

Abstract
Mitochondrial and bioenergetic function change with advancing age and may drive aging phenotypes. Mitochondrial and bioenergetic changes are also documented in various age-related neurodegenerative diseases, including Alzheimer’s disease (AD). In some instances AD mitochondrial and bioenergetic changes are reminiscent of those observed with advancing age, but are greater in magnitude. Mitochondrial and bioenergetic dysfunction could, therefore, link neurodegeneration to brain aging. Interestingly, mitochondrial defects in AD patients are not brain-limited, and mitochondrial function can be linked to classic AD histologic changes including amyloid precursor protein processing to beta amyloid. Also, transferring mitochondria from AD subjects to cell lines depleted of endogenous mitochondrial DNA (mtDNA) creates cytoplasmic hybrid (cybrid) cell lines that recapitulate specific biochemical, molecular, and histologic AD features. Such findings have led to the formulation of a “mitochondrial cascade hypothesis” that places mitochondrial dysfunction at the apex of the AD pathology pyramid. Data pertinent to this premise are reviewed.
Keywords: Aging, Alzheimer’s disease, bioenergetics cybrids, mitochondria
Introduction
Mitochondria were identified as cell organelles over 100 years ago [1]. Considerable time elapsed before their functions were fully appreciated. During the 1960’s it was determined that mitochondria contained their own genome, the mitochondrial DNA (mtDNA)[2, 3], and that they generated ATP according to a process defined as the chemiosmotic hypothesis [4]. The membranes that delineated these organelles were also identified, and the fact that these membrane boundaries created compartments that allowed for particular chemical reactions and indeed even pathways to reside was appreciated.
In the second half of the 20th century, a potential role for mitochondria in aging was widely postulated [5]. While mitochondria are neither central nor essential components of all aging hypotheses [6], their contribution to the aging process as either a primary or downstream contributor to this phenomenon is suspected under a variety of current paradigms [7]. The idea that mitochondria might also contribute to neurodegenerative diseases followed the emerging appreciation of their putative role in aging. This general concept was fueled by the observation that the more common neurodegenerative diseases are “age-related,” such that prevalence and incidence for diseases such as Alzheimer’s disease (AD) increase with advancing age [8]. Over the past three decades the contribution of mitochondria to neurodegenerative diseases in general, and to AD specifically, has been hotly debated with views ranging from a potential primary role to a mechanistically irrelevant artifact of cell death that arises due to other factors [9]. During this time, though, the debate has taken a notable turn and at this point the main question seems not so much whether mitochondrial dysfunction is important and relevant in selected neurodegenerative diseases, but rather how critical mitochondrial dysfunction will turn out to be in these selected neurodegenerative diseases [10].
This chapter will address ways in which perceptions of how mitochondria influence brain aging and neurodegeneration have evolved over time. In particular, it will focus on an evolving appreciation of how mitochondria may contribute to AD, and why modifying mitochondrial function is currently considered a viable AD therapeutic target.
Mitochondria and Aging
Overview
In the first half of the 20th century it was observed that caloric restriction enhanced rodent lifespan [11–15]. This contributed to the emerging view that metabolism in general, and perhaps energy metabolism specifically, could help to regulate lifespan. An appreciation of the idea that metabolism could influence aging came to form the basis of the “Rate of Living Hypothesis” that evolved through the early twentieth century [16, 17], which essentially stated species with higher metabolic rates had shorter life spans than species with lower metabolic rates. For example, rodents with high metabolic rates could survive for only a few years, while some reptiles such as turtles with presumably slow metabolic rates could live for many decades. This hypothesis, of course, could not account for long lived species with apparent high metabolic rates, such as birds.
The Rate of Living Hypothesis was eventually succeeded by the more mechanistically specific Free Radical Theory of Aging that assumed the production of oxygen radicals as a byproduct of biochemical reactions should be elevated in species with higher metabolic rates [18]. These free radicals, in turn, would react with molecules within cells and in doing so alter their structure and function. It was further believed that this accumulation of met abolism-generated oxidative stress would drive an aging phenotype.
By the 1970’s it was appreciated that within cells, mitochondria were a leading site of free radical generation. With this realization some circles rebranded the Free Radical Theory of Aging as the “Mitochondrial Theory of Aging” [5] As a corollary of this emerging mitochondria-centric approach to aging theory, some began to speculate mitochondria might possess some sort of aging clock [5, 19]. To this point mtDNA comprised a particularly attractive candidate, and by the late 1980’s it was proposed that an accumulation of somatic mtDNA mutations over time functions as the suspected aging clock [20]. Essentially, mtDNA mutations would accumulate as a consequence of oxidative damage to mtDNA and a modification of its bases, which would in turn compromise the production of respiratory chain subunits through either a lack of production or else through a production of miscoded peptides. In support of this view, an age-associated accumulation of mtDNA mutations, in either the form of deletions or point mutations, was recognized [21].
Mitochondrial aging was (and is) not central to all theories of aging [6], and a positive correlation between mtDNA somatic mutations and advancing age could also potentially reflect a consequence of aging as opposed to a driver of aging. Investigators began to critically assess this possibility in the early 21st century in genetically modified mice that were designed to accumulate mtDNA mutations at an accelerated pace. This was accomplished by creating mice with a mutated mtDNA polymerase gamma (mtPOLG), in which the innate proofreading ability of the enzyme was perturbed [22, 23]. This allowed for a rapid accumulation of somatic mtDNA mutations across a range of tissues. Phenotype characterizations of these mice showed changes consistent with accelerated aging, and were accepted as support for the view that increasing levels of somatic, heteroplasmic mtDNA mutations could drive an aging phenotype.
Mitochondrial Function and Homeostasis in Advancing Age
The predominant current view is that the functional capacity of an organism’s mitochondria declines with advancing age. Much of this thinking was informed by rodent studies, in which mitochondrial functional endpoints were assessed in mice or rats of different ages. For example, it has been reported that the maximum kinetic function of two mitochondrial respiratory chain enzymes, NADH: ubiquinone oxidoreductase (complex I) and cytochrome oxidase (COX; complex IV) declines with advancing age [24, 25]. Age-related declines in mitochondrial enzyme activities may represent a specific rather than generalized phenomenon, as the activities of other enzymes such as succinate dehydrogenase (complex II) appears to be preserved. In this respect it is perhaps of interest that complex I and IV are partially encoded by mtDNA, while complex II is entirely encoded by nuclear DNA.
An interesting parameter in the relationship between brain aging and mitochondria has to do with the number of mitochondria that are present, also referred to here as mitochondrial mass. Some studies have reported that in brains derived from subjects who were free of a neurodegenerative disease prior to death, mtDNA copy number increased with advancing age [26]. This increase in mtDNA was observed despite the fact that mRNA levels were reduced. Increased mtDNA content, therefore, was interpreted by the authors as potentially reflecting a compensatory response to a reduction in mtDNA transcription efficiency. As part of a related finding, another study reported protein levels of an mtDNA-encoded COX protein subunit, COX2, were increased in the brains of aged individuals when compared to the brains of young individuals [27]. These findings in humans were essentially reflected in a more recent study from 5,12, and 24 month old C57Bl/6 mice, in which synaptic mitochondria were found to demonstrate apparent adaptive changes at the protein level, which were arguably compensating for overall detrimental changes including an increase in mtDNA damage [28].
In general, relative to young organisms, mitochondria from aged organisms have been reported to show decreased ATP production, increased free radical production, depolarization of the mitochondrial membrane potential, and a reduced ability to buffer calcium [29]. Not all studies, though, have uniformly detected such changes, and to some extent attribute a possible preservation of mitochondrial functional indices to compensatory responses [30].
Mitochondria and Free Radical Production
Oxidative modifications of cell proteins are seen at increased levels in the aging brain, which could potentially reflect increased mitochondrial free radical production [31]. In corollary to this, levels of at least some mitochondrial antioxidant enzymes (for example manganese superoxide disputes, mitochondrial catalase, and periredoxin) increase with advancing age, presumably in response to increased mitochondrial free radical production [28].
When considering the significance of oxidative stress in aging, it must be kept in mind that oxidative stress is not uniformly a toxic event. Up to certain levels, it appears, oxidative stress functions as a signal transducer [32, 33]. For example, it has been reported that oxidative stress can facilitate retrograde signaling from the mitochondria to the nucleus [34, 35]. Free radicals, in fact, may promote mitochondrial biogenesis in situations where compensatory increases in mitochondrial mass could prove beneficial [36–38].
Mitochondrial DNA and Somatic Mutation
MtDNA differs from nuclear DNA in several notable ways. One unique characteristic is heteroplasmy, which is insome ways the mitochondrial correlate of nuclear heterozygosity. Nuclear heterozygosity infers one copy of a gene contains a particular variant while the other does not. MtDNA heteroplasmy is more complex because cells can each have hundreds to thousands of mtDNA. A state of homoplasmy exists when all mtDNA copies within a cell or organism are identical. Heteroplasmy is present when this is not the case and mtDNA copies diverge at a particular nucleotide or nucleotides. Heteroplasmy can be difficult to absolutely rule out, as doing so depends on the sensitivity of its ascertainment. For example, it appears that individual low-abundance mtDNA sequence deviations, or microheteroplasmies, are relatively common at the 1–3% level [39, 40]. They can be detected at levels even lower than 1%, although at extremely low percentages the reliability of the observed sequence deviation can be come questionable. In other words, does an extremely low-abundance deviation represent a bona fide sequence deviation, or could it perhaps represent a sequencing artifact?
Regardless, mtDNA reportedly accumulates mutations at approximately ten times the rate of nuclear DNA[41]. This has been attributed to a number of factors, including the lack of protective histone proteins, but also to the fact that mtDNA resides in close proximity to electron transport chain-derived free radical production [42]. In one scenario, cytosine can undergo an oxidative deamination to uracil, which pairs with an adenine rather than guanosine upon replication and results in a G-C base pair undergoing conversion to an A-T base pair (Figure 1A) [43, 44]. In another scenario, 2-deoxy guanosine is oxidized to 8-hydroxy-2-deoxy guanosine and then to 8-oxo-2-deoxyguanosine. 8-oxo-2-deoxyguanosine mismatches to an adenine nucleotide, eventually leading to the conversion of a G-C base pair to a T-A base pair (Figure 1B). 8-hyroxy-2-deoxy guanosine modifications increase with advancing age [45]. While substitutions in the mtDNA that are consistent with these patterns do appear to accumulate with advancing age, it is interesting to note that in one study of mtDNA POLG mutator mice, which over time accumulate such substitutions at an accelerated pace and show an accelerated aging phenotype, there was no evidence of an age-associated concomitant increase in oxidative stress [23].
Figure 1. Oxidation-mediated mtDNA mutation.

(A) A G-C pair is converted to a T-A pair following the oxidative deamination of a cytosine nucleotide to a uracil nucleotide. (B) A G-C-pair is converted to a T-A pair following the oxidative conversion of a guanosine to 8-hydroxy-2-deoxy guanosine and then to 8-oxo-2-deoxyguanosine.
As somatic mutations begin to accumulate they create heteroplasmies whose levels will likely initially reside below the level of detection. If they become fixed in the genome and are replicated over time, their percent of the total mtDNA copies increases and the percent heteroplasmy increases. Classically, it has been easier to detect somatic deletion mutations than it has been to detect somatic point mutations. Levels of some deletions, such as the ~5 KD common deletion, appear to increase in the brains of aging humans [46].
Could mtDNA Inheritance Affect Longevity?
It has been speculated that inherited mtDNA variations may influence aging and longevity. One prominent aging study, the Framingham Longevity Study, found that how many years either of an individual’s parents lived correlated with how long that individual would live, but the mother’s age at death correlated better with the age at death of the child [47]. One possible interpretation of this study is that a maternally inherited genetic factor, perhaps mtDNA, influences aging.
More specific mtDNA studies report particular mtDNA sequences also associate with life expectancy. An ND2 C5178A substitution is reportedly over represented in Japanese centenarians [48], while an ATPase6 G9055A substitution is reportedly over represented in French and Irish centenarians [49, 50]. Positive haplogroup association studies have also been published. MtDNA haplogroups are defined as patterns of nucleotide substitutions that tend to occur together within individuals, and which have arisen over the course of humanity and become fixed in different populations at different frequencies over the course of human migration and history [51, 52]. For example, the frequency of haplogroup J in Italian centenarians is reportedly higher than it is in the overall Italian population [53], and the frequency of haplogroups U and J is reportedly higher in Finnish centen arians than it is in the overall Finnish population [54].
Critical Questions about the Role Mitochondria Play in Aging
While there seems to be consensus that an organism’s mitochondria and bioenergetic performance change over time, concerns about the place and role of mitochondria in aging are frequently raised. For the case of somatic mutations, it can be pointed out that correlation and association do not prove causation; it is possible that the observed accumulation of mtDNA mutations over lifespans, including point mutations and deletions, represents a consequence of aging and are not actually driving aging. MtDNA POLG mutator mice experiments were done with the purpose of addressing this question [22, 23], but some have raised questions about how well this system mechanistically models actual human aging, as well as to how rigorously the phenotype changes observed in these mice truly reflect normal physiologic aging [55, 56]. For those accepting the mtDNA POLG mutator mice as a good model of aging more nuanced questions have spurred debate, such as whether point mutations or deletions are primarily responsible for driving age-related changes in the mice [57, 58].
In particular, questions have been raised about whether the magnitude of functional mitochondrial changes seen in aging organisms in general, and in humans specifically, is predictably profound enough to induce physiologic changes. As a corollary to this, it has certainly been reported that compensatory mechanisms are initiated in the face of declining mitochondrial function and it must be considered how well these compensations mitigate the potential consequences of age-related changes in mitochondrial function and cell bioenergetics [30].
Finally, animal experiments have been reported in which the pharmacological or genetic induction of mitochondrial dysfunction actually associated with increased lifespan [59]. At the very least, this suggests that even if mitochondria are a major driver of human aging, the overall picture of how and why they drive aging may turn out to be quite complex.
Mitochondria and Alzheimer’s Disesae
Overview
During the 1980’s fluorodeoxyglucose positron emission tomography (FDG PET) brain scans revealed cortical glucose utilization is reduced in AD subjects [60, 61]. Decreased glucose utilization was featured in neuroanatomically discrete regions, including the posterior temporal and parietal cortices, as well as the posterior cingulate-precuneus region (Figure 2). The underlying basis for this observation has to date remained uncertain. Proposed possibilities have postulated these declines may reflect an artifact of neuron loss, or a loss of synaptic connectivity. However, when glucose utilization is studied in homogenized brain tissue, where synaptic connectivity is no longer maintained and the amount of material in the assay is standardized, reduced glucose utilization still remains [62]. This raises the possibility that reduced glucose utilization on FDG PET could also possibly reflect perturbed glycolysis flux.
Figure 2. Fluorodeoxyglucose positron emission tomography (FDG PET) scan from an AD patient.
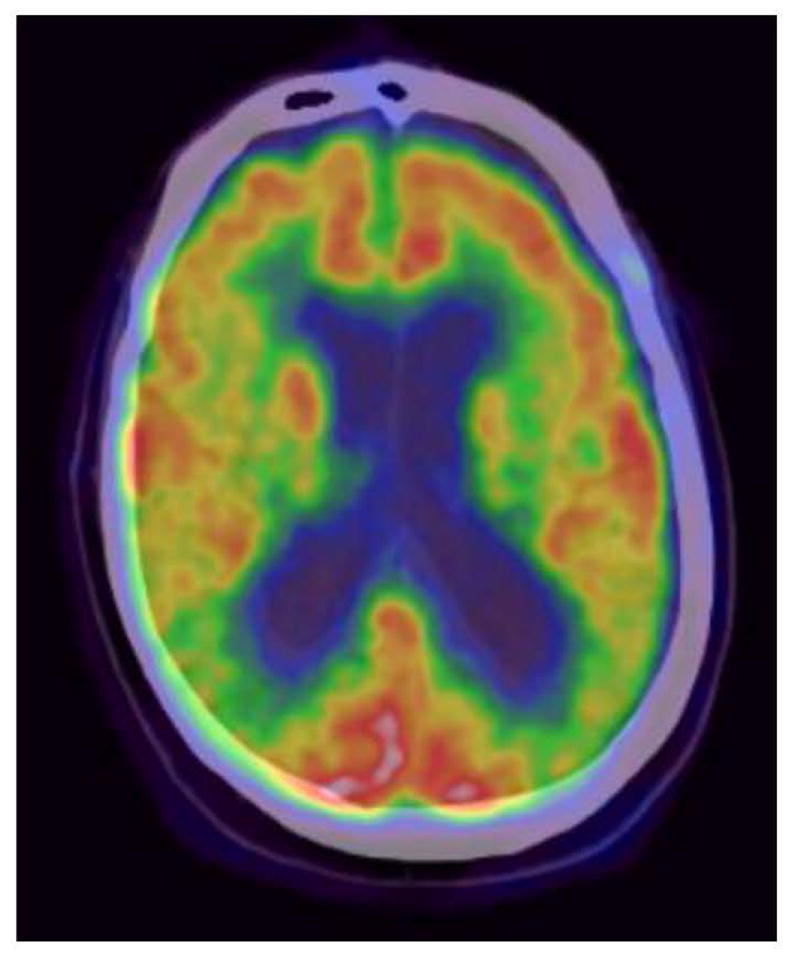
In the normal case the cortical region should show a consistent level of relatively high glucose uptake and, therefore, utilization. In in this FDG PET scan from an individual with AD there is attenuation of the high cortical glucose uptake/utilization signal (indicated by a red-orange color) in the region of the posterior temporal/inferior parietal cortical regions (indicated by a yellow-green color).
