

Abstract
Animals are routinely colonized by microorganisms. Despite many studies documenting the microbial taxa associated with animals, the pattern and ecological determinants of among-animal variation in microbial communities are poorly understood. This study quantified the bacterial communities associated with natural populations of Drosophila melanogaster. Across five collections, each fly bore 16–78 OTUs, predominantly of the Acetobacteraceae, Lactobacillaceae, and Enterobacteriaceae. Positive relationships, mostly among related OTUs, dominated both the significant co-occurrences and co-association networks among bacteria, and OTUs with important network positions were generally of intermediate abundance and prevalence. The prevalence of most OTUs was well predicted by a neutral model suggesting that ecological drift and passive dispersal contribute significantly to microbiome composition. However, some Acetobacteraceae and Lactobacillaceae were present in more flies than predicted, indicative of superior among-fly dispersal. These taxa may be well-adapted to the Drosophila habitat from the perspective of dispersal as the principal benefit of the association to the microbial partners. Taken together, these patterns indicate that both stochastic processes and deterministic processes relating to the differential capacity for persistence in the host habitat and transmission between hosts contribute to bacterial community assembly in Drosophila melanogaster.
Introduction
It is well-established that the microbial communities in the guts of animals influence host health and fitness, and numerous studies have attempted to understand how such communities are assembled. Research to date has tended to focus on characterizing the most common members of such host-associated communities (i.e., the core microbiome) and the role of selective processes, which result from relative fitness differences among taxa [1], in determining microbiome composition. However, the composition of gut microbial communities often varies significantly among individual host organisms and in a single host over time [2–5]. In addition, several studies have demonstrated that neutral processes, which are independent of species traits, may play an important role in shaping the composition of the gut microbiota [6–9]. Here we investigate microbiota composition in natural populations of D. melanogaster and use these patterns to identify the microbial taxa and underlying ecological processes that contribute to the assembly of these communities.
The fruit fly D. melanogaster is an excellent system to study community assembly of animal microbiomes. The gut microbiota of D. melanogaster is of low diversity, with positive and negative interactions between Acetobacter and Lactobacillus species that dominate the gut microbiota in the laboratory [10, 11]. Furthermore, microbiota variation among lines and individual flies maintained under uniform laboratory conditions [12, 13] suggests that stochastic processes contribute to community assembly, as investigated by Obadia et al. [14]. However, the gut communities in natural D. melanogaster populations are different and more diverse than in laboratory stocks [11, 15, 16], and the ecological processes shaping community composition in the laboratory may not be representative of the conditions encountered by natural populations.
Building on our previous research on gut microbiome assembly in mycophagous Drosophila species [17], we quantified the microbiome in individual flies from natural populations of the fruit fly D. melanogaster collected from kitchens. To our knowledge, all published studies to date on the microbiota of wild D. melanogaster have relied on pooled samples. We sequenced bacterial 16S rRNA gene amplicons of whole flies as a proxy for the microbial communities in the gut, following evidence, first, that > 95% of the reads in isolated gut metagenomes of D. melanogaster are of bacterial origin (the remainder is predominantly fungal) (Bost, Adair & Douglas, unpublished); and, second, that the D. melanogaster microbiota comprises two or three distinct communities: (i) the gut microbiota, (ii) the surface microbiota which is similar in composition to the gut microbiota though 10–100-fold lower in abundance and reduced further by washing the flies [18], and (iii) in some individuals, intracellular Wolbachia localized to the ovaries of females and various other organs, varying with multiple factors, including Drosophila and Wolbachia genotypes [19].
We surveyed the bacterial communities in 79 individual D. melanogaster collected from multiple sites to address three aims. First, we investigated the relative contributions of variation among flies within and among sites to the overall variation in composition of the bacterial communities in D. melanogaster. Second, we determined the co-occurrence patterns and co-association networks among the bacterial taxa, to gain insight into the nature of potential interactions and highlight the network roles of specific taxa within the gut microbiota community [20, 21]. Finally, we quantified the fit of microbiota composition at each site to the expectations of a neutral model, enabling us to assess the number and identity of taxa whose distribution was well-predicted by the neutral model as well as discordant taxa for which selective processes may be particularly important.
Materials and methods
Collections of Drosophila melanogaster from natural populations
Five collections of adult Drosophila melanogaster were made from compost bins or other food waste in domestic kitchens in Ithaca, NY, USA in August-September 2015 (Table 1). One kitchen was sampled twice (Collection-A and, 15 days later, Collection-B) and three kitchens were sampled once (Collection-C to Collection-E). The flies were starved for 2 h to ensure that any food in their guts was egested, and then frozen at −20 °C.
Table 1.
Diversity of bacterial communities in the collections of male D. melanogaster
| Collection | Collection date (2015) | Number of individuals | Number of OTUs in the collection | Number of OTUs per fly mean (sd) | Shannon index mean (sd) | Inverse Simpson index mean (sd) |
|---|---|---|---|---|---|---|
| A (site 1) | 2 August | 23 | 176 | 34.6 (15.6) | 1.55 (0.84) | 4.04 (3.75) |
| B (site 1) | 17 August | 19 | 156 | 53.3 (14.2) | 2.33 (0.48) | 6.50 (2.96) |
| C (site 2) | 9 September | 8 | 102 | 41.1 (8.4) | 2.19 (0.57) | 6.51 (3.56) |
| D (site 3) | 29 August | 14 | 116 | 37.9 (8.1) | 2.12 (0.43) | 5.51 (2.60) |
| E (site 4) | 8 September | 15 | 137 | 39.2 (10.6) | 2.18 (0.44) | 5.89 (2.06) |
DNA extraction, library preparation and sequencing
The frozen flies from each collection were transferred to a 9 cm Petri dish on ice for rapid separation of the sexes. The males were washed in 0.3% sodium hypochlorite in phosphate-buffered saline (PBS) and rinsed three times with sterile PBS. Washing solutions were ice-cold to anesthetize flies. Flies were transferred individually to sterile 2 ml screw-cap tubes containing zirconia/silica beads (250 μl of 0.1 mm diam. to disrupt fly tissue and 30 μl of 1 mm diam. to lyse bacterial cells) (BioSpec Products, Bartlesville, OK, USA), 678 μl cell lysis buffer (108 mM Tris-HCl, pH 8.0; 1.5 M NaCl; 21.6 mM EDTA), and 16 units proteinase K for DNA extraction. Females were discarded because our study site is within the range of D. simulans, and female D. melanogaster and D. simulans cannot be distinguished by external features. Each fly was homogenized at 4 m/s for 30 s with a FastPrep®-24 instrument (MP Biomedicals, Santa Ana, California, USA), incubated at 56 °C for 2 h, then 35 units RNaseA were added and incubated at 37 °C overnight. DNA was extracted with 750 μl phenol:chloroform:isoamyl alcohol (25:24:1). After centrifuging at 8000×g for 15 min at 4 °C, 500 μl aqueous phase was transferred to a new tube. To precipitate DNA, 37.5 μl 3 M sodium acetate and 500 μl isopropanol was added and samples were incubated at −20 °C overnight. Following centrifugation at 8000×g for 15 min at 4 °C, the pellet was washed in 750 μl cold 75% ethanol, air-dried, resuspended in 50 μl sterile endonuclease-free water and stored at −20 °C.
