

Abstract
The prosurvival Bcl-2 family proteins Mcl-1 and Bcl-xL inhibit apoptosis by sequestering BH3-only proteins such as Bid and Bim (MODE 1) or the effector proteins Bak and Bax (MODE 2). To better understand the contributions of MODE 1 and MODE 2 in blocking cell death, and thus how to bypass resistance to cell death, we examined prescribed mixtures of Bcl-2 family proteins. In a Bim and Bak mixture, Bcl-xL and Mcl-1 each sequestered not only Bim but also Bak as it became activated by Bim. In contrast, in a Bid and Bak mixture, Bcl-xL preferentially sequestered Bid while Mcl-1 preferentially sequestered Bak. Notably, Bcl-xL could sequester Bak in response to the BH3 mimetic ABT-737, despite this molecule targeting Bcl-xL. These findings highlight the importance of Bak sequestration in resistance to anti-cancer treatments, including BH3 mimetics.
Introduction
The mitochondrial pathway of apoptotic cell death is regulated by three subfamilies of Bcl-2 proteins: prosurvival proteins such as Bcl-2, Bcl-xL and Mcl-1; proapoptotic effectors Bak and Bax; and BH3-only proteins such as Bid and Bim [1, 2]. Defects in the mitochondrial pathway are permissive for tumorigenesis and most chemotherapeutic agents act via this pathway [3]. Small-molecule BH3 mimetics that specifically target Bcl-2 (venetoclax/ABT-199 (ref. [4])) or Mcl-1 (S63845 (ref. [5])) or multiple family members (ABT-737 and navitoclax/ABT-263 (refs. [6–8])) are proving effective as anti-cancer agents. However, chemoresistance can be caused by non-targeted prosurvival proteins, although the mechanisms remain poorly understood [4–9].
Apoptosis can be triggered by damage signals which either upregulate or activate BH3-only proteins, which then convert Bak and Bax into the activated forms capable of homo-oligomerisation and mitochondrial pore formation. The resulting cytochrome c release is the ‘point of no return’ for apoptotic cell death. The mechanism by which Bcl-2 family proteins interact to block cytochrome c release has been controversial, but in recent years a consensus has emerged [10–15]. In a unified model (Fig. S1), prosurvival proteins can sequester BH3-only proteins to block apoptosis upstream of Bak and Bax activation, called ‘MODE 1 inhibition’ [13]. Prosurvival proteins can also act downstream to sequester the activated forms of Bak or Bax, called ‘MODE 2 inhibition’ [13]. The relative contributions of MODE 1 and 2 to apoptosis resistance will depend on several factors including the Bcl-2 family proteins present and their relative concentrations, the specific binding affinities between them, and the activation status of Bak and Bax [13,16,17].
Protein interactions involved in MODE 1 inhibition have been widely studied in vitro, using BH3-only peptides and recombinant prosurvival proteins [16,18]. Analysis of MODE 2 inhibition has been more limited as the generation of MODE 2 complexes requires the additional components of Bak or Bax as well as membranes to allow Bak and Bax activation [19,20]. In mixtures of Bid or Bim, Bcl-xL or Mcl-1, and Bak or Bax, most resistance occurred via MODE 1 (refs. [21,22]). However, MODE 2 was enhanced when mutation decreased the affinity of BH3-only protein for prosurvival proteins [13,21]. Moreover, MODE 2 was decreased when mutations decreased the affinity of Bak or Bax for their prosurvival guardians, causing an increase in apoptosis [23–27]. Thus, several studies indicate that both MODE 1 and MODE 2 can contribute to apoptosis resistance, but that the relative contributions differ depending on context.
The present study sought to understand the relative contributions of MODE 1 and MODE 2 to prosurvival protein function by analysing defined mixtures of full-length Bcl-2 proteins at near-physiological concentrations in mitochondrial incubations and intact cells. In most combinations tested, both Mcl-1 and Bcl-xL functioned in part by sequestering Bak, i.e. MODE 2. Notably, in response to Bid, Mcl-1 caused particularly profound resistance via MODE 2 in mitochondria and in cells. We also found that the BH3 mimetic ABT-737 (that binds Bcl-xL, Bcl-2 and Bcl-w) was able to disrupt MODE 1 complexes to induce Bak:Bcl-xL MODE 2 complexes, with higher concentrations required to disrupt those complexes and permeabilise mitochondria. These studies highlight the importance of MODE 2 in the function of Mcl-1 and Bcl-xL to constrain Bak, and the potential importance of MODE 2 inhibition in chemoresistance.