Mitochondria are also different in AD patients than they are in age-matched control subjects. Overall mitochondrial size is reduced, although this is punctuated by the increased presence of overly swollen mitochondria with misshapen cristae [27, 63]. Activities of certain mitochondria-localized enzyme are also reduced. This includes a reduction in the activity of the Kreb’s cycle enzyme α-ketoglutarate dehydrogenase complex (KGDHC) and of pyruvate dehydrogenase complex [64–66], which gates the entry of glycolysis-derived, pyruvate-based carbon into the Kreb’s cycle. Interestingly, the activity of some Krebs cycle enzymes, specifically the activity of enzymes in the second half of the cycle, have been reported to be increased in brains from AD subjects [67].
COX activity also tends to be lower in AD subjects than it is in age-matched control subjects [68]. Interestingly, this COX activity reduction is not limited to the brain. In fact, it was first reported to be present in platelet mitochondria derived from AD subjects [69–74], and only after that was it assayed in the brains of AD subjects, where its activity was similarly and consistently found to be reduced [70, 75–84]. In addition to platelet and brain mitochondria, AD COX activity has also been reported in fibroblast cultures derived from sporadic AD subjects [85].
Determinations of mitochondrial number in AD are to some extent complicated. For the most part, in AD brain the number of normal mitochondria per neuron appears to be reduced, although the amount of mitochondrial material that seems to be undergoing digest ion within autophagosomesis increased [27]. Mitochondrial homeostasis is further perturbed in that there is an apparent shift towards increased mitochondrial fission, mitochondria are less likely to radiate from the perikaryon to neurite projections, and messenger RNA and protein levels of the transcriptional coactivator peroxisome-proliferater activated receptor γ coactivator 1α (PGC1α), which facilitates mitochondrial biogenesis, are reduced [86–90].
By some parameters mtDNA in AD subjects also seems to differ from that of age-matched control subjects. The amount of intact mtDNA is reportedly reduced, even though the amount of autophagosome-localized mtDNA may be increased [27, 91–93]. There appears to be increased numbers of some types of presumably somatic mutations, such as deletions and potentially also specific, presumably acquired point mutations [27, 92, 94–98]. Levels of mtDNA nucleotide oxidative damage are also higher in AD subjects than they are in control subjects [99].
Some studies claim inherited mtDNA variations are found in higher or lower frequencies in AD subjects as compared to non-AD subjects. Association studies have variably reported particular mtDNA haplogroups are statistically over or under-represented in AD cohorts, and that particular mtDNA single nucleotide polymorphisms (SNPs) are statistically over or under-represented in AD cohorts [100–113]. Results across different mtDNA association studies in AD have not been consistent across studies, though, which has led to some concern about the reliability of individual reports [114–118].
Could Aβ or APP Account for Differences in AD Mitochondria?
The plaques observed in AD subject brains contain a large percentage of the beta amyloid (Aβ) protein. A number of investigators have reported that Aβ localizes to mitochondria (in brains from human AD subjects and in APP transgenic mice), where it can bind to and interfere with the function of different intra-mitochondrial proteins included cyclophilin D and the Aβ-binding alcohol dehydrogenase (ABAD) protein [119–127]. Aβ has also been shown in experimental systems to interfere with mitochondrial respiratory chain function, and to specifically inhibit COX activity [122, 128–131].
When added to neuronal NT2 teratocarcinoma cells, Aβ induces increased oxidative stress and cell death. However, when Aβ is added to NT2 teratocarcinoma cells that have been depleted of their endogenous mtDNA (ρ0 cells), that do not produce mtDNA-encoded respiratory chain subunits and are unable to successfully perform oxidative phosphorylation, these toxic Aβ effects are not observed [132]. This suggests Aβ toxicity in in vitro systems is at least to some extent mediated through its effects on cell respiration.
Aβ also impacts other aspects of mitochondrial homeostasis. It appears able to reduce mitochondrial movement [133], and to shift the mitochondrial fission-fusion balance towards the fission end of the spectrum [86, 87, 90].
The Aβ protein derives from a larger parent protein called the amyloid precursor protein (APP). An elegant series of experiments has demonstrated APP itself localizes to mitochondria [134–137]. APP in fact contains an N-terminal mitochondrial targeting sequence of strategically placed, positively charged amino acids that lead it to enter mitochondria through the translocase of the outer mitochondrial membrane (TOMM) and translocase of the inner mitochondrial membrane (TIMM) protein import apparatus [134, 136]. However, APP also contains a peptide sequence that causes the process of mitochondrial import to prematurely arrest, leaving an intra-mitochondrial N-terminal end and a long extra-mitochondrial C-terminal end [134]. The presence of APP at the mitochondria appears to in general interfere with normal mitochondrial function, and to specifically reduce COX activity [134].
Much of the data showing Aβ-mitochondria and APP-mitochondria physical associations derives from model systems and model organisms, such as transgenic mice that over express a mutant human APP transgene [119–122, 124, 125, 127]. However, physical associations have also been demonstrated in brains from deceased AD subjects [119, 126, 134]. Still, it is not immediately clear how mitochondria-localized APP or Aβ might interfere with the function of mitochondria outside the brains of affected individuals.
Evidence of a Maternal Inheritance Contribution to AD
Epidemiologic studies suggest that although an individual’s AD risk is determined by both parents, maternal influence seems to be more profound than paternal influence [138–141]. This has been shown, for example, by the study of Edland et al. who found that among AD probands who also had a demented parent, the demented parent was more than twice as likely to be the mother [140]. Importantly, this relationship was observed even when the age of parental dementia onset was relatively young. This finding, therefore, is unlikely to simply reflect an artifact caused by greater longevity in women versus men, a factor that needs to be considered since survival of mothers to older ages than fathers might also increase their chance of developing a dementing disorder such as AD.
Endophenotype studies also support the presence of an AD maternal inheritance bias. Endophenotypes are incomplete manifestations of a disease; they can be defined by disease-consistent characteristics that are insufficient by themselves to qualify one for a diagnosis, or by biomarkers. AD endophenotype studies have consistently shown that the non-demented adult children of AD mothers are more likely to have AD-like biomarker changes than the non-demented children of AD fathers. The first of these studies was that of Mosconi et al., which reported that FDG PET scans from mostly middle aged children of AD-affected mothers were more likely to manifest AD-like glucose utilization patterns than the mostly middle aged children of AD-affected mothers [142]. Similarly, an arterial spin labeling (ASL) study found the middle aged children of AD-affected mothers were more likely to show decreased perfusion patterns than the middle aged children of AD-affected fathers [143]. Studies have also shown the children of AD-affected mothers tend to have more cerebral amyloid than the children of AD affected fathers, and are also more likely to have increased levels of cerebrospinal fluid oxidative stress markers [144–147]. Several studies have found children of AD mothers also tend to have greater amounts of neuroanatomically specific cerebral atrophy than do the children of AD fathers [148–152]. One study found platelet mitochondria COX activity was lower in the children of AD mothers than it was in the children of AD fathers [153]. Finally, it was reported in the Framingham Longevity study that middle aged, non-demented APOE4 carriers who also had an AD-affected mother had lower scores on a memory test than did middle aged, non-demented APOE4 carriers who also had an AD-affected father [154].
Could APOE Influence AD Risk by Affecting Mitochondrial Function?
The APOE gene, located at locus 19q13.2, represents the most extensively studied sporadic AD genetic risk factor [155–157]. The APOE gene encodes a protein, apolipoprotein E, that plays a role in lipid and cholesterol transport [158]. There are three relatively common polymorphism-defined APOE alleles, the APOE2, APOE3, and APOE4 variants [159]. The APOE4 version is associated with an increased lifetime risk of developing AD [155]. Several hypotheses have been proposed in an attempt to mechanistically explain the association between APOE variants and AD risk. One hypothesis has arisen from the observation that the apolipoprotein E4 isoform folds differently than the other versions, and that this folding difference causes it to be proteolyzed into a smaller peptide that displays a functional mitochondrial targeting sequence [160, 161]. This apolipoprotein E 4-derived peptide appears to have toxic effects on mitochondria, and in cell culture it even seems to reduce COX activity [162]. Human brains from young deceased APOE4 carriers were also found to have reduced cortical COX activity; this was observed despite an absence of concurrent Aβ accumulation [163].
As was pointed out in the previous section, a cognitive-based endophenotype study of the Framingham Longevity cohort found that among non-demented, middle-aged APOE4 carriers individuals with an AD-affected mother had less robust performance on a test of memory than did subjects with and AD-affected father [154]. If an apolipoprotein E4 degradation product truly functions as a mitochondrial toxin, and inherited mtDNA features do turn out to influence AD risk, it could be the case that APOE4 and particular mtDNA sequences could interact to give rise to a “double” mitochondrial hit that creates a particularly elevated AD risk. Potentially consistent with this possibility are reports that certain mtDNA haplogroups appear to modify the APOE4-associated increase in AD risk [106, 164].
While a reasonably strong case has been made that APOE genotype influences AD risk, it is nevertheless important to point out that the translocase of the outer mitochondrial membrane 40 kilodalton (TOMM40) subunit gene sits immediately adjacent to the APOE gene [165]. TOMM40 polymorphic variants have also been reported to associate with AD risk [166–174], although some of these TOMM40 variants are in linkage disequilibrium with the critical APOE isoform-defining variants [175]. This genetic confounding makes it difficult to prove or disprove whether the TOMM40 gene, through the TOMM40 protein, independently contributes to AD risk or contributes at least to some extent to the AD risk currently attributed in most circles to the APOE gene and apolipoprotein E protein [176].
Evidence for a Somatic mtDNA Mutation Contribution to AD
Some have hypothesized an accumulation of somatic mtDNA mutations could play a major role in the development of AD, and perhaps even represent a primary driving cause [21]. Data addressing this possibility have been mixed, and studies are limited to some extent because prior to the introduction of next generation sequencing approaches, it was technically difficult to resolve somatic single nucleotide changes at the microheteroplasmy level.
Perhaps for this reason initial studies emphasized quantification of large deletions. One early study, that of Corral-Debrinski et al., reported that brains from AD subjects who were less than 75 years of age had higher levels of the ~5 kb “common deletion” than brains from age-matched control subjects [94]. Hamblet et al. also found increased levels of the 5 kb common deletion in AD brain [96]. This finding was also essentially corroborated by Cottrell et al. and Krishnan et al., who initially found using a histochemical approach that AD brains were more likely to show COX-perturbed neurons in specifically examined areas, and later reported COX-perturbed neurons in AD brains had higher amounts of various large scale mtDNA deletions [177, 178].
Chang et al. used a sequential PCR amplification and restriction enzyme digestion approach to screen for the presence of mtDNA point mutations in AD brains [95]. This study did report evidence of increased AD brain mtDNA point mutations, which were felt to likely represent a consequence of mtDNA nucleotide oxidative damage. Interestingly, though, increased levels of the 5 kb common deletion were not detected in the AD brains from this study.
In a study by Coskun et al, the frequencies of several specific D-loop mutations were found to be profoundly increased in the brains of AD subjects [92]. This study further reported expression levels of ND6 were decreased and that the mtDNA to nuclear DNA ratio was reduced in the presence of these mtDNA control region mutations, which was interpreted as support for the view that these mutations interfered with mtDNA transcription and replication. It was also later reported by these investigators that the D-loop mutations that were found to be increased in AD subject brains were also increased in AD subject blood and lymphoblastoid cell line mtDNA [179].
On the other hand, in their study of AD and control brains Lin et al. did not detect a quantitative difference in the burden of microheteroplasmic point mutations [180]. To perform this study, the authors used a clonal sequencing analysis approach of PCR-amplified COX1 amplicons; in addition to analyzing brains from AD and age-matched control subjects, brains from a younger control group were also evaluated. While the microheteroplasmic mutation burdens were comparable between the AD and control groups, this study nevertheless presented several interesting findings: 1) microheteroplasmic mutations were relatively frequent; 2) there was a positive correlation between advancing age and the number of mutations per subject, so that it did appear that the mutation burden did increase with age; 3) there was a negative correlation between advancing age and COX enzyme activity, so that it did appear that COX activity did decline with age; and 4) there was a negative correlation between COX enzyme activity and COX1 mutation burden, so that it did appear that as COX1 mutations accumulated, COX activity fell.
Evidence of a Mitochondrial Link to Classic AD Histopathology Changes
More than one cell culture-based study has reported toxin-induced mitochondrial dysfunction, including toxin-mediated COX inhibition, reduces the processing of APP by the non-amyloidogenic α-secretase degradation pathway. This conclusion is based on the finding that cells treated with toxins such as sodium azide (a COX inhibitor) generate reduced levels of soluble APPα (sAPPα) [181, 182]. It has been further inferred by such studies that decreased α secretase-mediated processing of APP could reflect a shift in APP processing towards its amyloidogenic, β-secretase mediated processing pathway [183]. The cell culture study of Gabuzda et al. to some extent supports this inference, as these authors found that sodium azide-treated cells produced higher levels of an 11 kD APP cleavage product that was suspected to contain an intact Aβ peptide sequence [183].
A number of studies utilizing APP transgenic mice more directly suggest pertinent connections between brain energy metabolism and AD histopathology do exist. In the study of Scheffler et al., the investigators used a strategic strain inter-breeding approach to create groups of APP transgenic mice that ultimately differed primarily in their mtDNA sequences [184]. It was found that groups of mice with different mtDNA sequences developed profoundly different amounts of amyloid plaques.
Two mouse studies found reducing the amount of COX holoenzyme actually reduced amyloid plaque deposition. For the first of these studies, mice with APP and presenilin 1 (PS1) mutations were crossed with mice engineered for a Cre-loxP-mediated knock-out of the cytochrome oxidase 10 (COX10) gene [185]. The COX10 gene encodes a farnesyltransferase that is required for the synthesis of COX heme; eliminating this farnesyltransferase results in a dramatic reduction in COX holoenzyme production. COX activity accordingly declines, as does measureable markers of oxidative stress. In the second of these studies, APP/PS1 mutant mice were crossed with mice designed to express a mitochondria-targeted restriction enzyme that cleaves mtDNA[186]. This led to mtDNA depletion without generating evidence of oxidative stress, and a reduction in levels of the COX1 protein subunit. Increased levels of the APP-derived β-C terminal fragment (β-CTF) were detected, which did suggest a potential change to APP processing, although no increase in BACE activity was observed and similar to the findings of Fukui et al., plaque accumulation was reduced.
Kukreja et al. evaluated a different mouse model with a predictably different type of respiratory chain defect [187]. The mice in this study were generated by breeding mice expressing a mutant human APP transgene with mice designed to express a dysfunctional mtDNA polymerase γ (PolgA D257A mice) and which accumulate mtDNA mutations at an accelerated rate. In this model of accelerated aging, plaque accumulation was also accelerated. Plaque accumulation differences between the Korea et al. study and the studies of Fukui et al. and Pinto et al. are not entirely clear, although it seems reasonable to consider differences in the nature of the induced mitochondrial defects may prove pertinent. One potential distinction between these studies is that the Fukui et al. and Pinto et al. mice seem to have decreased amounts of respiratory chain enzymes (or at least decreased amounts of COX or COX protein subunits), while the Korea et al. mice may have made functionally abnormal respiratory chain enzymes rather than less respiratory chain enzymes.