A dual-indexing strategy modified from Kozich et al. [22] was used to generate amplicons of the V3 and V4 regions of bacterial 16 S rRNA genes. Indexed primers included the primer sequences 341 f (5′-CCTACGGGAGGCAGCAG-3′) and 806r (5′-GGACTACHVGGGTWTCTAAT-3′). Amplicons were generated in PCR reactions containing 1× buffer, 1.5 mM MgCl2, 0.2 mM dNTPs, 0.2 μM each primer, and 2 units Platinum Taq (Invitrogen, Carlsbad, California, USA) with: 94 °C for 30 s, 30 amplification cycles of 94 °C for 30 s, 55 °C for 60 s, and 68 °C for 90 s, with 5 min final extension at 68 °C. PCR products were purified using Agencourt Ampure XP beads (Beckman Coulter, Indianapolis, Indiana, USA), concentrations assayed by Qubit 2.0 fluorimeter (Invitrogen), and the samples were pooled to equimolar concentrations. Library quality was assessed with a Fragment Analyzer (Advanced Analytical Technologies, Ames, IA, USA). Libraries were subjected to 250 bp paired-end sequencing on MiSeq instrument (Illumina, San Diego, CA, USA). Sequence data are available at NCBI Short Read Archive under Bioproject PRJNA381755.
Sequence processing
The sequences were processed with Qiime 1.9.1 [23]. Paired-end reads were joined with fastq-join [24], demultiplexed and quality filtered with default parameters. Potentially chimeric sequences, identified by de novo chimera detection with USEARCH6.1 [25], and low coverage samples (<4670 reads) were removed, yielding 4,642,853 sequences with 409 bp average length. Sequences were assigned to operational taxonomic units (OTUs) at 97% sequence similarity via open-reference OTU picking [26]. The resultant OTU table (Supplementary Dataset 1a) was then filtered to exclude OTUs with <100 reads across all samples, generating a dataset of 251 bacterial OTUs across 101 flies (Supplementary Dataset 1b). Seven OTUs were assigned to the genus Wolbachia and they were excluded from all analyses. Samples with >95% Wolbachia reads were also removed because sequence coverage curves of non-Wolbachia reads indicated that their coverage was insufficient to represent the diversity present in these samples (Supplementary Figure 1). For the remaining 79 samples, non-Wolbachia reads were evenly subsampled to 1200 reads per sample. The saturation of OTU counts against read number in the sequence coverage plots indicated that this depth accounted for the bacterial diversity in these samples (Supplementary Figure 2). The non-Wolbachia OTUs for these 79 samples (Supplementary Dataset 1c) were used for analysis of bacterial diversity and composition.
Statistical analyses
All statistical analyses were conducted using R [27]. To generate sequence coverage plots, reads were subsampled at evenly spaced intervals and the number of OTUs detected at each step was calculated with the iNEXT package [28]. Bacterial diversity in each fly was calculated as the number of OTUs (OTU richness) and the Shannon-Weaver (H′) and inverse Simpson (1/D) diversity indices. To assess the variation in diversity measures among collections, random effects models were run with the lme function in the nlme package [29]. For each model, we calculated the intraclass correlation coefficient, which is the ratio of variance among collections to the total variance. The statistical significance of variation in diversity indices among collections was assessed by likelihood ratio tests comparing each model with collection as a random effect to the corresponding intercept-only model that did not include the random effect [30]. Analysis of deviance tested for differences in diversity among collections. The relationships between Wolbachia infection and diversity indices were determined by comparing mixed effects models that included proportion of Wolbachia reads as a fixed effect to collection-only models with likelihood ratio tests. Models used to test fixed effects were fitted using maximum likelihood. The strength and significance of relationships between proportion of Wolbachia reads and the relative abundance of bacterial orders and genera were assessed with Spearman correlations, using P-values adjusted for multiple tests.
Multivariate relationships among the microbiota of individual flies were visualized with principal coordinates analysis (PCoA) ordination plots of Jaccard indices (for the presence/absence data) and Bray–Curtis dissimilarities (for relative abundance data). The influence of collection and proportion of Wolbachia reads on these distance matrices was determined with distance-based redundancy analyses [31, 32] and significance assessed with permutation tests [33], using the dbrda and anova.cca functions in the vegan package [34].
Two approaches were taken to assess relationships among members of the D. melanogaster microbiota. First, co-occurrence patterns among pairs of bacterial OTUs based on presence/absence data were assessed with the probabilistic approach of Veech [35] with the “cooccur” package [36]. When the probability that two OTUs would co-occur more or less frequently than observed, if distributed randomly, was < 0.05, that OTU pair was considered to have significant positive or negative co-occurrence, respectively. Second, a co-association network was inferred from the read counts for bacterial OTUs with the sparse inverse covariance estimation for ecological association inference (SPIEC-EASI) method, which is robust to the challenges of compositional data and low sample number relative to OTUs [37]. The spiec.easi function was run with the ‘MB’ method applying the neighborhood selection framework in Meinshausen and Bühlmann [38]. The “igraph” package [39] was used to generate network plots, calculate node attributes, and determine community membership (modules) from edge betweenness. Co-occurrence and co-association patterns could potentially be biased by bacterial OTUs restricted to specific collections, so only OTUs present in ≥ 3 collections were used (Supplementary Dataset 1d). To verify that our conclusions were not unique to the OTU prevalence cutoff, we also conducted the co-occurrence and co-association analyses for the OTUs present in at least 37% of samples (following [37]) and found that the results did not differ between the two prevalence thresholds (data not shown).