Results
The concept of prosurvival proteins acting via two MODES to inhibit mitochondrial permeabilisation [13] allows a simplified description of interactions between Bcl-2 family proteins, and is continued here. For clarity, sometimes a distinction is made between the protein complexes (e.g. MODE 1 complexes) and the functional outcome of inhibiting cytochrome c release (e.g. MODE 1 inhibition). MODE 1 will mostly be considered in terms of MODE 1 inhibition because complexes are difficult to detect (and because MODE 1 complexes promote cytochrome c release if Bak has become activated; see below). MODE 2 will also mostly refer to MODE 2 inhibition, as although MODE 2 complexes are easier to estimate, they may not be sufficient for MODE 2 inhibition.
Mcl-1 blocks Bid signalling by sequestering activated Bak
To compare Mcl-1 binding of BH3-only proteins (MODE 1) with its binding of Bak (MODE 2), the first mixtures we analysed included Mcl-1 and Bak, together with Bid or the BidBim chimera which contains the BH3 domain of Bim [20]. To obtain Bak we used mouse liver mitochondria (MLM) which contain Bak but minimal other Bcl-2 family proteins [13,28,29]. We expected that Bid and BidBim might compete differently with activated Bak for binding to Mcl-1, as Mcl-1 binds the Bid BH3 peptide weakly compared to that of Bim (Fig. S1B and Fig. 1a). Importantly, however, Bid was only slightly more efficient than BidBim in activating Bak and releasing cytochrome c in the absence of Mcl-1 (Fig. 1b) [20], allowing us to test how altering MODE 1 affects MODE 2 for Mcl-1.
Fig. 1. MODE 2 complexes of Bak:Mcl-1 profoundly inhibit Bid signalling.

a. Diagram showing interactions between the Bcl-2 family members in the tripartite mixtures tested. The BH3-only proteins Bid and BidBim induce similar activation (arrows) of the apoptotic effector Bak. The prosurvival protein Mcl-1 can sequester the BH3-only protein or activated Bak. Note that Bid binds poorly (dotted lines) to Mcl-1 (MODE 1 complex), allowing Mcl-1 to bind activated Bak (MODE 2 complex). b, c Mouse liver mitochondria (MLM) supplemented with c or without b Mcl-1 were treated with Bid or BidBim at the indicated concentrations. Mitochondrial cytochrome c release was assessed by western blot (WB) of supernatant (sup) and pellet (pel) fractions. Bak conformation change (activation) was assessed by incubation with proteinase K (PK) followed by separation to supernatant and pellet. Protein–protein interactions were determined by solubilising samples (pre-IP), immunoprecipitating (IP) for Mcl-1, and western blotting for Bak, HA (Bid or BidBim) or Mcl-1. Samples in which Mcl-1 blocked the activation of Bak (MODE 1 inhibition, green) or blocked cytochrome c release by binding activated Bak (MODE 2 inhibition, blue) are highlighted. b and c are from the same experiment, and together with two independent experiments were quantified for MODE 1 and 2 inhibition (Fig. S2B). #Long exposure of supernatant blots compared to pellets. *Bands on Mcl-1 blots are due to previous probing for HA.
To test the effect of Mcl-1, MLM were pre-incubated with Mcl-1ΔNC at 35 nM (Fig. 1c). Incubations in which Mcl-1 acted via MODE 1 inhibition (to prevent Bak activation) are shown by green brackets, and were identified as those in which Bid or BidBim could trigger Bak activation in the absence, but not in the presence, of Mcl-1 (see proteinase K treatment; Fig. 1b and c). Incubations in which Mcl-1 was acting via MODE 2 inhibition are shown by blue brackets, and were identified in three ways: Bak activation without cytochrome c release; Bak cleavage by proteinase K to an ~10 kDa fragment; and Bak co-immunoprecipitation with Mcl-1 (Fig. 1c). A semi-quantitative comparison of MODE 1 and MODE 2 inhibition is provided in Fig. S2A and B.
We note that the ~10 kDa fragment of Bak generated by proteinase K is a new means of identifying Bak when sequestered in MODE 2. The fragment was recognised by antibodies that bind to the Bak BH3 domain (clone 4B5), and was released into the supernatant (Fig. 1c and Fig. S3), consistent with the Bak BH3 domain binding to C-terminally truncated Mcl-1. The strong correlation of the 10 kDa fragment with Bak:Mcl-1 co-immunoprecipitation also indicates that Bak:Mcl-1 heterodimers are stable in digitonin, as previously shown for Bak homodimers [30].