Another relevant mouse study is that of Dumont et al. [188]. This study crossed transgenic mice that expressed a mutant APP with transgenic mice that over expressed PGC1 α. Contrary to what was perhaps initially expected, the irenic mice showed increased amyloid plaque deposition. The irenic mice also demonstrated evidence of perturbed mitochondrial function, as the activities of several mitochondrial-localized enzymes (complex I, succinate dehydrogenase, and citrate syntheses) were diminished. Proteosome activity was also diminished in the irenic mice, and this was felt to play a role in the amyloid deposition increase.
Other data from transgenic mouse studies could be considered potentially consistent with a possible bioenergetics-amyloidosis relationship. It has been shown that in APP transgenic mice, Aβ secretion into brain interstitial fluid is higher when the mice are awake and lower when they are asleep [189]. When APP transgenic mice are manipulated into a state of sleep deprivation, interstitial fluid levels are further elevated [190]. Increasing the whisker stimulation of APP transgenic mice increases, while decreasing the whisker stimulation of these mice decreases, interstitial fluid Aβ levels [191]. When Yamamoto et al. used optogenetic stimulation to induce chronic neuronal excitability within the hippocampal performant pathways of APP transgenic mice, interstitial Aβ and Aβ plaque levels increased [192]. Taken together, studies such as these suggest synaptic activity increases Aβ production, and because synaptic activity creates a state of bioenergetic stress these data at least indirectly arguea link should exist between cell bioenergetics and APP processing/Aβ production.
In humans potential links between bioenergetics and APP processing/Aβ production are harder to establish, although a study of trauma victims did find that emergence from a coma state corresponded temporally with an increase in the brain’s interstitial Aβ level [193]. This would seem to be consistent with mouse data that report synaptic activity correlates with Aβ production.
Other relevant human data may be inferred from the study of Vlassenko et al., who reported that areas in which plaques initially present are parts of the brain’s default mode network (DMN)[194]. These regions show a unique bioenergetic pattern that features an increased reliance on aerobic glycolysis, defined by the authors as all glucose utilization that occurs in an adequately oxygenated tissue or adequately oxygenated cell that is not utilized in oxidative phosphorylation. Progressively lower amounts of glucose carbon released as CO2 in this study were interpreted as being indicative of a progressively increased metabolism of glucose through aerobic glycolysis; non-oxidative phosphorylation uses of glucose include metabolism of glucose to lactate, incorporation into glycogen, a contribution of carbon to fatty acid or cholesterol synthesis, or the entry of glucose into the pentose phosphate shunt. This study stresses that in considering the potential relationship between bioenergetics and APP/Aβ, in addition to considering how much energy metabolism is present, what energy fluxes are present as well as how and why particular fluxes are occurring warrants consideration.
Relationships between neuro fibrillary tangles and the tau protein they contain are also reported. Toxic perturbation of cell bioenergetics is certainly recognized to influence the activities of kinases that phosphorylate tau, and to increase tau phosphorylation. This has been demonstrated in both cell culture and animal-based experiments [195–197]. For example, administering the COX inhibitor sodium azide to rats increases tau phosphorylation [196], as does exposing wild type mice and mice that express a mutant tau transgene to annonacin, a complex I inhibitor [198, 199]. Links between tau phosphorylation and metabolism are also suggested by a study that reports prolonged fasting in mice induces brain tau phosphorylation [200].
A recent study by Zhao et al. demonstrated a potential link between mitochondria and the aggregation of tau into tangles [201]. In this study the authors evaluated the effects of a gene polymorphism in the myelin-associated oligodendrocyte basic protein (MOBP) gene that was previously associated with the risk of developing progressive supranuclear palsy (PSP), a neurodegenerative disease that features tangle accumulation [202]. MOBP is located relatively close to the gene that encodes a protein called appoptosin, anuclear-encoded protein that localizes to the mitochondrial inner membrane and participates in heme synthesis. The authors found that the MOBP polymorphism influenced appoptosin expression, which increased in the presence of the PSP-associated MOBP polymorphism [201]. Higher amounts of appoptosin lead to increased heme production, which in turn lead to increased production of cytochrome c. This resulted in an increase in the amount of cytochrome c protein that leaked into the cytoplasm, which in turn activated caspase 3. Caspase 3 then cleaved tau protein at a caspase cleavage site, which generated a tau fragment that aggregated to form tangles and also induced synaptic dysfunction.
Mitochondrial uncoupling has also been demonstrated to induce tau paired helical filament formation [203]. Interestingly, fibroblasts cultures prepared from sporadic AD subjects also show altered mitochondrial function [85], and are more likely to bind an antibody that recognizes paired helical filament tau than are fibroblast cultures prepared from non-AD subjects [204].
AD Cytoplasmic Hybrid (Cybrid) Studies
Overview
The cytoplasmic hybrid (cybrid) technique makes it possible to transfer mtDNA from one cell to another, and to then perpetuate that transfer (Figure 3)[205]. In some ways it is similar to forming cell hybrids [206], with an important distinction being that the resulting cell product contains nuclear DNA from only one source. Further, when mtDNA is transferred it is not transferred in its pureform. Rather, it is contained within the mitochondria from the donor source. Whole mitochondria, therefore, actually serve as a transfer vessel. Finally, mitochondrial transfer can be accomplished using different approaches that to some extent determine whether additional cell constituents are also transferred. For example, isolated mitochondria can be injected into the recipient cell, or donor and recipient cells can be mixed in the presence of a detergent that disrupts membrane integrity and allows for a more extensive mixing of cytosolic contents [207]. Ideally, though, how the mtDNA transfer is accomplished should ultimately lead to the same product because as the resultant cell undergoes subsequent growth and division non-perpetuating materials should degrade over time, and should dilute over the course of repeated cell divisions. The only obvious transferred component that can replicate and therefore perpetuate is the mtDNA. Conceptually, it could be possible that a templating protein could also be transferred and perpetuate, such as a prion protein, but to date this has not been described in the cybrid literature.
Figure 3. The cybrid technique.

A cell line’s endogenous mtDNA is removed to create a ρ0 cell line, which lacks respiratory-competence and must be maintained in medium supplemented with pyruvate and uridine. After mixing ρ0 cells with mitochondria-containing cytoplasts or platelets, and facilitating cytosolic mixing by addition of detergent, some ρ0 cells incorporate exogenous mitochondria and by extension their mtDNA. The transferred mtDNA allows for the restoration of respiratory competence, and the newly created cybrid cells can be selected for by removing pyruvate and uridine from the medium (leading to the removal of residual untransformed ρ0 cells). The cybrid cells that result from a single fusion can be grown as separate clonal colonies; in cases where the donor mtDNA carries a heteroplasmic mutation, the individual cybrid clonal lines can be analyzed to address issues of threshold. Alternatively, the cybrid cells that result from a single fusion can be expanded together, creating a single cybrid line that can be compared to other unique cybrid cell lines.
Although simply introducing isolated mitochondria to cultured cells is accompanied by some degree of intracellular mitochondrial internalization, an event referred to as “transformation” [208], the first intentional transfer of mitochondria to recipient cells (in the mid-1970’s) featured fusion of enucleated cytoplasts with nucleated cells. The scientific goal of these early studies (which introduced the cybrid term) was to test whether chloramphenicol resistance, a characteristic of some cell lines, was an mtDNA-determined trait [209, 210]. The investigators found that when mitochondria-containing cytoplasts from a chloramphenicol-resistant cell line were mixed and fused in culture with a chloramphenicol-sensitive cell line, some of the chloramphenicol-sensitive cells acquired chloramphenicol resistance. This lead the investigators to conclude that chloramphenicol resistance was indeed an mtDNA-determined trait.
It is important to note that as a result of this approach the resulting cybrid cells, at least initially, were presumably heteroplasmic as they should have contained the mtDNA that was endogenous to the accepting cell line, as well as the mtDNA from the donor cytoplast mitochondria. To refine the technique, King and Attardi subsequently developed the idea of using a ρ0 cell line as the accepting cell line [207]. ρ0 cells are cells that have undergone depletion of all detectable mtDNA. The development of ρ0 cell lines, in turn, followed the efforts of several groups to mimic in cultured cell lines the previously observed ability of yeast cells to deplete their mtDNA content under conditions that favored glycolysis [211–213]. These mtDNA-depleting yeast cells were called ρ petites, since prior to its identification as mtDNA cytosolic DNA was initially referred to as ρ DNA [214]. By using ρ0 cells as the accepting cell line, investigators gained the ability to create cell lines that contained only mtDNA from the mitochondrial donor.
Moving forward using ρ0 cell lines as the recipient cells, investigators began to study issues of heteroplasmy, threshold, and in general the biochemical consequences of known mtDNA mutations [215–220]. Mitochondria from human subjects with known homoplasmic or heteroplasmic mtDNA mutations were transferred to ρ0 cells. The resulting cybrid cells were expanded in culture. In instances where heteroplasmic mutations were transferred, the expanding cybrid cells were isolated in order to facilitate the creation of cybrid clones, which ultimately could be shown to contain different ratios of mutant to wild type mtDNA. These clones with different heteroplasmic ratios were then analyzed biochemically to determine how much of a mutational burden was required for a particular mutation to cause a change in biochemical function, and thereby estimate the percent of mutation that had to be present to reach a phenotypic threshold.
Interest in the cybrid approach to address mtDNA-related questions further developed as more ρ0 cell lines were created, and after it was shown that platelets could serve as mitochondrial donor cells [219, 221]. Platelets, which derive from megakaryocytes, lack nuclei and are easily accessed through routine phlebotomy. Through a simple procedure platelet-rich plasma can first be generated from a blood sample, and an enriched platelet fraction can then be prepared through centrifugation of the plasma. The enriched platelet fraction can then be mixed with the ρ0 line of choice to generate cybrid cells.
In the mid-1990s the cybrid approach was first used for a somewhat novel application that involved the utilization of mtDNAs whose sequences were unknown [222]. It was reasoned that biochemical differences between cybrid cell lines prepared from different mitochondrial donors could be used to infer differences existed in their mtDNA sequences. From the perspective of whether mtDNA sequences do indeed vary between individuals applying such an approach did not carry much risk; it was already recognized that mtDNA sequences from individuals that did not derive from a common maternal lineage typically deviate at multiple nucleotide positions. Whether bona fide biochemical differences would be too subtle to detect, and produce false negative results, though, was a concern. A second concern was that variation in biochemical measures could lead to false positive results or incorrect conclusions about the association of particular mtDNA genomes with a particular characteristic. Regarding these two points it was anticipated that by creating large enough groups of cybrid cell lines from large enough groups of mitochondrial donors with a particular biochemical characteristic, one could accurately ascertain whether the specific biochemical characteristic within that group was influenced by the mitochondrial genome [205].
At the time this strategy was defined it was felt to also offer particular experimental strengths. In the mid-1990s DNA sequencing was a far more expensive endeavor than it currently is, and the sequencing approaches of that time were not able to reliably detect low abundance heteroplasmic deviations. Plus, given the high degree of mtDNA polymorphic variation that exists between individuals, unless a particular “smoking gun” sequence mutation that reliably segregated with a cohort was identified, without functional data it would prove difficult to associate individual sequence deviations with a specific group. Doing so would presumably require analyzing very large numbers of individuals.
The first time this approach was utilized was in studies of cybrid cell lines prepared from a group of platelet mitochondria/mtDNA donors with Parkinson’s disease (PD)[222]. As a group, PD patients were already recognized to have platelet mitochondria complex I activities that were lower than those measured in age-matched control subjects [223–225]. Twenty-four cybrid lines were generated from PD subjects, and 28 cybrid lines were generated from 28 age-matched, control subjects. Platelets served as the mitochondria/mtDNA donor source. A human neuroblastoma SH-SY5Y ρ0 cell line that had previously been derived from the standard SH-SY5Y line served as the mitochondria/mtDNA acceptor [221]. The mean complex I activity was found to be ~20% lower in the PD subject-derived cybrid group (simply referred to as the “PD cybrid” group) than it was in the control subject-derived cybrid group (simply referred to as the “control cybrid” group). It was concluded that mtDNA, at least to some extent, contributes to reduced complex I activity in persons with PD.
AD Cybrid Experiments
The cybrid approach was deemed reasonable to address the specific question of why individuals with AD on average have a lower platelet mitochondria COX activity than age-matched, non-AD subjects [69–74]. Non-genetic explanations included the presence in the circulation of a factor that inhibits COX activity. Because COX contains 13 subunits, 10 of which are encoded by nuclear genes and 3 of which are encoded by mitochondrial genes, genetic explanations could alternatively implicate a nuclear DNA or mtDNA-dependent component. It was a priori hypothesized that mtDNA genes were more likely to contribute to lower COX activity in AD subjects than nuclear DNA genes, since late-onset AD (LOAD) rarely demonstrates recognizable Mendelian inheritance [226]. LOAD is generally considered to show sporadic epidemiology although nevertheless with a genetic influence, and in many ways the unique genetic rules of mtDNA, including heteroplasmy, threshold, mitotic segregation, and maternal inheritance uniquely position it to play a role in otherwise apparent sporadic diseases that also demonstrate altered mitochondrial function [226].
In terms of applying the cybrid technique to address this question, it was reasoned that if lower mean COX activities were caused by a circulating inhibitory factor, that factor would wash out over the course of expanding the cell lines generated using platelets obtained from AD subjects (herein referred to as “AD cybrids”). Presumably, low AD subject platelet mitochondria COX activity under this scenario would not perpetuate in culture as AD cybrid line COX activities would increase in culture to match that of the cybrid lines generated from platelets obtained from age-matched, control subjects (herein referred to as “control cybrids”). It was further reasoned that a nuclear DNA-dependent feature would be unlikely to account for a relative reduction in the AD cybrid COX activity because nuclei are not routinely transferred during the procedure, or if such a transfer did occur the transferred nuclei would be unlikely to perpetuate. Similar to toxin-induced activity reductions, nuclear DNA-dependent reductions in COX activity would predictably wash out after the transfer and selection process was completed.
The first published AD cybrid study was the one by Davis et al [227]. This study compared COX data from a group of 20 AD cybrid cell lines to a group of 45 control cybrid cell lines. Transferred mtDNA derived from subject platelet mitochondria, and the acceptor cell line was the SH-SY5Y ρ0 line. COX activity was ~20% lower in the AD cybrid group than it was in the control cybrid group. It is relevant to note that the Davis et al. report was subsequently retracted, although the reasons for the retraction were unrelated to the cybrid data that were presented [228].
Later in 1997 two other AD cybrid studies were reported. In the study of Sheehan et al., platelets served as the mitochondria/mtDNA donor source, and the acceptor cell line was the SH-SY5Y ρ0 line [229]. An ~50% lower COX activity in the AD cybrids was seen. In the other study, that of Swerdlow et al., platelets served as the mitochondria/mtDNA donor source and the acceptor cell line was an NT2 teratocarcinoma-derived ρ0 line [230]. Fifteen AD cybrid lines were compared to 9 control cybrid lines, and a relative 16% reduction in the AD cybrid group COX activity was observed.
Other studies of unique AD cybrid series have focused in particular on COX activity. In the study of Cardoso et al., the authors used platelet mitochondria to generate AD and control cybrid lines on an NT2 ρ0 nuclear background, and found that COX activity in the AD cybrid cell line (n=6) group was 22% lower than it was in the control cybrid cell line (n=5) group [231]. In the study of Silva et al, the authors used platelet mitochondria to generate AD and control cybrid lines on an SH-SY5Y ρ0 nuclear background, and found that COX activity in the AD cybrid cell line (n=8) group was ~30% lower than it was in the control cybrid cell line (n=7) group [232]. On the other hand, the study of Ito et al. also used COX activity as a primary endpoint, and found COX activity was comparable between the AD and control cybrid groups [233]. However, there are a number of notable methodologic differences between the Ito et al. study and the positive studies indicated above. The Ito et al. group used a HeLa cell ρ0 cell line to generate their cybrids, and the mitochondria/mtDNA donor source was mixed; four AD cybrid lines were prepared from platelet mitochondria, three control cybrid lines were prepared from platelet mitochondria, and two control cybrid lines were prepared from fibroblast mitochondria. Also included in the analysis were what were designated as an additional three AD cybrid lines, which were generated by mixing HeLa ρ0 cells with synaptosomes prepared from a brain that was acquired from a deceased AD subject after a 20-hour postmortem interval. The authors reported they were able to identify three cell colonies from this fusion that contained mtDNA, and COX activity data ascertained from each of these three colonies were individually included in the analysis. Due to these substantial methodologic differences, it is arguably difficult to conclude that the Ito et al. negative study contradicts the positive studies.