The contribution of neutral processes to fly microbiota assembly was assessed for each collection separately with the neutral community model of Sloan et al. [40], using the R code of Burns et al. [8]. This model predicts that less abundant taxa will be lost from individual flies due to ecological drift, while more abundant taxa are more likely to be dispersed by chance and therefore present in more individuals. Observed OTU distributions and mean relative abundances were fit to the neutral model by the parameter m, which estimates migration rate [40]. The fit of m for each collection was assessed with a generalized R-squared Burns et al. [8]. Neutral model fits were compared to the fit of binomial models with Akaike information criterion (AIC) scores. To distinguish bacterial taxa that were well-predicted by, or deviated from, the neutral model, we identified OTUs that fell within, above or below the 95% confidence interval around the neutral model prediction as per Burns et al. [8]. We considered taxa present exclusively within the 95% neutral model confidence intervals to be well-predicted by the neutral model. To test for differences in the composition of above and below neutral model partitions, distance-based redundancy analysis was conducted on Jaccard indices. Logistic regressions were run with the presence/absence of taxa and partition type to identify taxa indicative of above or below partitions.
Results
Composition and diversity of the microbiota
The 16S rRNA gene amplicons generated from individual male D. melanogaster were dominated by three orders of bacteria: lactic acid bacteria (Lactobacillales, phylum Firmicutes), acetic acid bacteria (Rhodospirillales, α-Proteobacteria), and enteric bacteria (Enterobacteriales, γ-Proteobacteria) (Fig. 1a). In total, 19 OTUs of these orders were either prevalent or abundant (Table 2). Four of the five most abundant OTUs were Acetobacter and Gluconobacter (family Acetobacteraceae in the Rhodospirillales), and these genera were also detected in at least 95% of the flies (Table 2). Other abundant or prevalent OTUs included Leuconostoc, Lactobacillus, and Enterococcus (Lactobacillales); and Tatumella, Providencia, Pantoea and Kluyvera (Enterobacteriales), and Stenotrophomonas (Xanthomonadales) within the γ-Proteobacteria (Table 2).
Fig. 1.

Composition and diversity of the microbiota of individual D. melanogaster. (a) Relative abundance of bacterial orders present in individual male D. melanogaster grouped by collection. The low abundance category includes all orders that contributed <5% of the reads in that individual. The number of OTUs (b) and genera (c) detected across each evenly subsampled collection (gray bars) and the mean number of taxa (with standard deviation) in the eight randomly chosen individuals (white bars)
Table 2.
Abundant and prevalent bacterial OTUs in male D. melanogaster. The OTUs listed are present in at least 50% of individuals or contribute to at least 2% of the reads averaged across all individuals. To obtain the nearest named isolate, the representative sequence for each OTU was compared to the GreenGenes database [66] and its taxonomic classification was generated with the Ribosomal Database Project (RDP) Classifier [67]. OTU IDs refer to Supplementary Dataset 1c
| OTU ID | Mean relative abundance (sd) | % of individuals | Nearest named isolates (identity)a | Taxonomic classification (confidence) |
|---|---|---|---|---|
| 161 | 12.1 (13.8) | 100 | Acetobacter fabarum (100%) | Acetobacteraceae (100%), Acetobacter (96%) |
| Acetobacter lovaniensis (100%) | ||||
| 10 | 11.0 (23.3) | 72 | Enterococcus termitis (99%) | Enterococcaceae (100%), Enterococcus (60%) |
| 49 | 9.7 (16.9) | 81 | Gluconobacter sp. (100%) | Acetobacteraceae (100%) |
| 124 | 6.4 (7.8) | 97 | Gluconobacter oxydans (100%) | Acetobacteraceae (100%), Gluconobacter (100%) |
| Gluconobacter frateurii (100%) | ||||
| Gluconobacter cerinus (100%) | ||||
| 235 | 5.0 (14.3) | 51 | Gluconobacter sp. (97%) | Acetobacteraceae (100%) |
| 167 | 4.8 (10.0) | 76 | Pantoea punctata (100%)b | Enterobacteriaceae (100%), Tatumella (98%) |
| 231c | 4.3 (8.6) | 68 | Enterobacterium str. (90%) | Orbaceae (85%) |
| 61 | 4.2 (8.3) | 59 | Providencia sneebia (100%) | Enterobacteriaceae (100%), Providencia (100%) |
| 111 | 3.8 (5.0) | 91 | Leuconostoc mesenteroides (100%) | Leuconostocaceae (100%), Leuconostoc (100%) |
| Leuconostoc pseudomesenteroides (100%) | ||||
| 64 | 3.6 (9.9) | 30 | Lactobacillus mali (100%) | Lactobacillaceae (100%), Lactobacillus (100%) |
| 242 | 3.1 (12.8) | 25 | Pseudomonas tuomuerensis (93%) | Pseudomonadaceae (100%) |
| 179 | 3.0 (9.7) | 81 | Pantoea agglomerans (99%) | Enterobacteriaceae (100%), Pantoea (98%) |
| Pantoea vagens (99%) | ||||
| 15 | 2.6 (4.7) | 61 | Stenotrophomonas maltophilia (100%) | Xanthomonadaceae (100%), Stenotrophomonas (100%) |
| Xanthomonas sp. (100%) | ||||
| Pseudomonas geniculate (100%) | ||||
| Pseudomonas hibiscicola (100%) | ||||
| 32 | 2.5 (3.4) | 95 | Gluconobacter oxydans (100%) | Acetobacteraceae (100%), Gluconobacter (100%) |
| Gluconobacter krungthepensis (100%) | ||||
| 96 | 1.1 (1.5) | 76 | Leuconostoc citreum (100%) | Leuconostocaceae (100%), Leuconostoc (100%) |
| Leuconostoc mesenteroides (100%) | ||||
| 101 | 0.9 (1.6) | 66 | Acetobacter syzygii (98%) | Acetobacteraceae (100%), Acetobacter (95%) |
| Acetobacter fabarum (98%) | ||||
| Acetobacter lovaniensis (98%) | ||||
| 142 | 0.9 (1.3) | 70 | Acetobacter indonesiensis (99%) | Acetobacteraceae (100%), Acetobacter (100%) |
| Acetobacter cibinongensis (99%) | ||||
| 108 | 0.8 (2.2) | 63 | Kluyvera intermedia (98%) | Enterobacteriaceae (100%) |
| 222 | 0.2 (0.3) | 62 | Gluconobacter cerinus (98%) | Acetobacteraceae (100%), Gluconobacter (82%) |
aSome OTUs had the same sequence identity to multiple named isolates, and all the isolates are shown
bThese species have been assigned to the genus Tatumella [65]
cThe taxonomy of this OTU is not settled. It is assigned to family Enterobacteriaceae (of order Enterobacteriales) by Greengenes (see Table), order Pasteurellales by QIIME, and the family Orbaceae (of order Orbales) by RDP
Overall, 244 non-Wolbachia bacterial OTUs and 87 genera were detected across the 79 flies, with 16–78 OTUs and 13–62 genera per fly. To investigate the relationship between the diversity in individual flies and collections, we randomly selected eight individuals from each collection (the number of flies in the smallest collection) and calculated the total number of OTUs and genera in the eight individuals (the pooled diversity). On average, the microbiota of an individual fly contained 25–40% of the OTUs (Fig. 1b) and 40–60% of the genera (Fig. 1c) in the pool.