Mcl-1 was very effective at blocking cytochrome c release in response to Bid, even at 300 nM Bid (Fig. 1c, left panels). MODE 1 inhibition was evident at low Bid concentrations (0.03–0.1 nM), while MODE 2 inhibition was evident at higher concentrations (0.3–300 nM). Thus, in isolated mitochondria Mcl-1 caused resistance to a wide range of Bid concentrations, but profound resistance was associated with sequestration of activated Bak.
Mcl-1 was less effective at blocking cytochrome c release in response to BidBim (Fig. 1b, c, right panels). MODE 1 inhibition was evident up to 1 nM BidBim, consistent with the high affinity of BidBim for Mcl-1. MODE 2 inhibition was evident at intermediate levels (~3–30 nM) of BidBim, consistent with activated Bak competing with BidBim for binding to Mcl-1. Cytochrome c was released at higher BidBim. Thus, Mcl-1 could function by binding both BidBim (MODE 1) and activated Bak (MODE 2), but strong binding of BidBim to Mcl-1 (seen by co-immunoprecipitation at ~30 nM BidBim) was able to overcome both resistance mechanisms.
In summary, both modes of inhibition contributed to the ability of Mcl-1 to block cytochrome c release induced by either Bid or BidBim. However, MODE 2 inhibition was dominant in response to Bid due to the low affinity of Bid for Mcl-1, protecting mitochondria from even super-stoichiometric concentrations of Bid.
Mcl-1 transiently blocks BidBim signalling by sequestering activated Bak
We next performed time-course experiments to test whether high BidBim was able to disrupt Bak:Mcl-1 MODE 2 complexes, or only prevent their formation. As in Fig. 1c, MLM were pre-incubated with or without Mcl-1, but Bid or BidBim concentrations were increased over time to mimic their up-regulation in cells during apoptotic signalling (Fig. 2a). In the absence of Mcl-1, ~3 nM of either Bid or BidBim activated Bak and released cytochrome c at around 30 min (Fig. 2b). In the presence of Mcl-1, Bid also rapidly activated Bak, but this was sequestered by Mcl-1, preventing cytochrome c release, even after 110 min at the highest Bid concentration (Fig. 2c). BidBim also rapidly activated Bak to generate Bak:Mcl-1 complexes, but cytochrome c was released after 80 min at the highest BidBim concentration, indicating that BidBim had disrupted the Bak:Mcl-1 complexes.
Fig. 2.

MODE 2 complexes of Bak:Mcl-1 can be disrupted by BidBim to cause cytochrome c release. a Timeline of protein addition and sampling. b, c Mouse liver mitochondria (MLM) supplemented with c or without b Mcl-1 were treated with increasing doses of Bid or BidBim according to the timeline in a. Samples collected at the indicated time points were assessed as in Fig. 1. b and c are from the same experiment, and together with two independent experiments were quantified for MODE 1 and MODE 2 inhibition (Fig. S2C). #Long exposure of supernatant blots compared to pellets. *Bands on HA blots are due to previous probing for Mcl-1
Thus, low affinity of Bid for Mcl-1 resulted in durable MODE 2 complexes, while higher affinity of BidBim for Mcl-1 allowed it to disrupt MODE 2 complexes (semi-quantitated in Fig. S2C). This disruption required at least 80 min with high levels of BidBim, suggesting a slow off-rate for activated Bak bound to Mcl-1. Therefore, MODE 2 sequestration may play a significant role in inhibiting apoptosis triggered by many BH3-only proteins, even for proteins like Bim that have a strong affinity for Mcl-1.
Full-length Mcl-1 also binds more Bak after Bid than after BidBim
We next examined how full-length Mcl-1 binds Bak in response to Bid or BidBim, since it was possible that Mcl-1 lacking the C-terminus has altered binding specificity, as we and others have recently found for Bcl-xL [20,31]. We took advantage of Mcl-1flox/flox mice which have slightly raised Mcl-1 levels [32]. Liver mitochondria from these mice were incubated with Bid or BidBim, as performed in Fig. 1c. As seen with truncated Mcl-1, more Bak:Mcl-1 complexes were detected after treatment with Bid than with BidBim (Fig. 3b), indicating that both truncated and endogenous full-length Mcl-1 have higher affinity for BidBim than for Bid.