A number of AD cybrid studies have evaluated various other aspects of mitochondrial function as well as parameters influenced by mitochondrial function [227, 229–232, 234–252]. In many cases changes are reported that recapitulate changes that are seen in the brains of AD subjects themselves [68]. Relative to control cybrid cell lines, in AD cybrid cell lines oxidative stress markers are increased [229–231, 234, 236, 240, 241, 245, 248, 249], inflammatory and stress signaling pathways are activated [234, 236, 239–241, 245, 250], Aβ levels are increased [238, 241], glucose utilization is decreased [232], oxygen consumption is decreased [232], there is a shift towards mitochondrial fission and a smaller average mitochondrial size [243, 247], numbers of ultrastructurally perturbed mitochondrial are increased [243, 252], PGC1α mRNA levels are reduced [232], HIF1α protein is reduced [232], mTOR protein is reduced [232], SIRT1 protein is reduced [232], and apoptotic markers are increased [231, 238–241, 245].
AD cybrids have also been used to model aspects of AD-specific, mitochondria-related function that are difficult or impossible to study in autopsy brain tissue [68]. For example, mitochondrial membrane potential analyses of AD cybrid lines shows a relative degree of depolarization [235, 238, 242, 252], and AD cybrid mitochondria appear to internalize less calcium and are less able to buffer calcium-mediated intracellular signaling activity than control cybrid lines [229]. AD cybrid ATP levels are reduced [231, 232]. It has been shown using differentiated AD cybrid cell lines that mitochondrial movement is relatively reduced [244]. AD cybrid cells are more sensitive to Aβ toxicity than are control cybrid cells [231, 250]. AD cybrids have also been used to screen the molecular effects of potential therapeutic interventions; pharmacologic inhibition of mitochondrial fission activity and antioxidants has been shown to benefit certain mitochondria-related functional parameters [241, 245, 247, 248].
Three studies, one performed using an NT2 ρ0 cell background and two performed using an SH-SY5Y ρ0 cell background, have reported mitochondria-relevant functional changes (including a reduction in COX activity) between cybrid lines generated from human subjects diagnosed with mild cognitive impairment (MCI; a frequent AD precursor state) and cybrids generated from age-matched control subjects [232, 246, 248]. To date, over 20 cybrid studies have been published that report at least one biochemical or molecular parameter that differs between groups of AD/MCI and control cybrid cell lines [227, 229–232, 234–252]. Most of these studies have in fact reported multiple divergent parameters. The only categorically negative AD cybrid study was that of Ito et al.[233], which evaluated just one biochemical parameter (COX activity), and which is notable for a variety of distinct methodologic differences that may have caused that study to differ from the other positive studies.
Implications and Limitations of AD Cybrid Studies
Data from AD cybrid studies support three important assumptions. First, they argue mtDNA accounts for, or at least to some extent contributes to, differences in mitochondrial function that reportedly exist between AD and non-AD subjects. Second, they argue that mitochondrial function can contribute to hallmark extra-mitochondrial histopathology changes, such as increases in oxidative stress markers [253] and Aβ production. Third, since cybrid cell lines in these studies have been almost exclusively generated through transfer of platelet mitochondria/mtDNA, the AD cybrid literature argues that at least at a molecular or biochemical level, AD changes are not strictly limited to the brain.
Cybrid studies also have limitations [205, 254]. Because mitochondria/mtDNA are transferred from platelets, it is possible that what drives mitochondrial dysfunction in AD cybrids is different from what drives mitochondrial dysfunction in AD brains. Given that the nature of mitochondrial dysfunction and its consequences seem to recapitulate so many biochemical, molecular, and physiologic features observed in AD brains, though [68], it would seem a stretch to propose that mitochondrial dysfunction in AD cybrids is entirely unrelated to mitochondrial dysfunction in AD brain.
The acceptor ρ0 cell lines are derived from tumor cell lines, which limits their ability to rigorously model the characteristics of a human brain. Therefore, when using cybrid cell lines to model AD mitochondrial functional defects, it is probably best not to extrapolate interpretations too far beyond the level of fundamental observation.
One general criticism of the cybrid approach is that although the approach was initially adapted to address questions about the contribution of mtDNA to mitochondrial function in AD, and subsequently used to model aspects of AD subject-specific mitochondrial function, cytosolic components other than mtDNA are transferred during the procedure. This is relevant because any transferred perpetuating component could theoretically lead to sustained changes in mitochondrial function. To date, though, no such component other than mtDNA has been identified. Also, since whole mitochondria are transferred from platelets to acceptor cells, and platelet mitochondria in AD show unique biochemical characteristics, the possibility that a pre-existing mitochondrial defect simply did not have adequate time to wash out requires consideration. This possibility was empirically addressed in one of the early AD cybrid studies, which found that the six-week selection and expansion period later used in most of the AD cybrid studies appears to provide an adequate wash-out period [230]. Moreover, one study reported that with extended time in active culture, a number of altered mitochondrial functional parameters appeared to become more profound [252]. This occurred despite an apparent activation of compensatory adaptations.
While cybrid studies implicate an mtDNA contribution to AD-associated mitochondrial dysfunction, they do not indicate what specific mtDNA features, characteristics, or sequences contribute to that dysfunction. They could reflect an accumulation of somatic mutations; to date data pertinent to the question of whether putative somatic mutations accumulate in circulating blood cells is mixed. One study did not find an obvious age-related accumulation of large deletions in a general population sample [255], which suggests large mtDNA deletions do not commonly accumulate in circulating cells as they do in the brain. Data addressing the question of whether somatic point mutations accumulate in AD subject blood cells do, however, suggest this may indeed occur [97, 98, 179].
Alternatively, it is possible that inherited mtDNA sequence differences may partly or exclusively contribute to AD cybrid line differences in mitochondrial function. Currently, the nature of such potential inherited mtDNA changes, if they in fact exist, is not clear. MtDNA sequence studies performed on AD thus far have not identified a single mutation or variant that distinguishes AD-affected from AD-unaffected individuals, although various studies have claimed common inherited mtDNA signatures, such as those defined by the different mtDNA haplogroups, influence AD risk [113]. No mtDNA study to date has definitively addressed the question of whether rare mtDNA mutations or sequence variants associate with AD; such studies would require extensive mtDNA sequence data from large numbers of AD and control subjects.
Finally, a considerable degree of polymorphic variation has been demonstrated to exist within human nuclear respiratory chain subunits [256]. Because the cybrid approach neutralizes the potential functional contributions of a subject’s nuclear DNA make-up, valuable information regarding the interplay between that individual’s nuclear and mitochondrial genomes is likely to be lost. Substituting the nuclear background of a ρ0 cell for the nuclear background of the mtDNA donor could theoretically magnify or mitigate a particular mtDNA sequence-associated functional consequence.
The Mitochondrial Cascade Hypothesis
Despite the aforementioned limitations of AD cybrid studies, it is tempting to try and integrate AD cybrid data with data generated from AD tissue, epidemiology, endophenotype, and genetic studies in order to define an overarching hypothesis of AD etiology. Because advancing age is the single greatest sporadic AD risk factor [8], it is recommended that any resulting hypothesis take into account applicable data and conceptual constructs generated through primary studies of human and animal aging.
A number of investigators have proposed mitochondrial dysfunction could play an important role in AD[21, 27, 62, 64, 69, 257–268]. Some have proposed mtDNA inheritance or an age-related somatic a ccumulation of mtDNA mutation could contribute [21, 226]. When these conceptual constructs are considered in conjunction with the observation that specific differences in non-brain mitochondrial function seem to exist between AD and non-AD subjects, and that differences in mitochondrial function and cell bioenergetics can influence AD histopathology changes, the case can be made that perhaps mitochondria and bioenergetic function play an upstream if not primary role in AD.
The “mitochondrial cascade hypothesis” represents one attempt to comprehensively synthesize data discussed throughout this chapter into a comprehensive hypothesis that tries to account for the development and progression of AD, as well as the relationship of AD to brain aging (Figure 4)[269–272]. The premise is that individuals inherit a baseline level of mitochondrial function and durability; this baseline is determined by genetic contributions from both parents, although the mother makes a greater contribution and has a greater influence because she contributes the mitochondrial genome [273]. Then, as the individual ages certain tissues, and especially the brain, either accumulates somatic mutations or else drifts towards increased levels of inherited microheteroplasmic mutations. The rate at which mutations accumulate would presumably be influenced by the individual’s genetic make-up, and also by lifestyle factors. These age-related changes would result in declining mitochondrial function, which up to a point could probably be accommodated and addressed through adaptive molecular changes, and thereby define a period of compensated aging. Eventually, though, a threshold of mitochondrial/bioenergetic dysfunction could be reached in which accommodation and compensation are no longer adequate, thus introducing a period of uncompensated brain aging. At this point, an AD phenotype would begin to emerge.
Figure 4. The AD mitochondrial cascade hypothesis.

Inheritance from both parents determine an individual’s bioenergetic set-point and durability, with the mother having the greater input due to her contribution of the mtDNA. Over time mitochondrial efficiency declines, likely due to accumulating damage to the mtDNA. At relatively low levels it is possible to compensate for this change (compensated brain aging), although the compensatory process may itself have consequences. More profound declines in mitochondria function, which may occur as further damage accumulates, can lead to a stage of uncompensated brain aging, which associates with other consequences as well as symptomatic AD.
The mitochondrial cascade hypothesis further presumes the classic histologic features of AD, including Aβ plaque and tau neurofibrillary tangle deposition, are a downstream consequence of changing mitochondrial and bioenergetic function. In its original form the hypothesis speculated Aβ plaques accumulated only after the state of uncompensated brain aging was reached [271]. Recent neuroimaging data from a human cohort longitudinal study now shows, though, that most Aβ plaque deposition occurs during the run-up to the AD clinical syndrome and dramatically slows during the symptomatic stages [274, 275]. In conjunction with experimental studies that show Aβ is generated as a byproduct of synaptic activity [191, 192], this suggests the possibility that Aβ plaque deposition may primarily represent a byproduct of the compensatory changes that accompany age-related mitochondrial functional declines. If correct, this modification to the mitochondrial cascade hypothesis could potentially account for why a substantial percentage of older adults develop Aβ plaques but do not develop the clinical disease [8]; such individuals may avoid making the transition from bioenergetically compensated to uncompensated brain aging.
Conclusions
Decades ago the aging field began to specifically postulate mitochondria and bioenergetic function contributed to aging [5, 19–21]. This was originally predicated on correlative and descriptive data. More recent experimental data have emerged, though, that are consistent with this possibility [22, 23].
Over an almost five-decade period it has become increasingly clear that bioenergetic and mitochondrial physical and functional changes also occur in AD[21, 27, 62, 64, 69, 257–268]. In many cases changes observed in AD are reminiscent of those seen in aging, and in some ways differ primarily in their magnitude [68]. While mitochondrial and bioenergetic changes in AD were initially felt to represent a consequence of the disease, their potential relevance to disease progression has increasingly been considered, and the view that such changes represent valid therapeutic targets has emerged [276–279].
When considering the hierarchy of biochemical, molecular, and physiologic events that result in AD, some have pointed out that in AD subjects bioenergetic and mitochondrial differences are found outside the brain, and that changes in bioenergetic and mitochondrial function can alter how cells and tissues handle other AD phenomena, including how APP is processed to Aβ and Aβ plaque deposition [9, 280]. Additional data pertinent to these points have been reported from studies of cybrid cell lines generated through the transfer of AD subject platelet mitochondria/mtDNA to ρ0 cell lines; results from these studies are consistent with the view that mtDNA contributes at least in part to AD mitochondrial and bioenergetic changes, and that these changes can drive or at least contribute to a variety of biochemical, molecular, and histologic phenomena observed in AD subject brains [68].
Synthesizing a spectrum of data from the aging, AD, and cybrid literature supports a conceptual construct that places mitochondrial function and bioenergetics at the apex of AD-associated molecular changes [269–272]. MtDNA would to some extent influence relevant mitochondrial and bioenergetic functional parameters. These molecular changes would similarly play out during the basic process of aging, and in some cases differences observed in both aging and AD would differ mostly by degree, with deficits being more prominent when clinical AD is present. Under this scenario some of the key histologic changes we now associate with AD, such as processing of APP to Aβ and Aβ plaque deposition, would represent downstream consequences of altered mitochondrial and bioenergetic function. Some of these histologic changes may arise during stages where declining brain mitochondrial and bioenergetic function could still be accommodated and compensated for, or could arise after declining brain mitochondrial and bioenergetic function have surpassed a critical level at which point successful compensation is no longer possible. In the brain, classic AD histology changes initiated by mitochondrial and bioenergetic dysfunction could in turn exacerbate failing mitochondrial and bioenergetic function. This mitochondrial cascade hypothesis makes testable predictions and suggests particular therapeutic strategies may be worth pursuing. It will be interesting to see how well the mitochondrial cascade hypothesis absorbs new current and future data generated by the AD research field.
Acknowledgments
Supported in part by the University of Kansas Alzheimer’s Disease Center (P30 AG035982).