The composition of the microbiota is variable
We then quantified among-fly variation in bacterial diversity (summary statistics in Table 1) and composition that could be attributed to collection. Intraclass correlation coefficients showed that collection explained ~22% of the variation in the number of OTUs detected in an individual fly (Likelihood ratio = 12.03, p < 0.0005) and ~18% of the variation in Shannon diversity indices (Likelihood ratio = 9.73, p = 0.0018). Collection explained only ~6% of the variation in the inverse Simpson diversity index and its effect was not significant (Likelihood ratio = 1.80, p = 0.1795). We also compared bacterial diversity between individuals from Collection-A and Collection-B at the same site 15 days apart (Table 1). For all indices tested, bacterial diversity differed significantly between these two collections (OTU richness, F1,40 = 22.01, p < 0.001; Shannon index, F1,40 = 24.88, p < 0.001; inverse Simpson index F1,40 = 18.19, p < 0.001), indicating that diversity of the microbiota can vary over relatively short timescales, as well as between different sites.
We assessed variation in the composition of the gut microbiota based on the presence/absence and relative abundance of bacterial OTUs (Fig. 2). Principal coordinates analysis ordinations show some clustering of individuals by collection, more distinctly for the OTU presence/absence (Fig. 2a) than relative abundance (Fig. 2b). Permutation tests of distance-based redundancy analyses (dbRDA) confirmed that collection significantly influenced the composition of the microbiota (presence/absence: F4,74 = 7.03, p < 0.001; relative abundance: F4,74 = 7.10, p < 0.001), explaining ~27% of the variation in both cases. Microbiota composition differed significantly between flies from Collection-A and Collection-B (made at the same site but 15 days apart) (presence/absence: F1,40 = 6.62, p < 0.001, relative abundance: F1,40 = 7.40, p < 0.001).
Fig. 2.

Principal coordinates analysis (PCoA) ordinations of pairwise Jaccard distances based on the presence/absence of bacterial OTUs (a) and Bray–Curtis distances based on relative abundance of bacterial OTUs (b) between samples. Collections are represented by different shapes. The variation explained by the PCoA axes is listed in parentheses
Wolbachia abundance does not affect the composition of the microbiota
Finally, we considered the relationship between the bacterial community and Wolbachia infection. We adopted the proportion of total reads from OTUs assigned to Wolbachia (Supplementary Dataset 1b) as an estimate of infection. Including the proportion of Wolbachia reads in the analysis did not significantly improve upon models that excluded Wolbachia for OTU richness (χ22 = 3.53, p = 0.171), but significantly affected microbiota composition (presence/absence: R2 = 0.02, F1,77 = 1.82, p = 0.017, relative abundance: R2 = 0.03, F1,77 = 2.52, p = 0.004). However, incorporating the Wolbachia reads into the dbRDA models increased the explained variation by only 1–2%. The relationships between the proportion of Wolbachia reads and relative abundance of bacterial orders or OTUs in the dataset that omitted Wolbachia reads were all non-significant (p > 0.05). Taken together, this analysis reveals no substantive biological relationship between Wolbachia abundance and the diversity or composition of the gut microbiota.
Microbial co-occurrences are predominantly random
Our analysis of relationships among bacterial taxa in Drosophila was conducted on OTUs detected in at least three collections (46 of the 244 OTUs in the dataset, Supplementary Dataset 1d). Just 196 (19%) of the 1035 pair-wise comparisons yielded statistically significant co-occurrence, comprising 155 (15%) positive and 41(4%) negative relationships (Fig. 3). Most bacterial taxa were not involved in either predominantly positively or negatively co-occurring pairs; exceptionally, the Xanthomonadaceae had an excess of positive correlations and Enterococcus OTU#26 and Staphylococcus aureus OTU#11 had an excess of negative correlations (Supplementary Table 1). Positively co-occurring pairs of OTUs were from the same bacterial family significantly more often than negatively co-occurring pairs: 52 (34%) of 155 pairs of significant positive correlations and one (2.5%) of 41 significant negative correlations were between OTUs of the same family (χ21 = 15.9, p < 0.001).
Fig. 3.

Pairwise co-occurrence patterns between the most prevalent bacterial OTUs. Each tick on the x- and y-axes refers to an OTU, ordered by family, order and phylum
Co-associations among microbial OTUs are predominantly positive
As a second approach to investigate co-association patterns in the microbiota, networks were inferred from read counts for bacterial OTUs in individual flies using the sparse inverse covariance estimation for ecological association inference (SPIEC-EASI) method [37]. Nodes correspond to OTUs and edges represent significant co-association between the two OTUs. Similar to co-occurrence patterns generated from the presence/absence data, the co-association network revealed more positive than negative co-associations (Fig. 4a). The abundant bacterial orders (Rhodospirillales, Enterobacteriales, Lactobacillales) showed distinct co-association patterns. The acetic acid bacteria (Rhodospirillales) and enteric bacteria (Enterobacteriales) tended to have more central roles in the network than the lactic acid bacteria (Lactobacillales), particularly OTUs from the Leuconostocaceae and Enterococaceae, which were peripheral and were not co-associated with other taxa (Fig. 4a). The modularity of the network was 0.67, and modules were dominated by OTUs from single bacterial families (Fig. 4b). The OTUs that were most highly connected (nodes with high degree) and that connect different parts of the network (nodes with high betweenness centrality) were Gluconobacter OTU#36, Staphylococcus aureus OTU#11, Providencia OTU#61 and Enterobacteriaceae OTU#212 (Fig. 4), only one of which (OTU#61) is among the 19 most abundant and prevalent OTUs in Table 2. These data suggest that the dominant taxa are not necessarily the most important in structuring the co-association network.
Fig. 4.