Fig. 3. Full-length Mcl-1 also binds more Bak after Bid than after BidBim.

a Diagram showing interactions between the Bcl-2 family members in the tripartite mixtures tested. b Mouse liver mitochondria (MLM) from Mcl-1flox/flox mice were treated with Bid or BidBim at the indicated concentrations. Samples were assessed as in Fig. 1. Blots are from the same experiment, representative of two independent experiments. Note that any Bid or BidBim co-immunoprecipitated with Mcl-1 (MODE 1) was below the limits of detection and therefore not shown.
Full-length forms of Bim promote MODE 2 while truncated forms do not
To examine whether BidBim was a faithful mimetic of Bim, full-length BimS generated by in vitro transcription/translation (IVTT) was incubated with mitochondria in the presence of Mcl-1, and examined for MODE 1 and MODE 2 (Fig. 4a). We tested IVTT BimS at 5% v/v and lowered the Mcl-1 concentration because >5% v/v IVTT reaction mixture inhibited Bak activation, as reported previously [33]. BimS partially activated Bak, which bound to Mcl-1 in MODE 2 (blue brackets, Fig. 4a), indicating that Bim activity is well represented by BidBim.
Fig. 4. Membrane-targeted Bim variants but not truncated Bim or Bim peptide cause MODE 2 complexes.

a Mouse liver mitochondria (MLM) supplemented with or without Mcl-1 were treated with four variants of Bim at the indicated concentrations (IVTT reagents were added at the indicated percentage v/v). Samples were assessed as in Fig. 1. Blots are from the same experiment, representative of at least two independent experiments. #Long exposure of supernatant blots compared to pellets. *Bands on Bim blots are due to previous probing for Mcl-1. b Diagram showing interactions between the Bcl-2 family members in the tripartite mixtures tested. Note that activators (e.g. BimS∆C) that do not target to mitochondria are not potent activators of Bak (broken arrow) and so underestimate the potential for MODE 2.
We also tested whether truncated forms of Bim (BimSΔC and the Bim BH3 peptide) could promote MODE 2 complexes. BimS and other BH3-only proteins contain a C-terminal membrane anchor [19,34,35] that is important for Bak activation [19,20]. Accordingly, both truncated forms required higher levels than BidBim to activate Bak at mitochondria (Fig. 4a), and only a minor portion of BimSΔC associated with the membrane fraction (Fig. S4). Notably, there was no evidence of MODE 2 complexes in response to either truncated Bim reagent (Fig. 4a). We conclude that the high levels of BimSΔC and BH3 peptide required to activate Bak at mitochondria also engaged a large portion of the Mcl-1 in MODE 1, depleting that available for MODE 2 (Fig. 4b).
In summary, BidBim is a more accurate mimic of the cellular Bim protein than is BimSΔC or Bim BH3 peptide. In addition, the contribution of MODE 2 may be grossly underestimated in studies using BH3 peptides or truncated BH3-only proteins.
Bcl-xL blocks BidBim signalling partly via MODE 2
We next examined MODE 1 and MODE 2 in mitochondrial mixtures containing Bcl-xL rather than Mcl-1. As both Bid and BidBim have high affinity for Bcl-xL [20], we included a Bid mutant (M97A) that has reduced affinity for Bcl-xL but retains the ability to activate Bak [27] (Fig. 5a). As expected, the Bid variants had similar ability to activate Bak and release cytochrome c in the absence of Bcl-xL (Fig. 5b), allowing us to test how altering MODE 1 affects MODE 2 for this prosurvival protein.
Fig. 5. Bcl-xL blocks BidBim signalling partly via MODE 2. .

a Diagram showing interactions between the Bcl-2 family members in the tripartite mixtures tested. b, c Mouse liver mitochondria (MLM) supplemented with c or without b full-length Bcl-xL were treated with Bid, BidBim or Bid M97A at the indicated concentrations. Samples were analysed as in Fig. 1, except that solubilised samples were immunoprecipitated for Bcl-xL rather than Mcl-1, and Bid M97A was detected by probing for Bid rather than HA. Blots from b and c are from the same experiment, and together with two independent experiments were quantified for MODE 1 and MODE 2 inhibition (Fig. S2D). #Long exposure of pellet fraction to observe the ‘MODE 2-specific fragment’ (unlike Mcl-1 experiments, Fig. S3)
Bcl-xL efficiently sequestered both Bid and BidBim via MODE 1, as 10 nM of Bid or BidBim was required to activate Bak in the presence of 10 nM Bcl-xL (Fig. 5c and Fig. S2D) compared to 0.03 nM in its absence. In fact, Bcl-xL blocked Bid only via MODE 1 inhibition, as no dose of Bid caused Bak activation without cytochrome c release, consistent with a previous report that Bcl-xL acted only via MODE 1 in a Bid and Bax mixture [21]. Bcl-xL did block one concentration of BidBim (10 nM) via MODE 2 inhibition. Thus, in these conditions Bcl-xL caused resistance to Bid- and BidBim-induced cytochrome c release predominantly by directly sequestering these proteins (MODE 1) rather than by sequestering activated Bak (MODE 2).