References
- 1.Lewis MR, Lewis WH. MITOCHONDRIA IN TISSUE CULTURE. Science (New York, NY) 1914;39:330–333. doi: 10.1126/science.39.1000.330. [DOI] [PubMed] [Google Scholar]
- 2.Nass MM, Nass S. INTRAMITOCHONDRIAL FIBERS WITH DNA CHARACTERISTICS. I. FIXATION AND ELECTRON STAINING REACTIONS. The Journal of cell biology. 1963;19:593–611. doi: 10.1083/jcb.19.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nass S, Nass MM. INTRAMITOCHONDRIAL FIBERS WITH DNA CHARACTERISTICS. II. ENZYMATIC AND OTHER HYDROLYTIC TREATMENTS. The Journal of cell biology. 1963;19:613–629. doi: 10.1083/jcb.19.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell P, Moyle J. Chemiosmotic hypothesis of oxidative phosphorylation. Nature. 1967;213:137–139. doi: 10.1038/213137a0. [DOI] [PubMed] [Google Scholar]
- 5.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muller FL, Lustgarten MS, Jang Y, Richardson A, Van Remmen H. Trends in oxidative aging theories. Free radical biology & medicine. 2007;43:477–503. doi: 10.1016/j.freeradbiomed.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 8.Swerdlow RH. Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging? Neurobiology of aging. 2007;28:1465–1480. doi: 10.1016/j.neurobiolaging.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 9.Swerdlow RH, Kish SJ. Mitochondria in Alzheimer’s disease. Int Rev Neurobiology. 2002;53:341–385. doi: 10.1016/s0074-7742(02)53013-0. [DOI] [PubMed] [Google Scholar]
- 10.Swerdlow RH. The neurodegenerative mitochondriopathies. Journal of Alzheimer’s disease: JAD. 2009;17:737–751. doi: 10.3233/JAD-2009-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osborne TB, Mendel LB, Ferry EL. THE EFFECT OF RETARDATION OF GROWTH UPON THE BREEDING PERIOD AND DURATION OF LIFE OF RATS. Science (New York, NY) 1917;45:294–295. doi: 10.1126/science.45.1160.294. [DOI] [PubMed] [Google Scholar]
- 12.McCay CM, Crowell MF, Maynard LA. The effect of retarded growth upon the length of life and upon the ultimate body size. J Nutr. 1935;10:63–79. [PubMed] [Google Scholar]
- 13.McCay CM, Maynard LA, Sperling G, Barnes LL. Retarded growth, lifespan, ultimate body size, and age changes in the albino rat after feeding diets restricted in calories. J Nutr. 1937;18:1–13. doi: 10.1111/j.1753-4887.1975.tb05227.x. [DOI] [PubMed] [Google Scholar]
- 14.Carlson AJ, Hoelzel F. Apparent prolongation of the lifespan of rats by intermittent fasting. The Journal of nutrition. 1946;31:363–375. doi: 10.1093/jn/31.3.363. [DOI] [PubMed] [Google Scholar]
- 15.Masoro EJ. Subfield history: caloric restriction, slowing aging, and extending life. Science of aging knowledge environment: SAGE KE. 2003;2003:Re2. doi: 10.1126/sageke.2003.8.re2. [DOI] [PubMed] [Google Scholar]
- 16.Rubner M. Das Problem deer Lebensdauer und seine Bezieung zu Wachstum und Ernehrung. Oldenburg, Munich: 1908. The problem of longevity and its relation to growth and nutrition. [Google Scholar]
- 17.Pearl R. Being an Account of Some Experimental Studies on the Biology of Life Duration. New York: Alfred A. Knopf; 1928. The Rate of Living. [Google Scholar]
- 18.Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- 19.Miquel J, Economos AC, Fleming J, Johnson JE., Jr Mitochondrial role in cell aging. Exp Gerontol. 1980;15:575–591. doi: 10.1016/0531-5565(80)90010-8. [DOI] [PubMed] [Google Scholar]
- 20.Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet (London, England) 1989;1:642–645. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 21.Wallace DC. Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science (New York, NY) 1992;256:628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 22.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 23.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science (New York, NY) 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 24.Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- 25.Boveris A, Navarro A. Brain mitochondrial dysfunction in aging. IUBMB Life. 2008;60:308–314. doi: 10.1002/iub.46. [DOI] [PubMed] [Google Scholar]
- 26.Barrientos A, Casademont J, Cardellach F, Estivill X, Urbano-Marquez A, Nunes V. Reduced steady-state levels of mitochondrial RNA and increased mitochondrial DNA amount in human brain with aging. Brain Res Mol Brain Res. 1997;52:284–289. doi: 10.1016/s0169-328x(97)00278-7. [DOI] [PubMed] [Google Scholar]
- 27.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21:3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stauch KL, Purnell PR, Fox HS. Aging synaptic mitochondria exhibit dynamic proteomic changes while maintaining bioenergetic function. Aging. 2014;6:320–334. doi: 10.18632/aging.100657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science (New York, NY) 2011;333:1109–1112. doi: 10.1126/science.1201940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stauch KL, Purnell PR, Villeneuve LM, Fox HS. Proteomic analysis and functional characterization of mouse brain mitochondria during aging reveal alterations in energy metabolism. Proteomics. 2015;15:1574–1586. doi: 10.1002/pmic.201400277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poon HF, Vaishnav RA, Getchell TV, Getchell ML, Butterfield DA. Quantitative proteomics analysis of differential protein expression and oxidative modification of specific proteins in the brains of old mice. Neurobiology of aging. 2006;27:1010–1019. doi: 10.1016/j.neurobiolaging.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 32.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 33.Finkel T. Reactive oxygen species and signal transduction. IUBMB Life. 2001;52:3–6. doi: 10.1080/15216540252774694. [DOI] [PubMed] [Google Scholar]
- 34.Haynes CM, Fiorese CJ, Lin YF. Evaluating and responding to mitochondrial dysfunction: the mitochondrial unfolded-protein response and beyond. Trends in cell biology. 2013;23:311–318. doi: 10.1016/j.tcb.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kotiadis VN, Duchen MR, Osellame LD. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochimica et biophysica acta. 2014;1840:1254–1265. doi: 10.1016/j.bbagen.2013.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007;6:280–293. doi: 10.1016/j.cmet.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 37.Gomez-Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, Sastre J, Vina J. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am J Clin Nutr. 2008;87:142–149. doi: 10.1093/ajcn/87.1.142. [DOI] [PubMed] [Google Scholar]
- 38.Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Payne BA, Wilson IJ, Yu-Wai-Man P, Coxhead J, Deehan D, Horvath R, Taylor RW, Samuels DC, Santibanez-Koref M, Chinnery PF. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2013;22:384–390. doi: 10.1093/hmg/dds435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye K, Lu J, Ma F, Keinan A, Gu Z. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:10654–10659. doi: 10.1073/pnas.1403521111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- 42.Bohr VA, Dianov GL. Oxidative DNA damage processing in nuclear and mitochondrial DNA. Biochimie. 1999;81:155–160. doi: 10.1016/s0300-9084(99)80048-0. [DOI] [PubMed] [Google Scholar]
- 43.Hudson EK, Hogue BA, Souza-Pinto NC, Croteau DL, Anson RM, Bohr VA, Hansford RG. Age-associated change in mitochondrial DNA damage. Free radical research. 1998;29:573–579. doi: 10.1080/10715769800300611. [DOI] [PubMed] [Google Scholar]
- 44.Richter C. Reactive oxygen and DNA damage in mitochondria. Mutation research. 1992;275:249–255. doi: 10.1016/0921-8734(92)90029-o. [DOI] [PubMed] [Google Scholar]
- 45.Nie B, Gan W, Shi F, Hu GX, Chen LG, Hayakawa H, Sekiguchi M, Cai JP. Age-dependent accumulation of 8-oxoguanine in the DNA and RNA in various rat tissues. Oxidative medicine and cellular longevity. 2013;2013:303181. doi: 10.1155/2013/303181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nature genetics. 1992;2:324–329. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- 47.Brand FN, Kiely DK, Kannel WB, Myers RH. Family patterns of coronary heart disease mortality: the Framingham Longevity Study. J Clin Epidemiol. 1992;45:169–174. doi: 10.1016/0895-4356(92)90009-c. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka M, Gong JS, Zhang J, Yoneda M, Yagi K. Mitochondrial genotype associated with longevity. Lancet (London, England) 1998;351:185–186. doi: 10.1016/S0140-6736(05)78211-8. [DOI] [PubMed] [Google Scholar]
- 49.Ross OA, McCormack R, Curran MD, Duguid RA, Barnett YA, Rea IM, Middleton D. Mitochondrial DNA polymorphism: its role in longevity of the Irish population. Experimental gerontology. 2001;36:1161–1178. doi: 10.1016/s0531-5565(01)00094-8. [DOI] [PubMed] [Google Scholar]
- 50.Ivanova R, Lepage V, Charron D, Schachter F. Mitochondrial genotype associated with French Caucasian centenarians. Gerontology. 1998;44:349. doi: 10.1159/000022041. [DOI] [PubMed] [Google Scholar]
- 51.Torroni A, Huoponen K, Francalacci P, Petrozzi M, Morelli L, Scozzari R, Obinu D, Savontaus ML, Wallace DC. Classification of European mtDNAs from an analysis of three European populations. Genetics. 1996;144:1835–1850. doi: 10.1093/genetics/144.4.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science (New York, NY) 2004;303:223–226. doi: 10.1126/science.1088434. [DOI] [PubMed] [Google Scholar]
- 53.De Benedictis G, Rose G, Carrieri G, De Luca M, Falcone E, Passarino G, Bonafe M, Monti D, Baggio G, Bertolini S, Mari D, Mattace R, Franceschi C. Mitochondrial DNA inherited variants are associated with successful aging and longevity in humans. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1999;13:1532–1536. doi: 10.1096/fasebj.13.12.1532. [DOI] [PubMed] [Google Scholar]
- 54.Niemi AK, Hervonen A, Hurme M, Karhunen PJ, Jylha M, Majamaa K. Mitochondrial DNA polymorphisms associated with longevity in a Finnish population. Human genetics. 2003;112:29–33. doi: 10.1007/s00439-002-0843-y. [DOI] [PubMed] [Google Scholar]
- 55.Khrapko K, Vijg J. Mitochondrial DNA mutations and aging: devils in the details? Trends in genetics: TIG. 2009;25:91–98. doi: 10.1016/j.tig.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nature genetics. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 57.Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nature genetics. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- 58.Kukat A, Trifunovic A. Somatic mtDNA mutations and aging--facts and fancies. Experimental gerontology. 2009;44:101–105. doi: 10.1016/j.exger.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 59.Hughes BG, Hekimi S. A mild impairment of mitochondrial electron transport has sex-specific effects on lifespan and aging in mice. PloS one. 2011;6:e26116. doi: 10.1371/journal.pone.0026116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foster NL, Chase TN, Fedio P, Patronas NJ, Brooks RA, Di Chiro G. Alzheimer’s disease: focal cortical changes shown by positron emission tomography. Neurology. 1983;33:961–965. doi: 10.1212/wnl.33.8.961. [DOI] [PubMed] [Google Scholar]
- 61.de Leon MJ, Ferris SH, George AE, Christman DR, Fowler JS, Gentes C, Reisberg B, Gee B, Emmerich M, Yonekura Y, Brodie J, Kricheff, Wolf AP. Positron emission tomographic studies of aging and Alzheimer disease. AJNR Am J Neuroradiol. 1983;4:568–571. [PMC free article] [PubMed] [Google Scholar]
- 62.Swerdlow R, Marcus DL, Landman J, Kooby D, Frey W, 2nd, Freedman ML. Brain glucose metabolism in Alzheimer’s disease. Am J Med Sci. 1994;308:141–144. doi: 10.1097/00000441-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 63.Baloyannis SJ. Mitochondrial alterations in Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2006;9:119–126. doi: 10.3233/jad-2006-9204. [DOI] [PubMed] [Google Scholar]
- 64.Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, Perrino P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Archives of neurology. 1988;45:836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
- 65.Gibson GE, Sheu KF, Blass JP. Abnormalities of mitochondrial enzymes in Alzheimer disease. J Neural Transm. 1998;105:855–870. doi: 10.1007/s007020050099. [DOI] [PubMed] [Google Scholar]
- 66.Sorbi S, Bird ED, Blass JP. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol. 1983;13:72–78. doi: 10.1002/ana.410130116. [DOI] [PubMed] [Google Scholar]
- 67.Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF. Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochimica et biophysica acta. 2010;1802:122–134. doi: 10.1016/j.bbadis.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Swerdlow RH. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid Redox Signal. 2012;16:1434–1455. doi: 10.1089/ars.2011.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Parker WD, Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology. 1990;40:1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 70.Bosetti F, Brizzi F, Barogi S, Mancuso M, Siciliano G, Tendi EA, Murri L, Rapoport SI, Solaini G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiology of aging. 2002;23:371–376. doi: 10.1016/s0197-4580(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 71.Mancuso M, Filosto M, Bosetti F, Ceravolo R, Rocchi A, Tognoni G, Manca ML, Solaini G, Siciliano G, Murri L. Decreased platelet cytochrome c oxidase activity is accompanied by increased blood lactate concentration during exercise in patients with Alzheimer disease. Experimental neurology. 2003;182:421–426. doi: 10.1016/s0014-4886(03)00092-x. [DOI] [PubMed] [Google Scholar]
- 72.Cardoso SM, Proenca MT, Santos S, Santana I, Oliveira CR. Cytochrome c oxidase is decreased in Alzheimer’s disease platelets. Neurobiology of aging. 2004;25:105–110. doi: 10.1016/s0197-4580(03)00033-2. [DOI] [PubMed] [Google Scholar]
- 73.Parker WD, Jr, Mahr NJ, Filley CM, Parks JK, Hughes D, Young DA, Cullum CM. Reduced platelet cytochrome c oxidase activity in Alzheimer’s disease. Neurology. 1994;44:1086–1090. doi: 10.1212/wnl.44.6.1086. [DOI] [PubMed] [Google Scholar]
- 74.Valla J, Schneider L, Niedzielko T, Coon KD, Caselli R, Sabbagh MN, Ahern GL, Baxter L, Alexander G, Walker DG, Reiman EM. Impaired platelet mitochondrial activity in Alzheimer’s disease and mild cognitive impairment. Mitochondrion. 2006;6:323–330. doi: 10.1016/j.mito.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kish SJ, Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, Chang LJ, Wilson JM, DiStefano LM, Nobrega JN. Brain cytochrome oxidase in Alzheimer’s disease. Journal of neurochemistry. 1992;59:776–779. doi: 10.1111/j.1471-4159.1992.tb09439.x. [DOI] [PubMed] [Google Scholar]
- 76.Parker WD, Jr, Parks J, Filley CM, Kleinschmidt-DeMasters BK. Electron transport chain defects in Alzheimer’s disease brain. Neurology. 1994;44:1090–1096. doi: 10.1212/wnl.44.6.1090. [DOI] [PubMed] [Google Scholar]
- 77.Mutisya EM, Bowling AC, Beal MF. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. Journal of neurochemistry. 1994;63:2179–2184. doi: 10.1046/j.1471-4159.1994.63062179.x. [DOI] [PubMed] [Google Scholar]
- 78.Valla J, Berndt JD, Gonzalez-Lima F. Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: superficial laminar cytochrome oxidase associated with disease duration. J Neurosci. 2001;21:4923–4930. doi: 10.1523/JNEUROSCI.21-13-04923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chagnon P, Betard C, Robitaille Y, Cholette A, Gauvreau D. Distribution of brain cytochrome oxidase activity in various neurodegenerative diseases. Neuroreport. 1995;6:711–715. doi: 10.1097/00001756-199503270-00002. [DOI] [PubMed] [Google Scholar]
- 80.Maurer I, Zierz S, Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiology of aging. 2000;21:455–462. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 81.Simonian NA, Hyman BT. Functional alterations in Alzheimer’s disease: diminution of cytochrome oxidase in the hippocampal formation. J Neuropathol Exp Neurol. 1993;52:580–585. [PubMed] [Google Scholar]
- 82.Verwer RW, Jansen KA, Sluiter AA, Pool CW, Kamphorst W, Swaab DF. Decreased hippocampal metabolic activity in Alzheimer patients is not reflected in the immunoreactivity of cytochrome oxidase subunits. Experimental neurology. 2000;163:440–451. doi: 10.1006/exnr.2000.7385. [DOI] [PubMed] [Google Scholar]
- 83.Wong-Riley M, Antuono P, Ho KC, Egan R, Hevner R, Liebl W, Huang Z, Rachel R, Jones J. Cytochrome oxidase in Alzheimer’s disease: biochemical, histochemical, and immunohistochemical analyses of the visual and other systems. Vision Res. 1997;37:3593–3608. doi: 10.1016/S0042-6989(96)00210-6. [DOI] [PubMed] [Google Scholar]
