors. Explicit memory is the intentional recollection of newly learned information. Implicit memory is the unconscious or unintentional remembering of material that was previously learned [38]. Implicit memory is studied in a variety of simple reflex systems, including those of invertebrates such as Aplysia californica, whereas explicit memory is best studied in mammals such as mice [39–41]. Long term storage of implicit and explicit memory in both mice and Aplysia use an essential core signaling pathway that includes c-AMP-dependent protein kinase (PKA), mitogen-activated protein kinase (MAPK), and the cAMP response element binding protein 1 (CREB-1) [38]. Importantly, CRBN, binds to an energy sensor, the alpha1 subunit of AMP-activated protein kinase [42], which in turn modulates CREB signaling [43]. Therefore, CREB signaling may be a potential molecular mechanism by which crbn affects HPC-mediated memory in mice. The results of our study suggest that crbn is an important intermediary in this pathway.
The present study describes a potential model to study how CRBN affects the cascade of developmental and molecular events that cause human ID. The major brain areas involved in contextual and cued fear conditioning include the amygdala, hippocampus, frontal cortex, and cingulate cortex. Although there is evidence that amygdala activity, especially the basolateral amygdala, is not necessary for associative learning due to contextual fear conditioning [44–47], the heterogeneous distribution of CamKII promoter activity within the basolateral complex, central nuclei, and the amygdalo–hippocampal area [48,49] does not exclude the amygdala as contributing to the behavioral phenotype in the CamKIIcre/+, crbn−/− mice. Heterozygous CamKIIcre/+, crbn+/− mice do not exhibit any overt abnormalities on the behavioral tests performed in this study suggesting that the expression a single crbn allele in the mouse forebrain is not sufficient to cause deficits in HPC-dependent memory, motor function, exploratory motivation, or affective behaviors. However, germ-line crbn−/−, like the CamKIIcre/+, crbn−/− mice, exhibit abnormal contextual fear conditioning suggesting that a homozygous deletion of crbn in forebrain neurons of postnatal mice selectively affects HPC-dependent memory and mimics the phenotype of humans with autosomal recessive non-syndromic ID (ARNSID) and a homozygous CRBN nonsense mutation [3,13,19].
There are only five other genes besides CRBN that cause ARNSID including neurotrypsin (PRSS12; OMIM #249500)[50], coiled-coil and C2 domain-containing 1A (CC2D1A;OMIM #608443)[51], glutamate receptor, ionotropic, kainate 2 (GRIK2; OMIM #138244)[52], and the tumor suppressor candidate 3 (TUSC3; OMIM #601385)[53]. A severe cognitive phenotype characterized by IQs less than 50 is caused by mutations in PRSS12, CC2D1A, GRIK2, and TUSC3. However, CRBN gene mutations results in a mild phenotype with IQs between 50 and 70 [1,3]. These five genes are expressed in the HPC but little is known of their precise role in the pathogenesis in ARNSID. Both PRSS12 and GRIK2 are found in the presynaptic nerve endings [50,54]. PRSS12 is a regulatory serine protease [50] whereas GRIK2 is glutamate receptor that acts to facilitate synaptic transmission [54]. CC2D1A regulates endocytosis and endosomal trafficking of Notch, a protein that regulates cell fate during development [55,56]. TUSC3 is involved in magnesium transport across the plasma membrane [53]. In summary, these four genetic causes of ARNSID directly disturb ion receptors or other synaptic processes including proteolysis, endocytosis, and ion transport. Similarly, CRBN regulates the synaptic expression of the BKCa in HCP neurons [15,17] and CIC-2 channels in retinal neurons [18]. In human lymphoblastoid cell lines expressing a nonsense CRBN mutation an immature BKCa channel isoform (STREX) that is normally found in fetal and early postnatal development persists in mutant cell lines compared to wild type [19]. This suggests that higher intracellular Ca2+ sensitivity, faster activation, and slower deactivation kinetics of STREX channels may contribute to cognitive impairments in individuals with mild ARNSID. Studying BKCa channel homeostasis in CamKIIcre/+, crbn−/− mice may show alterations in this type of Ca2+-mediated signal transduction accompanied by changes in synaptic maturation and connectivity.
5. Conclusions
Our data show that a decrease in crbn expression in the forebrain of mice mimics a human ID phenotype caused by a nonsense CRBN mutation. Based on these findings, it is evident that CamKIIcre/+, crbn−/− mice are a promising model to study the mechanisms whereby crbn leads to alterations in neuronal homeostasis, synaptic maturation, and connectivity. Results from studies using the CamKIIcre/+, crbn−/− mice will lead to a better understanding of the fundamental aspects of mammalian memory and learning, increase our knowledge of the pathogenesis of ID, and serve as models to test therapeutic agents that alter crbn homeostasis.