In the Bid M97A incubations, Bcl-xL could still act via MODE 1 to block low concentrations of the M97A variant (green brackets, Fig. 5c), indicating some affinity. However, MODE 2 inhibition was prominent, persisting even at the highest doses of Bid M97A (blue brackets, Fig. 5c) (semi-quantitated in Fig. S2D). Thus, Bcl-xL may be able to robustly inhibit apoptosis via MODE 2 if the upregulated BH3-only proteins can activate Bak but bind only poorly to Bcl-xL.
We note that in these Bcl-xL experiments the MODE 2-specific fragment of Bak generated by proteinase K was in the pellet fraction (Fig 5C and S3), rather than in the supernatant as in the Mcl-1 experiments (Fig 1C and S3). This is likely due to greater membrane-insertion of full-length Bcl-xL compared to C-terminally truncated Mcl-1.
In cells, endogenous Mcl-1 and Bcl-xL bind Bak in response to tBid or BimS signalling
To study the role of MODE 2 complexes formed by Mcl-1 and Bcl-xL in a cellular context, we used the DU145 prostate carcinoma cell line. These cells are expected to mimic the mitochondrial system used in Figs. 1 and 5, as apoptosis is dependent on Bak due to a frameshift mutation in Bax [36], and the cells have sufficient Mcl-1 and Bcl-xL to cause resistance to TRAIL [37]. To obtain rapid expression of tBid (p15 fragment of Bid) or BimS, DU145 cells were transduced with doxycycline-inducible expression constructs. Upon doxycycline treatment, Bak activation was evident at 2 h as indicated by immunoprecipitation (Fig. 6) [38]. Notably, the activated Bak formed MODE 2 complexes as indicated by its co-immunoprecipitation with Mcl-1 and Bcl-xL at 2 and 4 h. Moreover, the Bak:Mcl-1 and Bak:Bcl-xL complexes appeared transient in the BimS-expressing cells, as observed for those complexes formed in the BidBim mitochondria experiments (Figs. 1c and 5c). We also note that tBid preferentially generated Bak:Mcl-1 complexes while BimS generated both Bak:Mcl-1 and Bak:Bcl-xL complexes (Fig. 6). Although the levels of tBid and BimS were not equivalent, this finding reflects the distinct binding profiles of tBid and Bim observed in the mitochondria experiments comparing Bid and BidBim (Figs. 1c and 5c). In summary, MODE 2 complexes can form in cells without artificially over-expressing prosurvival proteins, and the complexes appear to be disrupted as BimS levels increase (Fig. 6, last lane).
Fig. 6. In cells, endogenous Mcl-1 and Bcl-xL bind Bak in response to either tBid or BimS signalling.