- 84.Parker WD, Jr, Parks JK. Cytochrome c oxidase in Alzheimer’s disease brain: purification and characterization. Neurology. 1995;45:482–486. doi: 10.1212/wnl.45.3.482. [DOI] [PubMed] [Google Scholar]
- 85.Curti D, Rognoni F, Gasparini L, Cattaneo A, Paolillo M, Racchi M, Zani L, Bianchetti A, Trabucchi M, Bergamaschi S, Govoni S. Oxidative metabolism in cultured fibroblasts derived from sporadic Alzheimer’s disease (AD) patients. Neurosci Lett. 1997;236:13–16. doi: 10.1016/s0304-3940(97)00741-6. [DOI] [PubMed] [Google Scholar]
- 86.Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet. 2011;20:2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. Journal of neurochemistry. 2012;120:419–429. doi: 10.1111/j.1471-4159.2011.07581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, Pasinetti GM. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Archives of neurology. 2009;66:352–361. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang X, Su B, Fujioka H, Zhu X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am J Pathol. 2008;173:470–482. doi: 10.2353/ajpath.2008.071208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de la Monte SM, Luong T, Neely TR, Robinson D, Wands JR. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Lab Invest. 2000;80:1323–1335. doi: 10.1038/labinvest.3780140. [DOI] [PubMed] [Google Scholar]
- 92.Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rodriguez-Santiago B, Casademont J, Nunes V. Is mitochondrial DNA depletion involved in Alzheimer’s disease? Eur J Hum Genet. 2001;9:279–285. doi: 10.1038/sj.ejhg.5200629. [DOI] [PubMed] [Google Scholar]
- 94.Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, McKee AC, Beal MF, Graham BH, Wallace DC. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics. 1994;23:471–476. doi: 10.1006/geno.1994.1525. [DOI] [PubMed] [Google Scholar]
- 95.Chang SW, Zhang D, Chung HD, Zassenhaus HP. The frequency of point mutations in mitochondrial DNA is elevated in the Alzheimer’s brain. Biochem Biophys Res Commun. 2000;273:203–208. doi: 10.1006/bbrc.2000.2885. [DOI] [PubMed] [Google Scholar]
- 96.Hamblet NS, Castora FJ. Elevated levels of the Kearns-Sayre syndrome mitochondrial DNA deletion in temporal cortex of Alzheimer’s patients. Mutation research. 1997;379:253–262. doi: 10.1016/s0027-5107(97)00158-9. [DOI] [PubMed] [Google Scholar]
- 97.Casoli T, Di Stefano G, Spazzafumo L, Balietti M, Giorgetti B, Giuli C, Postacchini D, Fattoretti P, Conti F. Contribution of non-reference alleles in mtDNA of Alzheimer’s disease patients. Annals of clinical and translational neurology. 2014;1:284–289. doi: 10.1002/acn3.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Casoli T, Spazzafumo L, Di Stefano G, Conti F. Role of diffuse low-level heteroplasmy of mitochondrial DNA in Alzheimer’s disease neurodegeneration. Frontiers in aging neuroscience. 2015;7:142. doi: 10.3389/fnagi.2015.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 100.Egensperger R, Kosel S, Schnopp NM, Mehraein P, Graeber MB. Association of the mitochondrial tRNA(A4336G) mutation with Alzheimer’s and Parkinson’s diseases. Neuropathol Appl Neurobiol. 1997;23:315–321. [PubMed] [Google Scholar]
- 101.Fesahat F, Houshmand M, Panahi MS, Gharagozli K, Mirzajani F. Do haplogroups H and U act to increase the penetrance of Alzheimer’s disease? Cell Mol Neurobiol. 2007;27:329–334. doi: 10.1007/s10571-006-9126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hutchin T, Cortopassi G. A mitochondrial DNA clone is associated with increased risk for Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:6892–6895. doi: 10.1073/pnas.92.15.6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ienco EC, Simoncini C, Orsucci D, Petrucci L, Filosto M, Mancuso M, Siciliano G. May “mitochondrial eve” and mitochondrial haplogroups play a role in neurodegeneration and Alzheimer’s disease? Int J Alzheimers Dis. 2011;2011:709061. doi: 10.4061/2011/709061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lakatos A, Derbeneva O, Younes D, Keator D, Bakken T, Lvova M, Brandon M, Guffanti G, Reglodi D, Saykin A, Weiner M, Macciardi F, Schork N, Wallace DC, Potkin SG. Association between mitochondrial DNA variations and Alzheimer’s disease in the ADNI cohort. Neurobiol Aging. 2010;31:1355–1363. doi: 10.1016/j.neurobiolaging.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Maruszak A, Canter JA, Styczynska M, Zekanowski C, Barcikowska M. Mitochondrial haplogroup H and Alzheimer’s disease--is there a connection? Neurobiology of aging. 2009;30:1749–1755. doi: 10.1016/j.neurobiolaging.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 106.Maruszak A, Safranow K, Branicki W, Gaweda-Walerych K, Pospiech E, Gabryelewicz T, Canter JA, Barcikowska M, Zekanowski C. The impact of mitochondrial and nuclear DNA variants on late-onset Alzheimer’s disease risk. Journal of Alzheimer’s disease: JAD. 2011;27:197–210. doi: 10.3233/JAD-2011-110710. [DOI] [PubMed] [Google Scholar]
- 107.Santoro A, Balbi V, Balducci E, Pirazzini C, Rosini F, Tavano F, Achilli A, Siviero P, Minicuci N, Bellavista E, Mishto M, Salvioli S, Marchegiani F, Cardelli M, Olivieri F, Nacmias B, Chiamenti AM, Benussi L, Ghidoni R, Rose G, Gabelli C, Binetti G, Sorbi S, Crepaldi G, Passarino G, Torroni A, Franceschi C. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. PloS one. 2010;5:e12037. doi: 10.1371/journal.pone.0012037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Shoffner JM, Brown MD, Torroni A, Lott MT, Cabell MF, Mirra SS, Beal MF, Yang CC, Gearing M, Salvo R, et al. Mitochondrial DNA variants observed in Alzheimer disease and Parkinson disease patients. Genomics. 1993;17:171–184. doi: 10.1006/geno.1993.1299. [DOI] [PubMed] [Google Scholar]
- 109.Takasaki S. Mitochondrial haplogroups associated with Japanese Alzheimer’s patients. J Bioenerg Biomembr. 2009;41:407–410. doi: 10.1007/s10863-009-9240-8. [DOI] [PubMed] [Google Scholar]
- 110.Tanaka N, Goto Y, Akanuma J, Kato M, Kinoshita T, Yamashita F, Tanaka M, Asada T. Mitochondrial DNA variants in a Japanese population of patients with Alzheimer’s disease. Mitochondrion. 2010;10:32–37. doi: 10.1016/j.mito.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 111.van der Walt JM, Dementieva YA, Martin ER, Scott WK, Nicodemus KK, Kroner CC, Welsh-Bohmer KA, Saunders AM, Roses AD, Small GW, Schmechel DE, Murali Doraiswamy P, Gilbert JR, Haines JL, Vance JM, Pericak-Vance MA. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci Lett. 2004;365:28–32. doi: 10.1016/j.neulet.2004.04.051. [DOI] [PubMed] [Google Scholar]
- 112.Ridge PG, Koop A, Maxwell TJ, Bailey MH, Swerdlow RH, Kauwe JS, Honea RA. Mitochondrial haplotypes associated with biomarkers for Alzheimer’s disease. PloS one. 2013;8:e74158. doi: 10.1371/journal.pone.0074158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang Y, Brinton RD. Triad of risk for late onset Alzheimer’s: Mitochondrial haplotype, APOE genotype and chromosomal sex. Frontiers Aging Neuroscience. 2016 doi: 10.3389/fnagi.2016.00232. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chinnery PF, Taylor GA, Howell N, Andrews RM, Morris CM, Taylor RW, McKeith IG, Perry RH, Edwardson JA, Turnbull DM. Mitochondrial DNA haplogroups and susceptibility to AD and dementia with Lewy bodies. Neurology. 2000;55:302–304. doi: 10.1212/wnl.55.2.302. [DOI] [PubMed] [Google Scholar]
- 115.Edland SD, Tobe VO, Rieder MJ, Bowen JD, McCormick W, Teri L, Schellenberg GD, Larson EB, Nickerson DA, Kukull WA. Mitochondrial genetic variants and Alzheimer disease: a case-control study of the T4336C and G5460A variants. Alzheimer Dis Assoc Disord. 2002;16:1–7. doi: 10.1097/00002093-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 116.Rodriguez Santiago B, Casademont J, Nunes V. Is there a relation between Alzheimers disease and defects of mitochondrial DNA? Rev Neurol. 2001;33:301–305. [PubMed] [Google Scholar]
- 117.Wragg MA, Talbot CJ, Morris JC, Lendon CL, Goate AM. No association found between Alzheimer’s disease and a mitochondrial tRNA glutamine gene variant. Neurosci Lett. 1995;201:107–110. doi: 10.1016/0304-3940(95)12146-3. [DOI] [PubMed] [Google Scholar]
- 118.Zsurka G, Kalman J, Csaszar A, Rasko I, Janka Z, Venetianer P. No mitochondrial haplotype was found to increase risk for Alzheimer’s disease. Biol Psychiatry. 1998;44:371–373. doi: 10.1016/s0006-3223(97)00461-7. [DOI] [PubMed] [Google Scholar]
- 119.Caspersen C, Wang N, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SD. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 120.Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science (New York, NY) 2004;304:448–452. doi: 10.1126/science.1091230. [DOI] [PubMed] [Google Scholar]
- 121.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 122.Crouch PJ, Blake R, Duce JA, Ciccotosto GD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad B, Glaser E, Ankarcrona M. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dragicevic N, Mamcarz M, Zhu Y, Buzzeo R, Tan J, Arendash GW, Bradshaw PC. Mitochondrial amyloid-beta levels are associated with the extent of mitochondrial dysfunction in different brain regions and the degree of cognitive impairment in Alzheimer’s transgenic mice. Journal of Alzheimer’s disease: JAD. 2010;20(Suppl 2):S535–550. doi: 10.3233/JAD-2010-100342. [DOI] [PubMed] [Google Scholar]
- 126.Yamaguchi H, Yamazaki T, Ishiguro K, Shoji M, Nakazato Y, Hirai S. Ultrastructural localization of Alzheimer amyloid beta/A4 protein precursor in the cytoplasm of neurons and senile plaque-associated astrocytes. Acta Neuropathol. 1992;85:15–22. doi: 10.1007/BF00304629. [DOI] [PubMed] [Google Scholar]
- 127.Yao J, Du H, Yan S, Fang F, Wang C, Lue LF, Guo L, Chen D, Stern DM, Gunn Moore FJ, Xi Chen J, Arancio O, Yan SS. Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31:2313–2320. doi: 10.1523/JNEUROSCI.4717-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Canevari L, Clark JB, Bates TE. beta-Amyloid fragment 25–35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett. 1999;457:131–134. doi: 10.1016/s0014-5793(99)01028-5. [DOI] [PubMed] [Google Scholar]
- 129.Casley CS, Canevari L, Land JM, Clark JB, Sharpe MA. Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. Journal of neurochemistry. 2002;80:91–100. doi: 10.1046/j.0022-3042.2001.00681.x. [DOI] [PubMed] [Google Scholar]
- 130.Parks JK, Smith TS, Trimmer PA, Bennett JP, Jr, Parker WD., Jr Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. Journal of neurochemistry. 2001;76:1050–1056. doi: 10.1046/j.1471-4159.2001.00112.x. [DOI] [PubMed] [Google Scholar]
- 131.Pereira C, Santos MS, Oliveira C. Mitochondrial function impairment induced by amyloid beta-peptide on PC12 cells. Neuroreport. 1998;9:1749–1755. doi: 10.1097/00001756-199806010-00015. [DOI] [PubMed] [Google Scholar]
- 132.Cardoso SM, Santos S, Swerdlow RH, Oliveira CR. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2001;15:1439–1441. doi: 10.1096/fj.00-0561fje. [DOI] [PubMed] [Google Scholar]
- 133.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. The Journal of cell biology. 2003;161:41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Anandatheerthavarada HK, Devi L. Amyloid precursor protein and mitochondrial dysfunction in Alzheimer’s disease. Neuroscientist. 2007;13:626–638. doi: 10.1177/1073858407303536. [DOI] [PubMed] [Google Scholar]
- 136.Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lin MT, Beal MF. Alzheimer’s APP mangles mitochondria. Nat Med. 2006;12:1241–1243. doi: 10.1038/nm1106-1241. [DOI] [PubMed] [Google Scholar]
- 138.Bassett SS, Avramopoulos D, Fallin D. Evidence for parent of origin effect in late-onset Alzheimer disease. Am J Med Genet. 2002;114:679–686. doi: 10.1002/ajmg.10648. [DOI] [PubMed] [Google Scholar]
- 139.Duara R, Lopez-Alberola RF, Barker WW, Loewenstein DA, Zatinsky M, Eisdorfer CE, Weinberg GB. A comparison of familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1377–1384. doi: 10.1212/wnl.43.7.1377. [DOI] [PubMed] [Google Scholar]
- 140.Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC. Increased risk of dementia in mothers of Alzheimer’s disease cases: evidence for maternal inheritance. Neurology. 1996;47:254–256. doi: 10.1212/wnl.47.1.254. [DOI] [PubMed] [Google Scholar]
- 141.Mosconi L, Berti V, Swerdlow RH, Pupi A, Duara R, de Leon M. Maternal transmission of Alzheimer’s disease: prodromal metabolic phenotype and the search for genes. Hum Genomics. 2010;4:170–193. doi: 10.1186/1479-7364-4-3-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mosconi L, Brys M, Switalski R, Mistur R, Glodzik L, Pirraglia E, Tsui W, De Santi S, de Leon MJ. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:19067–19072. doi: 10.1073/pnas.0705036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Okonkwo OC, Xu G, Oh JM, Dowling NM, Carlsson CM, Gallagher CL, Birdsill AC, Palotti M, Wharton W, Hermann BP, Larue A, Bendlin BB, Rowley HA, Asthana S, Sager MA, Johnson SC. Cerebral Blood Flow is Diminished in Asymptomatic Middle-Aged Adults with Maternal History of Alzheimer’s Disease. Cereb Cortex. 2012 doi: 10.1093/cercor/bhs381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mosconi L, Glodzik L, Mistur R, McHugh P, Rich KE, Javier E, Williams S, Pirraglia E, De Santi S, Mehta PD, Zinkowski R, Blennow K, Pratico D, de Leon MJ. Oxidative stress and amyloid-beta pathology in normal individuals with a maternal history of Alzheimer’s. Biol Psychiatry. 2010;68:913–921. doi: 10.1016/j.biopsych.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mosconi L, Rinne JO, Tsui WH, Berti V, Li Y, Wang H, Murray J, Scheinin N, Nagren K, Williams S, Glodzik L, De Santi S, Vallabhajosula S, de Leon MJ. Increased fibrillar amyloid-{beta} burden in normal individuals with a family history of late-onset Alzheimer’s. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:5949–5954. doi: 10.1073/pnas.0914141107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Honea RA, Vidoni ED, Swerdlow RH, Burns JM. Maternal family history is associated with Alzheimer’s disease biomarkers. Journal of Alzheimer’s disease: JAD. 2012;31:659–668. doi: 10.3233/JAD-2012-120676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liu Z, Chen HH, Li TL, Xu L, Du HQ. A cross-sectional study on cerebrospinal fluid biomarker levels in cognitively normal elderly subjects with or without a family history of Alzheimer’s disease. CNS Neurosci Ther. 2013;19:38–42. doi: 10.1111/cns.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Honea RA, Swerdlow RH, Vidoni E, Burns JM. Progressive regional atrophy in normal adults with a maternal history of Alzheimer disease. Neurology. 2011;76:822–829. doi: 10.1212/WNL.0b013e31820e7b74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Honea RA, Swerdlow RH, Vidoni ED, Goodwin J, Burns JM. Reduced gray matter volume in normal adults with a maternal family history of Alzheimer disease. Neurology. 2010;74:113–120. doi: 10.1212/WNL.0b013e3181c918cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Berti V, Mosconi L, Glodzik L, Li Y, Murray J, De Santi S, Pupi A, Tsui W, De Leon MJ. Structural brain changes in normal individuals with a maternal history of Alzheimer’s. Neurobiology of aging. 2011;32:2325.e2317–2326. doi: 10.1016/j.neurobiolaging.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Andrawis JP, Hwang KS, Green AE, Kotlerman J, Elashoff D, Morra JH, Cummings JL, Toga AW, Thompson PM, Apostolova LG. Effects of ApoE4 and maternal history of dementia on hippocampal atrophy. Neurobiology of aging. 2012;33:856–866. doi: 10.1016/j.neurobiolaging.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Reiter K, Alpert KI, Cobia DJ, Kwasny MJ, Morris JC, Csernansky JC, Wang L. Cognitively normal individuals with AD parents may be at risk for developing aging-related cortical thinning patterns characteristic of AD. Neuroimage. 2012;61:525–532. doi: 10.1016/j.neuroimage.2012.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mosconi L, de Leon M, Murray J, LE, Lu J, Javier E, McHugh P, Swerdlow RH. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2011;27:483–490. doi: 10.3233/JAD-2011-110866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Debette S, Wolf PA, Beiser A, Au R, Himali JJ, Pikula A, Auerbach S, Decarli C, Seshadri S. Association of parental dementia with cognitive and brain MRI measures in middle-aged adults. Neurology. 2009;73:2071–2078. doi: 10.1212/WNL.0b013e3181c67833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science (New York, NY) 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 156.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pericak-Vance MA, Bebout JL, Gaskell PC, Jr, Yamaoka LH, Hung WY, Alberts MJ, Walker AP, Bartlett RJ, Haynes CA, Welsh KA, et al. Linkage studies in familial Alzheimer disease: evidence for chromosome 19 linkage. Am J Hum Genet. 1991;48:1034–1050. [PMC free article] [PubMed] [Google Scholar]