Acknowledgments
This work was supported by the March of Dimes Birth Defects Foundation (12-FY08-152 to J.J.H.); and in part by The Hartwell Foundation (A.M.R., J.J.H.); and the Weill Cornell Medical College Clinical Translational Science Center (UL1-RR024996). The authors acknowledge Dr. Regina Allen from inGenious Targeting Labs, Inc., Stony Brook, New York, for her help in the construct design.
References
- 1.Higgins JJ, Rosen DR, Loveless JM, Clyman JC, Grau MJ. A gene for non-syndromic mental retardation maps to chromosome 3p25-pter. Neurology. 2000;55:335–40. doi: 10.1212/wnl.55.3.335. [DOI] [PubMed] [Google Scholar]
- 2.Higgins JJ, Pucilowska J, Lombardi RQ, Rooney JP. Candidate genes for recessive non-syndromic mental retardation on chromosome 3p (MRT2A) Clin Genet. 2004;65:496–500. doi: 10.1111/j.0009-9163.2004.00267.x. [DOI] [PubMed] [Google Scholar]
- 3.Higgins JJ, Pucilowska J, Lombardi RQ, Rooney JP. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology. 2004;63:1927–31. doi: 10.1212/01.wnl.0000146196.01316.a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–50. doi: 10.1126/science.1177319. [DOI] [PubMed] [Google Scholar]
- 5.Miller MT, Stromland K. Teratogen update: thalidomide: a review, with a focus on ocular findings and new potential uses. Teratology. 1999;60:306–21. doi: 10.1002/(SICI)1096-9926(199911)60:5<306::AID-TERA11>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 6.Melchert M, List A. The thalidomide saga. Int J Biochem Cell Biol. 2007;39:1489–99. doi: 10.1016/j.biocel.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 7.Knobloch J, Ruther U. Shedding light on an old mystery: thalidomide suppresses survival pathways to induce limb defects. Cell Cycle. 2008;7:1121–7. doi: 10.4161/cc.7.9.5793. [DOI] [PubMed] [Google Scholar]
- 8.Stromland K, Nordin V, Miller M, Akerstrom B, Gillberg C. Autism in thalidomide embryopathy: a population study. Dev Med Child Neurol. 1994;36:351–6. doi: 10.1111/j.1469-8749.1994.tb11856.x. [DOI] [PubMed] [Google Scholar]
- 9.Wu F, Xing J, Wang S, Li M, Zheng C. Screening and identification of host factors interacting with UL14 of herpes simplex virus 1. Med Microbiol Immunol. 2011;200:203–8. doi: 10.1007/s00430-011-0196-z. [DOI] [PubMed] [Google Scholar]
- 10.Ando Y, Kitayama H, Kawaguchi Y, Koyanagi Y. Primary target cells of herpes simplex virus type 1 in the hippocampus. Microbes Infect. 2008;10:1514–23. doi: 10.1016/j.micinf.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Greer MK, Lyons-Crews M, Mauldin LB, Brown FR., 3rd A case study of the cognitive and behavioral deficits of temporal lobe damage in herpes simplex encephalitis. J Autism Dev Disord. 1989;19:317–26. doi: 10.1007/BF02211849. [DOI] [PubMed] [Google Scholar]
- 12.Kapur N, Barker S, Burrows EH, Ellison D, Brice J, Illis LS, et al. Herpes simplex encephalitis: long term magnetic resonance imaging and neuropsychological profile. J Neurol Neurosurg Psychiatry. 1994;57:1334–42. doi: 10.1136/jnnp.57.11.1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higgins JJ, Tal AL, Sun X, Hauck SC, Hao J, Kosofosky BE, et al. Temporal and spatial mouse brain expression of cereblon, an ionic channel regulator involved in human intelligence. J Neurogenet. 2010;24:18–26. doi: 10.3109/01677060903567849. [DOI] [PubMed] [Google Scholar]
- 14.Lee KJ, Lee KM, Jo S, Kang KW, Park CS. Induction of cereblon by NF-E2-related factor 2 in neuroblastoma cells exposed to hypoxia-reoxygenation. Biochem Biophys Res Commun. 2010;399:711–5. doi: 10.1016/j.bbrc.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 15.Jo S, Lee KH, Song S, Jung YK, Park CS. Identification and functional characterization of cereblon as a binding protein for large-conductance calcium-activated potassium channel in rat brain. J Neurochem. 2005;94:1212–24. doi: 10.1111/j.1471-4159.2005.03344.x. [DOI] [PubMed] [Google Scholar]
- 16.Xin W, Xiaohua N, Peilin C, Xin C, Yaqiong S, Qihan W. Primary function analysis of human mental retardation related gene CRBN. Mol Biol Rep. 2008;35:251–6. doi: 10.1007/s11033-007-9077-3. [DOI] [PubMed] [Google Scholar]
- 17.Berkefeld H, Sailer CA, Bildl W, Rohde V, Thumfart JO, Eble S, et al. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science. 2006;314:615–20. doi: 10.1126/science.1132915. [DOI] [PubMed] [Google Scholar]