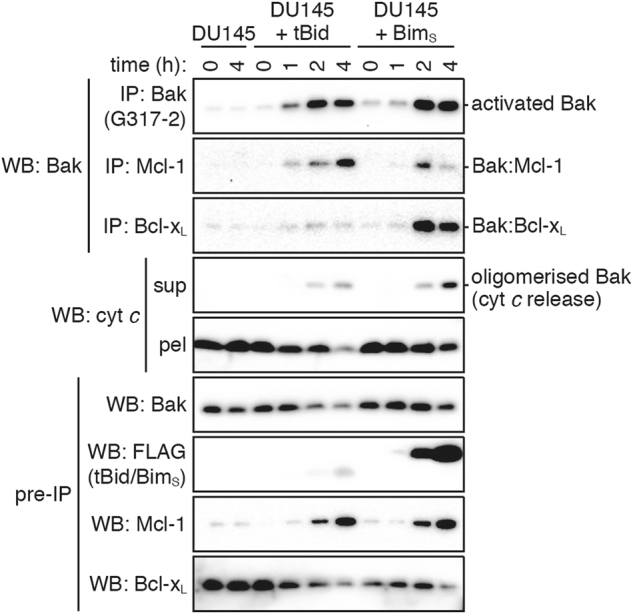
DU145 cells or cells infected with tBid or BimS lentivirus were treated with doxycycline for up to 4 h. Bak activation and protein–protein interactions were determined by solubilising samples with 1% digitonin (pre-IP) and immunoprecipitating (IP) with antibodies against Mcl-1, Bcl-xL or activated Bak (G317-2). Cytochrome c release was determined by permeabilising samples with 0.025% digitonin and centrifuging to separate supernatant (sup) and pellet (pel). Blots are from the same experiment, representative of three independent experiments
BH3 mimetics must overcome MODE 2 to cause cytochrome c release, and can also induce MODE 2
Having established robust conditions for generating MODE 1 and MODE 2 complexes containing Bcl-xL (Fig. 5c), we next compared their disruption by ABT-737, a BH3 mimetic that targets Bcl-xL [6]. As illustrated in Fig. 7a, the incubations involved a 1 h incubation with Bid or Bid M97A to generate MODE 1 or MODE 2 complexes, respectively, followed by a 60 min incubation with increasing concentrations of ABT-737 to disrupt the complexes. The 1 h samples were run in lane 2 (Fig. 7b) and show MODE 1 inhibition (left panel) and MODE 2 inhibition (middle panel). The 2 h samples in the left panel (Fig. 7b) show that MODE 2 inhibition could be induced by low levels of ABT-737 (blue brackets). For example, 0.3 μM ABT-737 activated Bak (presumably by displacing Bid from MODE 1 complexes), which then became sequestered by Bcl-xL. Much higher levels of ABT-737 (10 μM) could prevent MODE 2 and release cytochrome c. A similarly high level of ABT-737 (30 μM) could also disrupt pre-formed MODE 2 complexes in the Bid M97A incubations (Fig. 7b, middle panel). Thus, in this mixture of Bcl-2 family members, cytochrome c release occurred when ABT-737 could circumvent (i.e. either prevent or disrupt) both MODE 1 and MODE 2 complexes. In addition, the concentrations of ABT-737 that either prevent or disrupt MODE 2 complexes appear similar.
Fig. 7. ABT-737 can both induce and disrupt MODE 2 complexes of Bak:Bcl-xL.

. a Timeline of protein addition and sampling. Note that prior to the addition of ABT-737 at 60 min, MODE 1 inhibition was generated by incubation with Bid or BidBak, or MODE 2 inhibition was generated by incubation with Bid M97A (also see 1 h samples in lane 2 in b). b Mouse liver mitochondria (MLM) supplemented with full-length Bcl-xL were treated as in a, and samples analysed as in Fig. 5. Blots are from the same experiment and representative of at least two independent experiments. #Longer exposure
We next used the BidBak chimera [20] to activate Bak, and create a system in which the MODE 1 and MODE 2 complexes both involved the Bak BH3 domain binding to Bcl-xL. A 1 h incubation with BidBak resulted in MODE 1 inhibition (Fig. 7b, right panel), consistent with significant affinity between BidBak and Bcl-xL [20]. Subsequent ABT-737 addition had the same effect as in the Bid incubations, with intermediate ABT-737 causing MODE 2 and higher levels releasing cytochrome c. Therefore, even when the same BH3 domain (from Bak) mediated MODE 1 and MODE 2 complexes with Bcl-xL, ABT-737 was more efficient at overcoming MODE 1 inhibition.
Thus, regions other than the Bak BH3 domain may be involved in binding to Bcl-xL resulting in higher affinity binding in the MODE 2 complex, as suggested previously [13]. However, an alternative explanation is that only a fraction of the activator needs to be displaced from MODE 1 to activate all of Bak (e.g. 0.03 nM Bid in Fig., 1a,c and 5b,c), whereas around half of Bak needs to be displaced from the Bak:Bcl-xL complexes to allow cytochrome c release (Fig. 7b).
In summary, in each of the three types of experiments in Fig. 7, ABT-737 needed to circumvent MODE 2 to allow cytochrome c release. Indeed, intermediate levels of ABT-737 (0.3–3 μM) induced MODE 2 in the Bid and BidBak incubations. Thus, resistance to ABT-737 and other BH3 mimetics may involve the targeted Bcl-2 prosurvival protein (i.e. Bcl-xL) sequestering Bak or Bax in MODE 2, especially if intermediate levels of drug are used.