Co-association network of bacterial OTUs in the gut microbiota of individual male D. melanogaster. Nodes represent the 46 OTUs that are present in at least three collections. For all plots, gray edges indicate positive co-associations, red edges represent negative co-associations, and edge thickness corresponds to confidence in the association. (a) Node color represents bacterial family and node labels refer to OTU IDs from Supplementary Dataset 1d. (b) Modules based on edge betweenness are represented with convex hulls. (c) Intensity of blue color indicates greater node degree and increasing size indicates higher betweenness centrality. (d) Intensity of green color indicates greater OTU prevalence and increasing size indicates higher mean relative abundance across all individuals
Most OTUs fit to predictions of the neutral model
We investigated the contribution of neutral processes to the assembly of the fly microbiota. As indicated by lower AIC scores, the neutral model predicted the relationship between OTU distribution and mean relative abundance better than the binomial distribution for all collections (Fig. 5a), suggesting that the processes of ecological drift and passive dispersal may play a role in community assembly. We found that the distributions of 68–84% of the OTUs in a collection were predicted by the neutral model (Fig. 5b). For Collection-A and Collection-D, nearly twice as many OTUs fell above the neutral model prediction as below, while similar proportions (~7–8%) were above and below the neutral model for the other three collections. Taxa found exclusively within the 95% confidence limits and present in at least three collections included the phyla Verrucomicrobia and TM7, orders Pasteurellales, Sphingomonadales and Sphingobacteriales, several families of Actinomycetales and the genera Achromobacter and Comamonas (family Burkholderiales) (Supplementary Table 2), with the implication that the distribution of these taxa can be explained by neutral processes. The OTUs in the above- and below- partitions beyond the 95% confidence limits of each neutral model separated on a PCoA plot (Fig. 5c) and differed significantly (F1,9 = 2.44, p < 0.01). The Comamonadaceae (order Burkholderiales) and Holosporaceae (order Rickettsiales) occurred significantly more often in the below partition meaning they were present in fewer individuals than predicted by the neutral model, while Acetobacter, Gluconobacter, and Leuconostoc occurred significantly more often in the above partition as they were present in more individuals than predicted by the neutral model (p < 0.05) (Fig. 6).
Fig. 5.
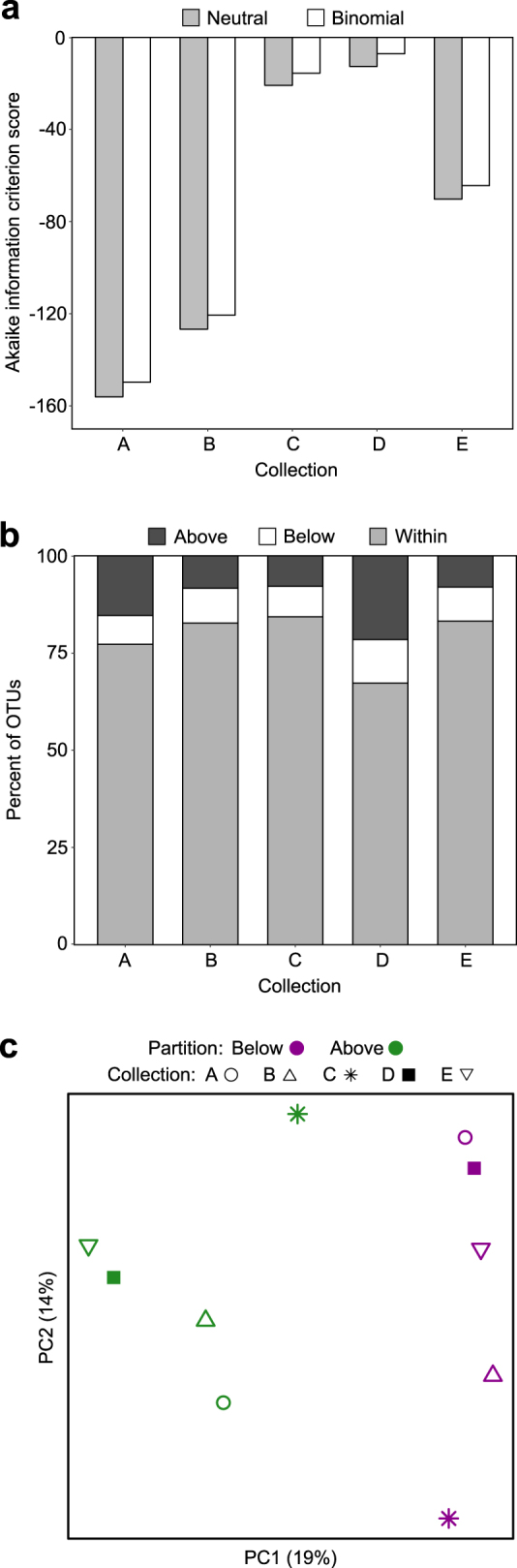
Characteristics of neutral models for collections of D. melanogaster. (a) Akaike information criterion (AIC) scores comparing neutral model fit to fit of a binomial model. (b) The percent of OTUs from each collection that fall above, below and within neutral model prediction. (c) Principal coordinates analysis plot of partitions above and below neutral model predictions based on Jaccard index calculated from the presence/absence of OTUs
Fig. 6.

Fit of neutral model for Collection-A (a), Collection-B (b), Collection-C (c), Collection-D (d), and Collection-E (e). The predicted occurrence frequency is shown as a solid gray line and dashed lines indicate the 95% confidence interval around the neutral model prediction. Each point represents an OTU and taxa whose presence differs significantly between above and below neutral model partitions are highlighted
Discussion
Drosophila melanogaster has a cosmopolitan distribution and is found in a wide diversity of habitats, many created or strongly influenced by human activities, e.g., composters, wineries, fruit orchards [41], and the bacterial communities in the fly gut have been investigated in natural populations of D. melanogaster from multiple habitats ([11, 15, 16, 42]; this study). A common conclusion from these studies is that the composition of the bacterial communities can vary widely at the level of OTU or species, but tends to be dominated by three higher-order taxa, the Acetobacteraceae, Enterobacteriaceae and Lactobacillales. To our knowledge, all previous studies of the microbiota of wild D. melanogaster have relied on pooled samples. This first analysis of individual flies from natural populations reveals that, although the OTU composition of the microbiota varies significantly among fly populations sampled from different locations or at different times, the greater part of the total variation is among individual hosts from a single sample. The high individual variation cannot be attributed simply to variation in microbial composition of recently-ingested food because all analyses were conducted on flies that had been starved for longer than the food transit time through the gut (see methods). More generally, our finding of substantial among-individual variation is consistent with data on gut microbiota composition in natural populations of other animals, including mycophagous Drosophila species [17], mosquitoes [3], fish [4], horses [5] and apes [2], as well as humans [43]. Insight into the processes contributing to among-host variation in microbiota composition can be gained from analysis of the co-occurrence and co-association patterns of different taxa in individual hosts [44, 45] and by accounting for neutral, in addition to selective, processes [7–9].