- 158.Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science (New York, NY) 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 159.Mahley RW, Rall SC., Jr Apolipoprotein E: far more than a lipid transport protein. Annual review of genomics and human genetics. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 160.Mahley RW, Huang Y. Apolipoproteine sets the stage: response to injury triggers neuropathology. Neuron. 2012;76:871–885. doi: 10.1016/j.neuron.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor, therapeutic target in neuropathology, including Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chen HK, Ji ZS, Dodson SE, Miranda RD, Rosenblum CI, Reynolds IJ, Freedman SB, Weisgraber KH, Huang Y, Mahley RW. Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J Biol Chem. 2011;286:5215–5221. doi: 10.1074/jbc.M110.151084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Valla J, Yaari R, Wolf AB, Kusne Y, Beach TG, Roher AE, Corneveaux JJ, Huentelman MJ, Caselli RJ, Reiman EM. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer’s susceptibility gene. Journal of Alzheimer’s disease: JAD. 2010;22:307–313. doi: 10.3233/JAD-2010-100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Carrieri G, Bonafe M, De Luca M, Rose G, Varcasia O, Bruni A, Maletta R, Nacmias B, Sorbi S, Corsonello F, Feraco E, Andreev KF, Yashin AI, Franceschi C, De Benedictis G. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s disease. Human genetics. 2001;108:194–198. doi: 10.1007/s004390100463. [DOI] [PubMed] [Google Scholar]
- 165.Bekris LM, Galloway NM, Montine TJ, Schellenberg GD, Yu CE. APOE mRNA and protein expression in postmortem brain are modulated by an extended haplotype structure. Am J Med Genet B Neuropsychiatr Genet. 2009;153B:409–417. doi: 10.1002/ajmg.b.30993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Cervantes S, Samaranch L, Vidal-Taboada JM, Lamet I, Bullido MJ, Frank-Garcia A, Coria F, Lleo A, Clarimon J, Lorenzo E, Alonso E, Sanchez-Juan P, Rodriguez-Rodriguez E, Combarros O, Rosich M, Vilella E, Pastor P. Genetic variation in APOE cluster region and Alzheimer’s disease risk. Neurobiology of aging. 2011;32:2107 e2107–2117. doi: 10.1016/j.neurobiolaging.2011.05.023. [DOI] [PubMed] [Google Scholar]
- 167.Hayden KM, McEvoy JM, Linnertz C, Attix D, Kuchibhatla M, Saunders AM, Lutz MW, Welsh-Bohmer KA, Roses AD, Chiba-Falek O. A homopolymer polymorphism in the TOMM40 gene contributes to cognitive performance in aging. Alzheimers Dement. 2012;8:381–388. doi: 10.1016/j.jalz.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Johnson SC, La Rue A, Hermann BP, Xu G, Koscik RL, Jonaitis EM, Bendlin BB, Hogan KJ, Roses AD, Saunders AM, Lutz MW, Asthana S, Green RC, Sager MA. The effect of TOMM40 poly-T length on gray matter volume and cognition in middle-aged persons with APOE epsilon3/epsilon3 genotype. Alzheimers Dement. 2011;7:456–465. doi: 10.1016/j.jalz.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li G, Bekris LM, Leong L, Steinbart EJ, Shofer JB, Crane PK, Larson EB, Peskind ER, Bird TD, Yu CE. TOMM40 intron 6 poly-T length, age at onset, and neuropathology of AD in individuals with APOE varepsilon3/varepsilon3. Alzheimers Dement. 2012 doi: 10.1016/j.jalz.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Maruszak A, Peplonska B, Safranow K, Chodakowska-Zebrowska M, Barcikowska M, Zekanowski C. TOMM40 rs10524523 polymorphism’s role in late-onset Alzheimer’s disease and in longevity. Journal of Alzheimer’s disease: JAD. 2012;28:309–322. doi: 10.3233/JAD-2011-110743. [DOI] [PubMed] [Google Scholar]
- 171.Potkin SG, Guffanti G, Lakatos A, Turner JA, Kruggel F, Fallon JH, Saykin AJ, Orro A, Lupoli S, Salvi E, Weiner M, Macciardi F. Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer’s disease. PloS one. 2009;4:e6501. doi: 10.1371/journal.pone.0006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Roses AD. An inherited variable poly-T repeat genotype in TOMM40 in Alzheimer disease. Archives of neurology. 2010;67:536–541. doi: 10.1001/archneurol.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, Huentelman MJ, Welsh-Bohmer KA, Reiman EM. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010;10:375–384. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Takei N, Miyashita A, Tsukie T, Arai H, Asada T, Imagawa M, Shoji M, Higuchi S, Urakami K, Kimura H, Kakita A, Takahashi H, Tsuji S, Kanazawa I, Ihara Y, Odani S, Kuwano R. Genetic association study on in and around the APOE in late-onset Alzheimer disease in Japanese. Genomics. 2009;93:441–448. doi: 10.1016/j.ygeno.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 175.Guerreiro RJ, Hardy J. TOMM40 association with Alzheimer disease: tales of APOE and linkage disequilibrium. Archives of neurology. 2012;69:1243–1244. doi: 10.1001/archneurol.2012.1935. [DOI] [PubMed] [Google Scholar]
- 176.Cruchaga C, Nowotny P, Kauwe JS, Ridge PG, Mayo K, Bertelsen S, Hinrichs A, Fagan AM, Holtzman DM, Morris JC, Goate AM. Association and expression analyses with single-nucleotide polymorphisms in TOMM40 in Alzheimer disease. Archives of neurology. 2011;68:1013–1019. doi: 10.1001/archneurol.2011.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nature genetics. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 178.Krishnan KJ, Ratnaike TE, De Gruyter HL, Jaros E, Turnbull DM. Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiology of aging. 2012;33:2210–2214. doi: 10.1016/j.neurobiolaging.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 179.Coskun PE, Wyrembak J, Derbereva O, Melkonian G, Doran E, Lott IT, Head E, Cotman CW, Wallace DC. Systemic mitochondrial dysfunction and the etiology of Alzheimer’s disease and down syndrome dementia. Journalof Alzheimer’s disease: JAD. 2010;20(Suppl 2):310. doi: 10.3233/JAD-2010-100351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Lin MT, Simon DK, Ahn CH, Kim LM, Beal MF. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum Mol Genet. 2002;11:133–145. doi: 10.1093/hmg/11.2.133. [DOI] [PubMed] [Google Scholar]
- 181.Webster MT, Pearce BR, Bowen DM, Francis PT. The effects of perturbed energy metabolism on the processing of amyloid precursor protein in PC12 cells. J Neural Transm. 1998;105:839–853. doi: 10.1007/s007020050098. [DOI] [PubMed] [Google Scholar]
- 182.Gasparini L, Racchi M, Benussi L, Curti D, Binetti G, Bianchetti A, Trabucchi M, Govoni S. Effect of energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neurosci Lett. 1997;231:113–117. doi: 10.1016/s0304-3940(97)00536-3. [DOI] [PubMed] [Google Scholar]
- 183.Gabuzda D, Busciglio J, Chen LB, Matsudaira P, Yankner BA. Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem. 1994;269:13623–13628. [PubMed] [Google Scholar]
- 184.Scheffler K, Krohn M, Dunkelmann T, Stenzel J, Miroux B, Ibrahim S, von Bohlen Und Halbach O, Heinze HJ, Walker LC, Gsponer JA, Pahnke J. Mitochondrial DNA polymorphisms specifically modify cerebral beta-amyloid proteostasis. Acta Neuropathol. 2012;124:199–208. doi: 10.1007/s00401-012-0980-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Fukui H, Diaz F, Garcia S, Moraes CT. Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:14163–14168. doi: 10.1073/pnas.0705738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Pinto M, Pickrell AM, Fukui H, Moraes CT. Mitochondrial DNA damage in a mouse model of Alzheimer’s disease decreases amyloid beta plaque formation. Neurobiology of aging. 2013;34:2399–2407. doi: 10.1016/j.neurobiolaging.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kukreja L, Kujoth GC, Prolla TA, Van Leuven F, Vassar R. Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Molecular neurodegeneration. 2014;9:16. doi: 10.1186/1750-1326-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Dumont M, Stack C, Elipenahli C, Jainuddin S, Launay N, Gerges M, Starkova N, Starkov AA, Calingasan NY, Tampellini D, Pujol A, Beal MF. PGC-1alpha overexpression exacerbates beta-amyloid and tau deposition in a transgenic mouse model of Alzheimer’s disease. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2014;28:1745–1755. doi: 10.1096/fj.13-236331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science (New York, NY) 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Roh JH, Jiang H, Finn MB, Stewart FR, Mahan TE, Cirrito JR, Heda A, Snider BJ, Li M, Yanagisawa M, de Lecea L, Holtzman DM. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. The Journal of experimental medicine. 2014;211:2487–2496. doi: 10.1084/jem.20141788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Yamamoto K, Tanei Z, Hashimoto T, Wakabayashi T, Okuno H, Naka Y, Yizhar O, Fenno LE, Fukayama M, Bito H, Cirrito JR, Holtzman DM, Deisseroth K, Iwatsubo T. Chronic optogenetic activation augments abeta pathology in a mouse model of Alzheimer disease. Cell reports. 2015;11:859–865. doi: 10.1016/j.celrep.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 193.Brody DL, Magnoni S, Schwetye KE, Spinner ML, Esparza TJ, Stocchetti N, Zipfel GJ, Holtzman DM. Amyloid-beta dynamics correlate with neurological status in the injured human brain. Science (New York, NY) 2008;321:1221–1224. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Vlassenko AG, Vaishnavi SN, Couture L, Sacco D, Shannon BJ, Mach RH, Morris JC, Raichle ME, Mintun MA. Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta ) deposition. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17763–17767. doi: 10.1073/pnas.1010461107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Escobar-Khondiker M, Hollerhage M, Muriel MP, Champy P, Bach A, Depienne C, Respondek G, Yamada ES, Lannuzel A, Yagi T, Hirsch EC, Oertel WH, Jacob R, Michel PP, Ruberg M, Hoglinger GU. Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J Neurosci. 2007;27:7827–7837. doi: 10.1523/JNEUROSCI.1644-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Szabados T, Dul C, Majtenyi K, Hargitai J, Penzes Z, Urbanics R. A chronic Alzheimer’s model evoked by mitochondrial poison sodium azide for pharmacological investigations. Behav Brain Res. 2004;154:31–40. doi: 10.1016/j.bbr.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 197.Hoglinger GU, Lannuzel A, Khondiker ME, Michel PP, Duyckaerts C, Feger J, Champy P, Prigent A, Medja F, Lombes A, Oertel WH, Ruberg M, Hirsch EC. The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. Journal of neurochemistry. 2005;95:930–939. doi: 10.1111/j.1471-4159.2005.03493.x. [DOI] [PubMed] [Google Scholar]
- 198.Rottscholl R, Haegele M, Jainsch B, Xu H, Respondek G, Hollerhage M, Rosler TW, Bony E, Le Ven J, Guerineau V, Schmitz-Afonso I, Champy P, Oertel WH, Yamada ES, Hoglinger GU. Chronic consumption of Annona muricata juice triggers and aggravates cerebral tau phosphorylation in wild-type and MAPT transgenic mice. Journal of neurochemistry. 2016 doi: 10.1111/jnc.13835. [DOI] [PubMed] [Google Scholar]
- 199.Yamada ES, Respondek G, Mussner S, de Andrade A, Hollerhage M, Depienne C, Rastetter A, Tarze A, Friguet B, Salama M, Champy P, Oertel WH, Hoglinger GU. Annonacin, a natural lipophilic mitochondrial complex I inhibitor, increases phosphorylation of tau in the brain of FTDP-17 transgenic mice. Experimental neurology. 2014;253:113–125. doi: 10.1016/j.expneurol.2013.12.017. [DOI] [PubMed] [Google Scholar]
- 200.Yanagisawa M, Planel E, Ishiguro K, Fujita SC. Starvation induces tau hyperphosphorylation in mouse brain: implications for Alzheimer’s disease. FEBS Lett. 1999;461:329–333. doi: 10.1016/s0014-5793(99)01480-5. [DOI] [PubMed] [Google Scholar]
- 201.Zhao Y, Tseng IC, Heyser CJ, Rockenstein E, Mante M, Adame A, Zheng Q, Huang T, Wang X, Arslan PE, Chakrabarty P, Wu C, Bu G, Mobley WC, Zhang YW, St George-Hyslop P, Masliah E, Fraser P, Xu H. Appoptosin-Mediated Caspase Cleavage of Tau Contributes to Progressive Supranuclear Palsy Pathogenesis. Neuron. 2015;87:963–975. doi: 10.1016/j.neuron.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Hoglinger GU, Melhem NM, Dickson DW, Sleiman PM, Wang LS, Klei L, Rademakers R, de Silva R, Litvan I, Riley DE, van Swieten JC, Heutink P, Wszolek ZK, Uitti RJ, Vandrovcova J, Hurtig HI, Gross RG, Maetzler W, Goldwurm S, Tolosa E, Borroni B, Pastor P, Cantwell LB, Han MR, Dillman A, van der Brug MP, Gibbs JR, Cookson MR, Hernandez DG, Singleton AB, Farrer MJ, Yu CE, Golbe LI, Revesz T, Hardy J, Lees AJ, Devlin B, Hakonarson H, Muller U, Schellenberg GD. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nature genetics. 2011;43:699–705. doi: 10.1038/ng.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Blass JP, Baker AC, Ko L, Black RS. Induction of Alzheimer antigens by an uncoupler of oxidative phosphorylation. Archives of neurology. 1990;47:864–869. doi: 10.1001/archneur.1990.00530080046009. [DOI] [PubMed] [Google Scholar]
- 204.Blass JP, Baker AC, Ko L, Sheu RK, Black RS. Expression of ‘Alzheimer antigens’ in cultured skin fibroblasts. Archives of neurology. 1991;48:709–717. doi: 10.1001/archneur.1991.00530190055016. [DOI] [PubMed] [Google Scholar]
- 205.Swerdlow RH. Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J Neurosci Res. 2007;85:3416–3428. doi: 10.1002/jnr.21167. [DOI] [PubMed] [Google Scholar]
- 206.Poste G, Reeve P. Enucleation of mammalian cells by cytochalasin B. II. Formation of hybrid cells and heterokaryons by fusion of anucleate and nucleated cells. Exp Cell Res. 1972;73:287–294. doi: 10.1016/0014-4827(72)90050-x. [DOI] [PubMed] [Google Scholar]
- 207.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science (New York, NY) 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 208.Clark MA, Shay JW. Mitochondrial transformation of mammalian cells. Nature. 1982;295:605–607. doi: 10.1038/295605a0. [DOI] [PubMed] [Google Scholar]
- 209.Bunn CL, Wallace DC, Eisenstadt JM. Cytoplasmic inheritance of chloramphenicol resistance in mouse tissue culture cells. Proceedings of the National Academy of Sciences of the United States of America. 1974;71:1681–1685. doi: 10.1073/pnas.71.5.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wallace DC, Bunn CL, Eisenstadt JM. Cytoplasmic transfer of chloramphenicol resistance in human tissue culture cells. The Journal of cell biology. 1975;67:174–188. doi: 10.1083/jcb.67.1.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Morais R, Desjardins P, Turmel C, Zinkewich-Peotti K. Development and characterization of continuous avian cell lines depleted of mitochondrial DNA. In Vitro Cell Dev Biol. 1988;24:649–658. doi: 10.1007/BF02623602. [DOI] [PubMed] [Google Scholar]
- 212.Desjardins P, de Muys JM, Morais R. An established avian fibroblast cell line without mitochondrial DNA. Somat Cell Mol Genet. 1986;12:133–139. doi: 10.1007/BF01560660. [DOI] [PubMed] [Google Scholar]
- 213.Desjardins P, Frost E, Morais R. Ethidium bromide-induced loss of mitochondrial DNA from primary chicken embryo fibroblasts. Mol Cell Biol. 1985;5:1163–1169. doi: 10.1128/mcb.5.5.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ephrussi B, Hottinger H, Chimenes A. Action de l’acriflavine sur les levures, I: la mutation “petite clonie”. Ann Inst Pasteur. 1949;76:531. [Google Scholar]
- 215.King MP, Koga Y, Davidson M, Schon EA. Defects in mitochondrial protein synthesis and respiratory chain activity segregate with the tRNA(Leu(UUR)) mutation associated with mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes. Mol Cell Biol. 1992;12:480–490. doi: 10.1128/mcb.12.2.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Masucci JP, Davidson M, Koga Y, Schon EA, King MP. In vitro analysis of mutations causing myoclonus epilepsy with ragged-red fibers in the mitochondrial tRNA(Lys) gene: two genotypes produce similar phenotypes. Mol Cell Biol. 1995;15:2872–2881. doi: 10.1128/mcb.15.5.2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Michikawa Y, Mazzucchelli F, Bresolin N, Scarlato G, Attardi G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science (New York, NY) 1999;286:774–779. doi: 10.1126/science.286.5440.774. [DOI] [PubMed] [Google Scholar]