Discussion
Our studies clarify how specific binding of prosurvival proteins to their proapoptotic relatives directs their ability to block apoptosis. Titration experiments showed that MODE 1 inhibition predominates at low levels of BH3-only reagents, while MODE 2 inhibition predominates at higher levels. If sufficient Mcl-1 or Bcl-xL is present, all Bak can be engaged in MODE 2 complexes, especially if the Bak activator competes poorly with activated Bak for binding to prosurvival proteins. In addition, Bcl-xL sequestration of Bak could block ABT-737 signalling, despite ABT-737 having significant affinity for Bcl-xL. As proposed previously [39], time-course experiments confirm that neither the MODE 1 or MODE 2 means of sequestration involved ‘dead-end’ complexes, as BH3-only proteins released from MODE 1 sequestration could activate Bak, and Bak released from MODE 2 sequestration could release cytochrome c.
Our findings support the proposition that MODE 2 is more efficient and less easily derepressed than MODE 1 (ref. [13]). One possible explanation is that activated Bak has high affinity for Mcl-1, perhaps due to interactions beyond the BH3 domain. An alternative argument is that different thresholds apply for circumventing MODE 1 and MODE 2. For example, circumventing MODE 1 inhibition required displacement of only a fraction of the activator from Mcl-1 to 'catalytically' activate Bak (e.g. 0.03 nM compared to 0.3 nM Bid in Fig. 1b, c). In contrast, circumventing MODE 2 inhibition required much more of Bak to be freed from Mcl-1 to mediate cytochrome c release (e.g. the 100 nM BidBim sample in Fig. 1c).
We found that MODE 2 failed to occur when BH3 peptide or truncated BH3-only protein was used to trigger apoptotic signalling. Of four Bim reagents that could bind to Mcl-1 (MODE 1), only the membrane-targeted reagents (IVTT BimS and BidBim) could cause Bak activation and still allow its binding to Mcl-1 (MODE 2). Most BH3-only proteins have hydrophobic membrane anchors that target them to mitochondria [34] and enhance their ability to activate Bak and Bax [19,20,40]. As BH3 peptides are sometimes used to assess responsiveness to anti-cancer therapies, replacement with BH3 reagents that target to mitochondria may improve their accuracy.
MODE 2 sequestration contributed significantly to blockade of Bid signalling by both truncated and full-length Mcl-1, in mitochondrial and cell-based assays, and was explained by the low affinity of Bid for Mcl-1. The low affinity of Bid for Mcl-1 is consistent with several studies using peptides or recombinant or cellular proteins [13,16,18,20,41,42], although not all [26,43]. Thus, it is possible that Bid-induced MODE 2 may contribute to resistance to TRAIL signalling, granzyme B or other Bid-dependent stimuli [44–46], with resistance more likely where Bax is low or absent [47,48]. A recent study concluded that Bid has a preference for activating Bak while Bim has a preference for activating Bax [49]. As those activation preferences were not apparent in the mitochondrial system used in the present study [20] or a related model system [50], it remains possible that Bid-related MODE 2 explains the reduced effect on mitochondria.
Notably, MODE 2 inhibition occurred even when the BH3 reagent bound strongly to a prosurvival protein, but was applied at intermediate concentrations. For example, BidBim and ABT-737 each have high affinity for Bcl-xL, but generated MODE 2 resistance when applied at intermediate concentrations. Thus, MODE 2 complexes may form regularly in both physiological and pathological settings. In keeping with this, we recently reported defects in T-cell and blood platelet survival in mice expressing a variant of Bak that bound poorly to Bcl-xL [27]. In addition, MODE 2 complexes may contribute to resistance to chemotherapies, including BH3 mimetics, that target Bcl-2 and Mcl-1 (refs. [4,5,13,51]). In our model systems, the complexes were only generated after treatment. However, MODE 2 complexes have been reported in untreated cells, including certain cancer cell lines [24], possibly related to the ability of Bak to become activated in the absence of known activators [52,53].
In conclusion, our studies validate the importance of MODE 2 in the ability of prosurvival proteins to inhibit apoptosis, and provide important insight into targeting these proteins for therapeutic benefit in cancer and other diseases. As MODE 2 complexes become better characterised, ready detection in patient samples may help tailor therapy in a way that minimises resistance due to MODE 2.