A striking feature of the bacterial communities in D. melanogaster studied here is the small number of significant co-occurrence relationships among pairs of bacterial OTUs and also the paucity of significant edges, resulting in a weakly structured co-association network. Furthermore, the co-occurrence patterns that are significant are predominantly positive, as are the edges in the co-association network. The lack of co-occurrence and co-associations suggests a lack of interaction among members of the D. melanogaster gut microbiota, a pattern also observed by Rivett et al. [46] in aquatic microcosms. Positive associations are open to two alternative explanations: niche overlap, usually among related bacterial taxa; and mutualistic interactions, including metabolic cross-feeding [47, 48], usually (but not invaraibly) between phylogenetically-distant taxa ([49], but see refs. [50, 51]). Our finding that many of the co-occurring pairs of OTUs and network modules in the D. melanogaster communities comprised bacteria of the same family is indicative niche overlap among members of the gut microbiota; and this hypothesis can be tested by experimental study. This analysis should consider processes operating both in the Drosophila gut and in the food on which the insect feeds, in the light of evidence from laboratory studies that the Drosophila gut microbiota can drive the microbial composition of the food [52] and vice versa [53]. Interestingly, co-association networks in the human gut microbiota also tend to show associations between phylogenetically related taxa [37]. Further research is required to assess the generality of these patterns and whether they are underpinned by similar processes across host taxa.
We additionally investigated the relationship between the composition and diversity of bacterial communities and the relative abundance of Wolbachia. Our rationale was that, although direct interactions between gut bacteria and Wolbachia are most unlikely because the gut microorganisms are restricted to the gut lumen and Wolbachia is intracellular [19], indirect interactions are possible, mediated for example by microbial effects on the immunological function or nutrition of the Drosophila host. However, we obtained no strong correspondence between microbiota composition and Wolbachia infection in this study of wild D. melanogaster. This result is consistent with the absence of significant differences in gut microbiota composition between laboratory D. melanogaster lines that bear and lack Wolbachia [54].
Inspection of the co-association network of bacterial OTUs reveals that the most abundant and prevalent bacterial OTUs are not necessarily the most connected. In particular, the lactic acid bacteria, which are common members of the D. melanogaster gut microbiota [11, 15, 42, 52], are peripheral to the co-association network suggesting that they are not sensitive to the composition of the rest of the microbiota. The finding that Gluconobacter OTU#36, with a mean relative abundance across individuals of just 0.15%, and several potentially pathogenic taxa (Providencia, Erwinia, and Staphylococcus) are particularly well-connected is consistent with previous studies demonstrating that low abundance taxa can hold key network positions and that pathogens can play an important role in shaping microbial community networks [45, 55–57]. Furthermore, our analysis excluded eukaryotic members of the microbiota [58–60]. Although these taxa are much less abundant than bacteria in Drosophila, their community assembly is of intrinsic interest and may interact with bacterial members of the microbiome [61, 62].
Complementary insights into the processes shaping bacterial community assembly in D. melanogaster come from the fit of the bacterial communities to predictions of the neutral model. For the many OTUs that conform to the neutral model, the processes of ecological drift and passive dispersal, which are independent of bacterial functional traits, define abundance in individual flies and distribution across the host population. The taxa that deviate from the neutral model may be indicative of ability to proliferate within and disperse among hosts. Specifically, representatives of the Comamonadaceae and Holosporaceae are detected in fewer individual flies than would be predicted by their abundance, while the several members of the Acetobacteraceae and Lactobacilli, which have a disproportionately high occurrence frequency, may be adapted for dispersal among host individuals. The latter finding is consistent with the hypothesis that fly-mediated dispersal is the key the benefit of associating with Drosophila for Acetobacter and Lactobacilli, suggesting that these bacteria may possess specific adaptations that promote fecal-oral cycling by the Drosophila host [17]. The nature of these adaptations is an interesting area for future research.
In summary, the patterns of abundance and distribution of bacterial taxa associated with wild populations of Drosophila reported in this study builds on laboratory evidence that the Drosophila gut-microbial association is permissive [11, 14, 63], revealing considerable among-host variation in microbiota composition, generally weak patterns of among-taxon co-association, and many taxa with prevalence that can be explained by neutral processes independent of their phenotypic traits. The important implication of these results is that microbiota community composition is not shaped entirely by the traits of the individual microbial taxa or their current host, but also by ecological processes of microbial ecological drift and passive dispersal, as well as the population biology of the host. A quantitative understanding of these ecological processes is a priority to establish the determinants of community assembly of host-associated microorganisms. The exceptions to these generalities are also important because they likely include taxa that interact strongly with other members of the microbial community and the host. In particular, the relative importance of niche and among-microbe interactions in shaping the positive associations between different bacteria, especially related taxa, is an unresolved issue. A further open question is whether bacterial taxa with higher occurrence frequency in hosts than predicted by neutral models have superior dispersal capabilities and derive fitness benefits from these traits. The evidence for elevated frequency of motility in bacteria associated with wild Drosophila than laboratory Drosophila (the latter impose minimal barriers to among-host transmission) [64] provides an excellent starting point to address the significance of dispersal to the ecology and fitness of Drosophila-associated microorganisms. Because the Drosophila-gut microbiota system is amenable to study in both the laboratory and field conditions, it offers a superb model to investigate the ecological principles governing microbial community assembly in animals.
Electronic supplementary material
Acknowledgements
We thank John McMullen for assistance with fly collections and Vince Martinson for technical advice. This research was funded by NSF grant BIO1241099.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Electronic supplementary material
The online version of this article (10.1038/s41396-017-0020-x) contains supplementary material, which is available to authorized users.