- 218.Chomyn A, Martinuzzi A, Yoneda M, Daga A, Hurko O, Johns D, Lai ST, Nonaka I, Angelini C, Attardi G. MELAS mutation in mtDNA binding site for transcription termination factor causes defects in protein synthesis and in respiration but no change in levels of upstream and downstream mature transcripts. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:4221–4225. doi: 10.1073/pnas.89.10.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Chomyn A, Lai ST, Shakeley R, Bresolin N, Scarlato G, Attardi G. Platelet-mediated transformation of mtDNA-less human cells: analysis of phenotypic variability among clones from normal individuals--and complementation behavior of the tRNALys mutation causing myoclonic epilepsy and ragged red fibers. Am J Hum Genet. 1994;54:966–974. [PMC free article] [PubMed] [Google Scholar]
- 220.Hofhaus G, Johns DR, Hurko O, Attardi G, Chomyn A. Respiration and growth defects in transmitochondrial cell lines carrying the 11778 mutation associated with Leber’s hereditary optic neuropathy. J Biol Chem. 1996;271:13155–13161. doi: 10.1074/jbc.271.22.13155. [DOI] [PubMed] [Google Scholar]
- 221.Miller SW, Trimmer PA, Parker WD, Jr, Davis RE. Creation and characterization of mitochondrial DNA-depleted cell lines with “neuronal-like” properties. Journal of neurochemistry. 1996;67:1897–1907. doi: 10.1046/j.1471-4159.1996.67051897.x. [DOI] [PubMed] [Google Scholar]
- 222.Swerdlow RH, Parks JK, Miller SW, Tuttle JB, Trimmer PA, Sheehan JP, Bennett JP, Jr, Davis RE, Parker WD., Jr Origin and functional consequences of the complex I defect in Parkinson’s disease. Ann Neurol. 1996;40:663–671. doi: 10.1002/ana.410400417. [DOI] [PubMed] [Google Scholar]
- 223.Parker WD, Jr, Boyson SJ, Parks JK. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann Neurol. 1989;26:719–723. doi: 10.1002/ana.410260606. [DOI] [PubMed] [Google Scholar]
- 224.Krige D, Carroll MT, Cooper JM, Marsden CD, Schapira AH. Platelet mitochondrial function in Parkinson’s disease. The Royal Kings and Queens Parkinson Disease Research Group. Ann Neurol. 1992;32:782–788. doi: 10.1002/ana.410320612. [DOI] [PubMed] [Google Scholar]
- 225.Benecke R, Strumper P, Weiss H. Electron transfer complexes I and IV of platelets are abnormal in Parkinson’s disease but normal in Parkinson-plus syndromes. Brain. 1993;116(Pt 6):1451–1463. doi: 10.1093/brain/116.6.1451. [DOI] [PubMed] [Google Scholar]
- 226.Parker WD. Sporadic neurologic disease and the electron transport chain: a hypothesis. In: Pascuzzi RM, editor. Proceedings of the 1989 Scientfic Meeting of the American Society for Neurological Investigation: New Developments in Neuromuscular Disease. Indiana University Printing Services; Bloomington, Indiana: 1990. [Google Scholar]
- 227.Davis RE, Miller S, Herrnstadt C, Ghosh SS, Fahy E, Shinobu LA, Galasko D, Thal LJ, Beal MF, Howell N, Parker WD., Jr Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:4526–4531. doi: 10.1073/pnas.94.9.4526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 228.Davis RE, Miller S, Herrnstadt C, Ghosh SS, Fahy E, Shinobu LA, Galasko D, Thal LJ, Beal MF, Howell N, Parker WD. Retraction. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:12069. doi: 10.1073/pnas.95.20.12069-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sheehan JP, Swerdlow RH, Miller SW, Davis RE, Parks JK, Parker WD, Tuttle JB. Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s disease. J Neurosci. 1997;17:4612–4622. doi: 10.1523/JNEUROSCI.17-12-04612.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Swerdlow RH, Parks JK, Cassarino DS, Maguire DJ, Maguire RS, Bennett JP, Jr, Davis RE, Parker WD., Jr Cybrids in Alzheimer’s disease: a cellular model of the disease? Neurology. 1997;49:918–925. doi: 10.1212/wnl.49.4.918. [DOI] [PubMed] [Google Scholar]
- 231.Cardoso SM, Santana I, Swerdlow RH, Oliveira CR. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. Journal of neurochemistry. 2004;89:1417–1426. doi: 10.1111/j.1471-4159.2004.02438.x. [DOI] [PubMed] [Google Scholar]
- 232.Silva DF, Selfridge JE, Lu J, LE, Roy N, Hutfles L, Burns JM, Michaelis EK, Yan S, Cardoso SM, Swerdlow RH. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum Mol Genet. 2013;22:3931–3946. doi: 10.1093/hmg/ddt247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Ito S, Ohta S, Nishimaki K, Kagawa Y, Soma R, Kuno SY, Komatsuzaki Y, Mizusawa H, Hayashi J. Functional integrity of mitochondrial genomes in human platelets and autopsied brain tissues from elderly patients with Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:2099–2103. doi: 10.1073/pnas.96.5.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Bijur GN, Davis RE, Jope RS. Rapid activation of heat shock factor-1 DNA binding by H2O2 and modulation by glutathione in human neuroblastoma and Alzheimer’s disease cybrid cells. Brain Res Mol Brain Res. 1999;71:69–77. doi: 10.1016/s0169-328x(99)00168-0. [DOI] [PubMed] [Google Scholar]
- 235.Cassarino DS, Swerdlow RH, Parks JK, Parker WD, Jr, Bennett JP., Jr Cyclosporin A increases resting mitochondrial membrane potential in SY5Y cells and reverses the depressed mitochondrial membrane potential of Alzheimer’s disease cybrids. Biochem Biophys Res Commun. 1998;248:168–173. doi: 10.1006/bbrc.1998.8866. [DOI] [PubMed] [Google Scholar]
- 236.De Sarno P, Bijur GN, Lu R, Davis RE, Jope RS. Alterations in muscarinic receptor-coupled phosphoinositide hydrolysis and AP-1 activation in Alzheimer’s disease cybrid cells. Neurobiology of aging. 2000;21:31–38. doi: 10.1016/s0197-4580(00)00095-6. [DOI] [PubMed] [Google Scholar]
- 237.Ghosh SS, Swerdlow RH, Miller SW, Sheeman B, Parker WD, Jr, Davis RE. Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimer’s disease and Parkinson’s disease. Ann N Y Acad Sci. 1999;893:176–191. doi: 10.1111/j.1749-6632.1999.tb07825.x. [DOI] [PubMed] [Google Scholar]
- 238.Khan SM, Cassarino DS, Abramova NN, Keeney PM, Borland MK, Trimmer PA, Krebs CT, Bennett JC, Parks JK, Swerdlow RH, Parker WD, Jr, Bennett JP., Jr Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann Neurol. 2000;48:148–155. [PubMed] [Google Scholar]
- 239.Onyango IG, Tuttle JB, Bennett JP., Jr Altered intracellular signaling and reduced viability of Alzheimer’s disease neuronal cybrids is reproduced by beta-amyloid peptide acting through receptor for advanced glycation end products (RAGE) Mol Cell Neurosci. 2005;29:333–343. doi: 10.1016/j.mcn.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 240.Onyango IG, Bennett JP, Jr, Tuttle JB. Endogenous oxidative stress in sporadic Alzheimer’s disease neuronal cybrids reduces viability by increasing apoptosis through pro-death signaling pathways and is mimicked by oxidant exposure of control cybrids. Neurobiol Dis. 2005;19:312–322. doi: 10.1016/j.nbd.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 241.Onyango IG, Ahn JY, Tuttle JB, Bennett JP, Jr, Swerdlow RH. Nerve growth factor attenuates oxidant-induced beta-amyloid neurotoxicity in sporadic Alzheimer’s disease cybrids. Journal of neurochemistry. 2010;114:1605–1618. doi: 10.1111/j.1471-4159.2010.06871.x. [DOI] [PubMed] [Google Scholar]
- 242.Thiffault C, Bennett JP., Jr Cyclical mitochondrial deltapsiM fluctuations linked to electron transport, F0F1 ATP-synthase and mitochondrial Na+/Ca+2 exchange are reduced in Alzheimer’s disease cybrids. Mitochondrion. 2005;5:109–119. doi: 10.1016/j.mito.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 243.Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP, Jr, Miller SW, Davis RE, Parker WD., Jr Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Experimental neurology. 2000;162:37–50. doi: 10.1006/exnr.2000.7333. [DOI] [PubMed] [Google Scholar]
- 244.Trimmer PA, Borland MK. Differentiated Alzheimer’s disease transmitochondrial cybrid cell lines exhibit reduced organelle movement. Antioxid Redox Signal. 2005;7:1101–1109. doi: 10.1089/ars.2005.7.1101. [DOI] [PubMed] [Google Scholar]
- 245.Zhang H, Liu Y, Lao M, Ma Z, Yi X. Puerarin protects Alzheimer’s disease neuronal cybrids from oxidant-stress induced apoptosis by inhibiting pro-death signaling pathways. Experimental gerontology. 2011;46:30–37. doi: 10.1016/j.exger.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 246.Silva DF, Santana I, Esteves AR, Baldeiras I, Arduino DM, Oliveira CR, Cardoso SM. Prodromal metabolic phenotype in MCI cybrids: implications for Alzheimer’s disease. Current Alzheimer research. 2013;10:180–190. doi: 10.2174/1567205011310020008. [DOI] [PubMed] [Google Scholar]
- 247.Gan X, Huang S, Wu L, Wang Y, Hu G, Li G, Zhang H, Yu H, Swerdlow RH, Chen JX, Yan SS. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim Biophys Acta. 2014;1842:220–231. doi: 10.1016/j.bbadis.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Gan X, Wu L, Huang S, Zhong C, Shi H, Li G, Yu H, Howard Swerdlow R, Xi Chen J, Yan SS. Oxidative stress-mediated activation of extracellular signal-regulated kinase contributes to mild cognitive impairment-related mitochondrial dysfunction. Free radical biology & medicine. 2014;75:230–240. doi: 10.1016/j.freeradbiomed.2014.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Yu Q, Fang D, Swerdlow RH, Yu H, Chen JX, Yan SS. Antioxidants Rescue Mitochondrial Transport in Differentiated Alzheimer’s Disease Trans-Mitochondrial Cybrid Cells. Journal of Alzheimer’s disease: JAD. 2016;54:679–690. doi: 10.3233/JAD-160532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Costa RO, Ferreiro E, Martins I, Santana I, Cardoso SM, Oliveira CR, Pereira CM. Amyloid beta-induced ER stress is enhanced under mitochondrial dysfunction conditions. Neurobiology of aging. 2012;33:824.e825–816. doi: 10.1016/j.neurobiolaging.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 251.Jeong JH, Yum KS, Chang JY, Kim M, Ahn JY, Kim S, Lapchak PA, Han MK. Dose-specific effect of simvastatin on hypoxia-induced HIF-1alpha and BACE expression in Alzheimer’s disease cybrid cells. BMC neurology. 2015;15:127. doi: 10.1186/s12883-015-0390-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Trimmer PA, Keeney PM, Borland MK, Simon FA, Almeida J, Swerdlow RH, Parks JP, Parker WD, Jr, Bennett JP., Jr Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer’s disease worsen with passage in culture. Neurobiol Dis. 2004;15:29–39. doi: 10.1016/j.nbd.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 253.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 254.Khan SM, Smigrodzki RM, Swerdlow RH. Cell and animal models of mtDNA biology: progress and prospects. Am J Physiol Cell Physiol. 2007;292:C658–669. doi: 10.1152/ajpcell.00224.2006. [DOI] [PubMed] [Google Scholar]
- 255.Meissner C, Mohamed SA, Klueter H, Hamann K, von Wurmb N, Oehmichen M. Quantification of mitochondrial DNA in human blood cells using an automated detection system. Forensic science international. 2000;113:109–112. doi: 10.1016/s0379-0738(00)00249-8. [DOI] [PubMed] [Google Scholar]
- 256.Lu J, Wang K, Rodova M, Esteves R, Berry D, LE, Crafter A, Barrett M, Cardoso SM, Onyango I, Parker WD, Fontes J, Burns JM, Swerdlow RH. Polymorphic variation in cytochrome oxidase subunit genes. Journal of Alzheimer’s disease: JAD. 2010;21:141–154. doi: 10.3233/JAD-2010-100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Sims NR, Finegan JM, Blass JP. Altered glucose metabolism in fibroblasts from patients with Alzheimer’s disease. N Engl J Med. 1985;313:638–639. doi: 10.1056/NEJM198509053131013. [DOI] [PubMed] [Google Scholar]
- 258.Sims NR, Finegan JM, Blass JP. Altered metabolic properties of cultured skin fibroblasts in Alzheimer’s disease. Ann Neurol. 1987;21:451–457. doi: 10.1002/ana.410210507. [DOI] [PubMed] [Google Scholar]
- 259.Sims NR, Finegan JM, Blass JP, Bowen DM, Neary D. Mitochondrial function in brain tissue in primary degenerative dementia. Brain Res. 1987;436:30–38. doi: 10.1016/0006-8993(87)91553-8. [DOI] [PubMed] [Google Scholar]
- 260.Blass JP, Zemcov A. Alzheimer’s disease. A metabolic systems degeneration? Neurochem Pathol. 1984;2:103–114. doi: 10.1007/BF02834249. [DOI] [PubMed] [Google Scholar]
- 261.Hoyer S. Brain oxidative energy and related metabolism, neuronal stress, and Alzheimer’s disease: a speculative synthesis. J Geriatr Psychiatry Neurol. 1993;6:3–13. doi: 10.1177/002383099300600101. [DOI] [PubMed] [Google Scholar]
- 262.Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 263.Reddy PH, Beal MF. Are mitochondria critical in the pathogenesis of Alzheimer’s disease? Brain Res Brain Res Rev. 2005;49:618–632. doi: 10.1016/j.brainresrev.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 264.Moreira PI, Carvalho C, Zhu X, Smith MA, Perry G. Mitochondrial dysfunction is a trigger of Alzheimer’s disease pathophysiology. Biochimica et biophysica acta. 2010;1802:2–10. doi: 10.1016/j.bbadis.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 265.Moreira PI, Cardoso SM, Santos MS, Oliveira CR. The key role of mitochondria in Alzheimer’s disease. Journal of Alzheimer’s disease: JAD. 2006;9:101–110. doi: 10.3233/jad-2006-9202. [DOI] [PubMed] [Google Scholar]
- 266.Zhu X, Perry G, Moreira PI, Aliev G, Cash AD, Hirai K, Smith MA. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. Journal of Alzheimer’s disease: JAD. 2006;9:147–153. doi: 10.3233/jad-2006-9207. [DOI] [PubMed] [Google Scholar]
- 267.Brinton RD. The healthy cell bias of estrogen action: mitochondrial bioenergetics and neurological implications. Trends in neurosciences. 2008;31:529–537. doi: 10.1016/j.tins.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Mancuso M, Siciliano G, Filosto M, Murri L. Mitochondrial dysfunction and Alzheimer’s disease: new developments. Journal of Alzheimer’s disease: JAD. 2006;9:111–117. doi: 10.3233/jad-2006-9203. [DOI] [PubMed] [Google Scholar]
- 269.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis. Journal of Alzheimer’s disease: JAD. 2010;20(Suppl 2):S265–279. doi: 10.3233/JAD-2010-100339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Swerdlow RH, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: an update. Experimental neurology. 2009;218:308–315. doi: 10.1016/j.expneurol.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses. 2004;63:8–20. doi: 10.1016/j.mehy.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 272.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta. 2014;1842:1219–1231. doi: 10.1016/j.bbadis.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Giles RE, Blanc H, Cann HM, Wallace DC. Maternal inheritance of human mitochondrial DNA. Proceedings of the National Academy of Sciences of the United States of America. 1980;77:6715–6719. doi: 10.1073/pnas.77.11.6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Jack CR, Jr, Wiste HJ, Lesnick TG, Weigand SD, Knopman DS, Vemuri P, Pankratz VS, Senjem ML, Gunter JL, Mielke MM, Lowe VJ, Boeve BF, Petersen RC. Brain beta-amyloid load approaches a plateau. Neurology. 2013;80:890–896. doi: 10.1212/WNL.0b013e3182840bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Burns JM, Swerdlow RH. Backwaters and rapids on the amyloid river. Neurology. 2013;80:878–879. doi: 10.1212/WNL.0b013e3182840d14. [DOI] [PubMed] [Google Scholar]
- 276.Swerdlow RH. Bioenergetic medicine. Br J Pharmacol. 2014;171:1854–1869. doi: 10.1111/bph.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Swerdlow RH. Mitochondrial medicine and the neurodegenerative mitochondriopathies. Pharmaceuticals. 2009;2:150–167. doi: 10.3390/ph2030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Swerdlow RH. Role and treatment of mitochondrial DNA-related mitochondrial dysfunction in sporadic neurodegenerative diseases. Curr Pharm Des. 2011;17:3356–3373. doi: 10.2174/138161211798072535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Swerdlow RH. Treating neurodegeneration by modifying mitochondria: potential solutions to a “complex” problem. Antioxid Redox Signal. 2007;9:1591–1603. doi: 10.1089/ars.2007.1676. [DOI] [PubMed] [Google Scholar]
- 280.Swerdlow RH. Alzheimer’s disease pathologic cascades: who comes first, what drives what. Neurotoxicity Research. 2012;22:182–194. doi: 10.1007/s12640-011-9272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]