- 18.Hohberger B, Enz R. Cereblon is expressed in the retina and binds to voltage-gated chloride channels. FEBS Lett. 2009;583:633–7. doi: 10.1016/j.febslet.2009.01.018. [DOI] [PubMed] [Google Scholar]
- 19.Higgins JJ, Hao J, Kosofsky BE, Rajadhyaksha AM. Dysregulation of large-conductance Ca2+-activated K+ channel expression in nonsyndromal mental retardation due to a cereblon p.R419X mutation. Neurogenetics. 2008;9:219–23. doi: 10.1007/s10048-008-0128-2. [DOI] [PubMed] [Google Scholar]
- 20.MacDonald SH, Ruth P, Knaus HG, Shipston MJ. Increased large conductance calcium-activated potassium (BK) channel expression accompanied by STREX variant downregulation in the developing mouse CNS. BMC Dev Biol. 2006;6:37. doi: 10.1186/1471-213X-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayford M, Wang J, Kandel ER, O’Dell TJ. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 22.Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, et al. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–26. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Buchholz F, Muyrers JP, Stewart AF. A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet. 1998;20:123–8. doi: 10.1038/2417. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Muyrers JP, Testa G, Stewart AF. DNA cloning by homologous recombination in Escherichia coli. Nat Biotechnol. 2000;18:1314–7. doi: 10.1038/82449. [DOI] [PubMed] [Google Scholar]
- 25.Muyrers JP, Zhang Y, Testa G, Stewart AF. Rapid modification of bacterial artificial chromosomes by ET-recombination. Nucleic Acids Res. 1999;27:1555–7. doi: 10.1093/nar/27.6.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–84. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakai K, Miyazaki J. A transgenic mouse line that retains Cre recombinase activity in mature oocytes irrespective of the cre transgene transmission. Biochem Biophys Res Commun. 1997;237:318–24. doi: 10.1006/bbrc.1997.7111. [DOI] [PubMed] [Google Scholar]
- 28.Franklin K, Paxinos G. The mouse brain in stereotaxic coordinates. San Diego: Academic Press; 1997. [Google Scholar]
- 29.Brown SD, Chambon P, de Angelis MH. EMPReSS: standardized phenotype screens for functional annotation of the mouse genome. Nat Genet. 2005;37:1155. doi: 10.1038/ng1105-1155. [DOI] [PubMed] [Google Scholar]
- 30.Brown SD, Hancock JM, Gates H. Understanding mammalian genetic systems: the challenge of phenotyping in the mouse. PLoS Genet. 2006;2:e118. doi: 10.1371/journal.pgen.0020118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrell AV, Allan AM. Improvements in hippocampal-dependent learning and decremental attention in 5-HT(3) receptor overexpressing mice. Learn Mem. 2003;10:410–9. doi: 10.1101/lm.56103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paz R, Barsness B, Martenson T, Tanner D, Allan AM. Behavioral teratogenicity induced by nonforced maternal nicotine consumption. Neuropsychopharmacology. 2007;32:693–9. doi: 10.1038/sj.npp.1301066. [DOI] [PubMed] [Google Scholar]
- 33.Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, et al. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science. 2006;314:140–3. doi: 10.1126/science.1129663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holmes A, Yang RJ, Crawley JN. Evaluation of an anxiety-related phenotype in galanin overexpressing transgenic mice. J Mol Neurosci. 2002;18:151–65. doi: 10.1385/JMN:18:1-2:151. [DOI] [PubMed] [Google Scholar]
- 35.Holmes A, Kinney JW, Wrenn CC, Li Q, Yang RJ, Ma L, et al. Galanin GAL-R1 receptor null mutant mice display increased anxiety-like behavior specific to the elevated plus-maze. Neuropsychopharmacology. 2003;28:1031–44. doi: 10.1038/sj.npp.1300164. [DOI] [PubMed] [Google Scholar]
- 36.Mayford M, Bach ME, Huang YY, Wang L, Hawkins RD, Kandel ER. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–83. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 37.Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–85. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 38.Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–8. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- 39.Carew TJ, Sahley CL. Invertebrate learning and memory: from behavior to molecules. Annu Rev Neurosci. 1986;9:435–87. doi: 10.1146/annurev.ne.09.030186.002251. [DOI] [PubMed] [Google Scholar]