References
- 1.Vellend M. Conceptual synthesis in community ecology. Q Rev Biol. 2010;85:183–206. doi: 10.1086/652373. [DOI] [PubMed] [Google Scholar]
- 2.Degnan PH, Pusey AE, Lonsdorf EV, Goodall J, Wroblewski EE, Wilson ML, et al. Factors associated with the diversification of the gut microbial communities within chimpanzees from Gombe National Park. Proc Natl Acad Sci USA. 2012;109:13034–9. doi: 10.1073/pnas.1110994109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Osei-Poku J, Mbogo CM, Palmer WJ, Jiggins FM. Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol Ecol. 2012;21:5138–50. doi: 10.1111/j.1365-294X.2012.05759.x. [DOI] [PubMed] [Google Scholar]
- 4.Smith CCR, Snowberg LK, Caporaso GJ, Knight R, Bolnick DI. Dietary input of microbes and host genetic variation shape among-population differences in stickleback gut microbiota. ISME J. 2015;9:1–12. doi: 10.1038/ismej.2015.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steelman SM, Chowdhary BP, Dowd S, Suchodolski J, Janečka JE. Pyrosequencing of 16S rRNA genes in fecal samples reveals high diversity of hindgut microflora in horses and potential links to chronic laminitis. BMC Vet Res. 2012;8:231. doi: 10.1186/1746-6148-8-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeraldo P, Sipos M, Chia N, Brulc JM, Dhillon AS, Konkel ME, et al. Quantification of the relative roles of niche and neutral processes in structuring gastrointestinal microbiomes. Proc Natl Acad Sci USA. 2012;109:9692–8. doi: 10.1073/pnas.1206721109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L, Ma Z. Testing the neutral theory of biodiversity with human microbiome datasets. Sci Rep. 2016;6:31448. doi: 10.1038/srep31448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burns AR, Stephens WZ, Stagaman K, Wong S, Rawls JF, Guillemin K, et al. Contribution of neutral processes to the assembly of gut microbial communities in the zebrafish over host development. ISME J. 2016;10:655–64. doi: 10.1038/ismej.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Venkataraman A, Bassis CM, Beck JM, Young VB, Curtis JL, Huffnagle GB, et al. Application of a neutral community model to assess structuring of the human lung microbiome. mBio. 2015;6:e02284–14. doi: 10.1128/mBio.02284-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newell PD, Douglas AE. Interspecies interactions determine the impact of the gut microbiota on nutrient allocation in Drosophila melanogaster. Appl Environ Microbiol. 2014;80:788–96. doi: 10.1128/AEM.02742-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong ACN, Chaston JM, Douglas AE. The inconstant gut microbiota of Drosophila species revealed by 16S rRNA gene analysis. ISME J. 2013;7:1922–32. doi: 10.1038/ismej.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chaston JM, Dobson AJ, Newell PD, Douglas AE. Host genetic control of the microbiota mediates the Drosophila nutritional phenotype. Appl Environ Microbiol. 2016;82:671–9. doi: 10.1128/AEM.03301-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broderick NA, Buchon N, Lemaitre B. Microbiota-induced changes in Drosophila melanogaster host gene expression and gut morphology. mBio. 2014;5:e01117–14. doi: 10.1128/mBio.01117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Obadia B, Guvener ZY, Zhang V, Ceja-Navarro JA, Brodie EL, Ja WW, Ludington WB. Probabilistic invasion underlies natural gut microbiome stability. Curr Biol. 2017;27:1999–2006.e8. doi: 10.1016/j.cub.2017.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chandler JA, Morgan Lang J, Bhatnagar S, Eisen JA, Kopp A. Bacterial communities of diverse Drosophila species: ecological context of a host–microbe model system. PLoS Genet. 2011;7:e1002272. doi: 10.1371/journal.pgen.1002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Staubach F, Baines JF, Künzel S, Bik EM, Petrov DA. Host species and environmental effects on bacterial communities associated with Drosophila in the laboratory and in the natural environment. PLoS ONE. 2013;8:e70749. doi: 10.1371/journal.pone.0070749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinson VG, Douglas AE, Jaenike J. Community structure of the gut microbiota in sympatric species of wild Drosophila. Ecol Lett. 2017;20:629–39. doi: 10.1111/ele.12761. [DOI] [PubMed] [Google Scholar]
- 18.Ren C, Webster P, Finkel SE, Tower J. Increased internal and external bacterial load during Drosophila aging without life-span trade-off. Cell Metab. 2007;6:144–52. doi: 10.1016/j.cmet.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Pietri JE, DeBruhl H, Sullivan W. The rich somatic life of Wolbachia. Microbiol Open. 2016;5:923–36. doi: 10.1002/mbo3.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Layeghifard M, Hwang DM, Guttman DS. Disentangling interactions in the microbiome: a network perspective. Trends Microbiol. 2017;25:217–28. doi: 10.1016/j.tim.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cardona C, Weisenhorn P, Henry C, Gilbert JA. Network-based metabolic analysis and microbial community modeling. Curr Opin Microbiol. 2016;31:124–31. doi: 10.1016/j.mib.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–20. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aronesty E. Comparison of sequencing utility programs. Open Bioinf J. 2013;7:1–8. doi: 10.2174/1875036201307010001. [DOI] [Google Scholar]
- 25.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 26.Rideout JR, He Y, Navas-Molina JA, Walters WA, Ursell LK, Gibbons SM, et al. Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. PeerJ. 2014;2:e545. doi: 10.7717/peerj.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.R Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2015. [Google Scholar]
- 28.Hsieh TC, Ma KH, Chao A. iNEXT: iNterpolation and EXTrapolation for species diversity. R package version 2.0.8; 2016.
- 29.Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team. nlme: linear and nonlinear mixed effects models. R package version 3; 2016: 1–128.
- 30.Zuur AF, Ieno EN, Walker NJ, Saveliev AA, Smith GM. Mixed effects models and extensions in ecology with R. New York, NY; 2009.
- 31.Legendre P, Anderson M. Distance-based redundancy analysis: testing multispecies responses in multifactorial ecological experiments. Ecol Monogr. 1999;69:1–24. doi: 10.1890/0012-9615(1999)069[0001:DBRATM]2.0.CO;2. [DOI] [Google Scholar]
- 32.McArdle B, Anderson M. Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology. 2001;82:290–7. doi: 10.1890/0012-9658(2001)082[0290:FMMTCD]2.0.CO;2. [DOI] [Google Scholar]
- 33.Legendre P, Oksanen J, ter Braak CJF. Testing the significance of canonical axes in redundancy analysis. Methods Ecol Evol. 2011;2:269–77. doi: 10.1111/j.2041-210X.2010.00078.x. [DOI] [Google Scholar]
- 34.Oksanen J, Guillaume Blanchet F, Kindt R, Legendre P, Minchin PR, O’Hara RB et al. Vegan: community ecology package. R package version 2. 2016: 3–4.
- 35.Veech JA. A probabilistic model for analysing species co-occurrence. Glob Ecol Biogeogr. 2013;22:252–60. doi: 10.1111/j.1466-8238.2012.00789.x. [DOI] [Google Scholar]
- 36.Griffith DM, Veech JA, Marsh CJ. cooccur: probabilistic species co-occurrence analysis in R. J Stat Softw. 2016;69:1–17. doi: 10.18637/jss.v069.c02. [DOI] [Google Scholar]
- 37.Kurtz ZD, Müller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput Biol. 2015;11:1–25. doi: 10.1371/journal.pcbi.1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meinshausen N, Bühlmann P. High-dimensional graphs and variable selection with the Lasso. Ann Stat. 2006;34:1436–62. doi: 10.1214/009053606000000281. [DOI] [Google Scholar]
- 39.Csardi G, Nepusz T. The igraph software package for complex network research. Inter J Complex Syst: 2006: 1695.
- 40.Sloan WT, Lunn M, Woodcock S, Head IM, Nee S, Curtis TP. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ Microbiol. 2006;8:732–40. doi: 10.1111/j.1462-2920.2005.00956.x. [DOI] [PubMed] [Google Scholar]
- 41.Keller A. Drosophila melanogaster’s history as a human commensal. Curr Biol. 2007;17:R77–81. doi: 10.1016/j.cub.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 42.Corby-Harris V, Pontaroli AC, Shimkets LJ, Bennetzen JL, Habel KE, Promislow DEL. Geographical distribution and diversity of bacteria associated with natural populations of Drosophila melanogaster. Appl Environ Microbiol. 2007;73:3470–9. doi: 10.1128/AEM.02120-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–30. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faust K, Raes J. Microbial interactions: from networks to models. Nat Rev Microbiol. 2012;10:538–50. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 45.Berry D, Widder S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front Microbiol. 2014;5:1–14. doi: 10.3389/fmicb.2014.00219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rivett DW, Scheuerl T, Culbert CT, Mombrikotb SB, Johnstone E, Barraclough TG, et al. Resource-dependent attenuation of species interactions during bacterial succession. ISME J. 2016;10:2259–68. doi: 10.1038/ismej.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris BEL, Henneberger R, Huber H, Moissl-Eichinger C. Microbial syntrophy: interaction for the common good. FEMS Microbiol Rev. 2013;37:384–406. doi: 10.1111/1574-6976.12019. [DOI] [PubMed] [Google Scholar]
- 48.Ponomarova O, Patil KR. Metabolic interactions in microbial communities: untangling the Gordian knot. Curr Opin Microbiol. 2015;27:37–44. doi: 10.1016/j.mib.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 49.Zelezniak A, Andrejev S, Ponomarova O, Mende DR, Bork P, Patil KR. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc Natl Acad Sci USA. 2015;112:E7156. doi: 10.1073/pnas.1421834112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Turroni F, Milani C, Duranti S, Mancabelli L, Mangifesta M, Viappiani A, et al. Deciphering bifidobacterial-mediated metabolic interactions and their impact on gut microbiota by a multi-omics approach. ISME J. 2016;10:1–13. doi: 10.1038/ismej.2015.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rakoff-Nahoum S, Foster KR, Comstock LE. The evolution of cooperation within the gut microbiota. Nature. 2016;533:255–9. doi: 10.1038/nature17626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong ACN, Luo Y, Jing X, Franzenburg S, Bost A, Douglas AE. The host as the driver of the microbiota in the gut and external environment of Drosophila melanogaster. Appl Environ Microbiol. 2015;81:6232–40. doi: 10.1128/AEM.01442-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blum JE, Fischer CN, Miles J, Handelsman J. Frequent replenishment sustains the beneficial microbiome of Drosophila melanogaster. mBio. 2013;4:e00860-13–e00860-13. doi: 10.1128/mBio.00860-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobson AJ, Chaston JM, Newell PD, Donahue L, Hermann SL, Sannino DR, et al. Host genetic determinants of microbiota-dependent nutrition revealed by genome-wide analysis of Drosophila melanogaster. Nat Commun. 2015;6:6312. doi: 10.1038/ncomms7312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Agler MT, Ruhe J, Kroll S, Morhenn C, Kim ST, Weigel D, et al. Microbial hub taxa link host and abiotic factors to plant microbiome variation. PLoS Biol. 2016;14:e1002352. doi: 10.1371/journal.pbio.1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hajishengallis G, Darveau R, Curtis M. The keystone pathogen hypothesis. Nat Rev Microbiol. 2012;10:717–25. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jordán F, Lauria M, Scotti M, Nguyen TP, Praveen P, Morine M, et al. Diversity of key players in the microbial ecosystems of the human body. Sci Rep. 2015;5:15920. doi: 10.1038/srep15920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoang D, Kopp A, Chandler JA. Interactions between Drosophila and its natural yeast symbionts - is Saccharomyces cerevisiae a good model for studying the fly-yeast relationship? PeerJ. 2015;3:e1116. doi: 10.7717/peerj.1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stamps JA, Yang LH, Morales VM, Boundy-Mills KL. Drosophila regulate yeast density and increase yeast community similarity in a natural substrate. PLoS ONE. 2012;7:e42238. doi: 10.1371/journal.pone.0042238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chandler JA, Eisen JA, Kopp A. Yeast communities of diverse Drosophila species: comparison of two symbiont groups in the same hosts. Appl Environ Microbiol. 2012;78:7327–36. doi: 10.1128/AEM.01741-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huffnagle GB, Noverr MC. The emerging world of the fungal microbiome. Trends Microbiol. 2013;21:334–41. doi: 10.1016/j.tim.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sam QH, Chang MW, Chai LYA. The fungal mycobiome and its interaction with gut bacteria in the host. Int J Mol Sci. 2017;18:330. doi: 10.3390/ijms18020330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chaston JM, Newell PD, Douglas AE. Metagenome-wide association of microbial determinants of host phenotype in Drosophila melanogaster. mBio. 2014;5:e01631–14. doi: 10.1128/mBio.01631-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Winans NJ, Walter A, Chouaia B, Chaston JC, Douglas AE, Newell PD. A genomic investigation of ecological differentiation between free-living and Drosophila-associated bacteria. Mol Ecol. 2017;26:4536–50. doi: 10.1111/mec.14232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brady CL, et al. Transfer of Pantoea citrea, Pantoea punctata and Pantoea terrea to the genus Tatumella emend. as Tatumella citrea comb. nov., Tatumella punctata comb. nov. and Tatumella terrea comb. nov. and description of Tatumella morbirosei sp. nov. Int J Syst Evol Microbiol. 2010;60:484–94. [DOI] [PubMed]
- 66.DeSantis TZ, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–72. [DOI] [PMC free article] [PubMed]
- 67.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl Environ Microbiol. 2007;73(16):5261–7. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.