- 40.Byrne JH. Cellular analysis of associative learning. Physiol Rev. 1987;67:329–439. doi: 10.1152/physrev.1987.67.2.329. [DOI] [PubMed] [Google Scholar]
- 41.Hawkins RD, Kandel ER, Siegelbaum SA. Learning to modulate transmitter release: themes and variations in synaptic plasticity. Annu Rev Neurosci. 1993;16:625–65. doi: 10.1146/annurev.ne.16.030193.003205. [DOI] [PubMed] [Google Scholar]
- 42.Lee KM, Jo S, Kim H, Lee J, Park CS. Functional modulation of AMP-activated protein kinase by cereblon. Biochim Biophys Acta. 2011;1813:448–55. doi: 10.1016/j.bbamcr.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 43.Thomson DM, Herway ST, Fillmore N, Kim H, Brown JD, Barrow JR, et al. AMP-activated protein kinase phosphorylates transcription factors of the CREB family. J Appl Physiol. 2008;104:429–38. doi: 10.1152/japplphysiol.00900.2007. [DOI] [PubMed] [Google Scholar]
- 44.Maren S. Neurotoxic or electrolytic lesions of the ventral subiculum produce deficits in the acquisition and expression of Pavlovian fear conditioning in rats. Behav Neurosci. 1999;113:283–90. doi: 10.1037//0735-7044.113.2.283. [DOI] [PubMed] [Google Scholar]
- 45.Ponnusamy R, Poulos AM, Fanselow MS. Amygdala-dependent and amygdala-independent pathways for contextual fear conditioning. Neuroscience. 2007;147:919–27. doi: 10.1016/j.neuroscience.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vazdarjanova A, McGaugh JL. Basolateral amygdala is not critical for cognitive memory of contextual fear conditioning. Proc Natl Acad Sci USA. 1998;95:15003–7. doi: 10.1073/pnas.95.25.15003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lepicard EM, Mizuno K, Antunes-Martins A, von Hertzen LS, Giese KP. An endogenous inhibitor of calcium/calmodulin-dependent kinase II is up-regulated during consolidation of fear memory. Eur J Neurosci. 2006;23:3063–70. doi: 10.1111/j.1460-9568.2006.04830.x. [DOI] [PubMed] [Google Scholar]
- 48.Odeh F, Leergaard TB, Boy J, Schmidt T, Riess O, Bjaalie JG. Atlas of transgenic Tet-Off Ca2+/calmodulin-dependent protein kinase II and prion protein promoter activity in the mouse brain. Neuroimage. 2011;54:2603–11. doi: 10.1016/j.neuroimage.2010.11.032. [DOI] [PubMed] [Google Scholar]
- 49.Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, et al. Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol Psychiatry. 2007;61:187–97. doi: 10.1016/j.biopsych.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 50.Molinari F, Rio M, Meskenaite V, Encha-Razavi F, Auge J, Bacq D, et al. Truncating neurotrypsin mutation in autosomal recessive nonsyndromic mental retardation. Science. 2002;298:1779–81. doi: 10.1126/science.1076521. [DOI] [PubMed] [Google Scholar]
- 51.Basel-Vanagaite L, Attia R, Yahav M, Ferland RJ, Anteki L, Walsh CA, et al. The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive non-syndromic mental retardation. J Med Genet. 2006;43:203–10. doi: 10.1136/jmg.2005.035709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Motazacker MM, Rost BR, Hucho T, Garshasbi M, Kahrizi K, Ullmann R, et al. A defect in the ionotropic glutamate receptor 6 gene (GRIK2) is associated with autosomal recessive mental retardation. Am J Hum Genet. 2007;81:792–8. doi: 10.1086/521275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou H, Clapham DE. Mammalian MagT1 and TUSC3 are required for cellular magnesium uptake and vertebrate embryonic development. Proc Natl Acad Sci USA. 2009;106:15750–5. doi: 10.1073/pnas.0908332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29:209–16. doi: 10.1016/s0896-6273(01)00191-x. [DOI] [PubMed] [Google Scholar]
- 55.Gallagher CM, Knoblich JA. The conserved c2 domain protein lethal (2) giant discs regulates protein trafficking in Drosophila. Dev Cell. 2006;11:641–53. doi: 10.1016/j.devcel.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 56.Jaekel R, Klein T. The Drosophila Notch inhibitor and tumor suppressor gene lethal (2) giant discs encodes a conserved regulator of endosomal trafficking. Dev Cell. 2006;11:655–69. doi: 10.1016/j.devcel.2006.09.019. [DOI] [PubMed] [Google Scholar]