Materials and methods
Materials
Thrombin-cleaved HA-tagged human Bid and BidBim chimera [20], mouse Mcl-1ΔN151C23 (ref. [16]) and human Bcl-xL (ref. [20]) were prepared as previously described. Caspase 8-cleaved human Bid M97A was prepared as described for wild-type Bid (ref. [27]). Recombinant human BimSΔC27 was expressed as an N-terminal hexaHis fusion protein in BL21 DE3 Escherichia coli cells following induction with isopropyl β-d-thiogalactopyranoside (IPTG) for 3 h at 37 °C. The cell pellet was resuspended in Tris-buffered saline (TBS) pH 8, and lysed by addition of lysozyme (8 mg/g cell pellet) and deoxycholic acid (4 mg/g cell pellet) for ~30 min at room temperature. After the removal of cell debris by centrifugation, BimSΔC27 was purified from the supernatant by nickel-affinity chromatography using a HiTrap chelating column (Amersham Biosciences, Buckinghamshire, UK) charged with nickel chloride. The eluate was further purified by gel-filtration chromatography on a Superdex 75 16/600 column (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) pre-equilibrated and run in TBS. The pooled peak fractions used for all experiments were >90% pure as assessed by Coomassie Blue staining. To help accurate dilution of recombinant proteins, stock solutions were prepared in buffer containing 1% bovine serum albumin (Sigma-Aldrich, St Louis, MO, USA), which minimises binding of proteins to tubes [20]. Human Bim peptide (DMRPEIWIAQELRRIGDEFNAYYARR) was synthesised by Mimotopes (Notting Hill, VIC, Australia) [16]. Stock solutions of Bim peptide and ABT-737 (a gift from D.C.S. Huang) were prepared with dimethyl sulfoxide.
In vitro transcription/translation was performed with the TnT T7 Coupled Wheat Germ Extract System (Promega, Madison, WI, USA). Reaction mixtures with luciferase control DNA (supplied in kit) or mouse BimS cDNA cloned into pBluescript II (+) linearised with ScaI were prepared according to the manufacturer’s recommendation, using complete amino acid mixture (Promega). Reactions were incubated at 30 °C for 45 min before being added to isolated mitochondria.
Mitochondrial assays
MLM were prepared from C57BL/6 wild-type, Bak−/− or Mcl-1flox/flox mice as described [20]. MLM were diluted to 1 mg/ml in MELB (100 mM KCl, 2.5 mM MgCl2, 100 mM sucrose, 20 mM HEPES/KOH pH 7.5) supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland) and 4 mg/ml pepstatin A (Sigma-Aldrich). MLM were pre-incubated for 20 min at 37 °C with or without Mcl-1 or Bcl-xL as indicated (except Mcl-1flox/flox MLM which had no pre-incubation step). BH3-only stimuli were added as indicated and incubated at 37 °C for 2 h except where otherwise indicated. In the time-course experiments, samples were collected just prior to Bid or BidBim addition at each time point.
To assess cytochrome c release, supernatant and pellet were separated by centrifugation at 16,000 × g for 5 min. To measure Bak activation, pre-chilled samples were incubated with proteinase K (30 μg/ml; Roche) for 20 min on ice. Proteinase K was quenched with 2 mM PMSF (Sigma-Aldrich) and supernatant and pellet fractions obtained as described above. To immunoprecipitate Bcl-2 proteins, pre-chilled samples were solubilised by the addition of 1% w/v digitonin (Biosynth AG, Staad, Switzerland) and incubated on ice for 30 min before pelleting debris by centrifugation at 16,000 × g for 5 min. The resulting clarified lysates were immunoprecipitated with antibodies as described [30] using anti-Mcl-1 rat monoclonal 14C11 (WEHI mAb Facility, Bundoora, VIC, Australia [54]), anti-Bcl-x rat monoclonal IC2 (WEHI mAb Facility [16]) or anti-Bak mouse monoclonal G317-2 (BD Biosciences, San Jose, CA, USA) and Protein G Sepharose 4 Fast Flow resin (GE Healthcare Bio-Sciences AB, Uppsala, Sweden).
Sodium dodecyl sulphate polyacrylamide gel electrophoresis and western blotting
Sodium dodecyl sulphate polyacrylamide gel electrophoresis and western blotting were performed as described [55], except that for immunoprecipitated samples the nitrocellulose membranes were blocked with 5–20% (v/v) horse serum (SAFC Biosciences, St Louis, MO, USA) in addition to the usual 5% (w/v) skimmed milk powder in TBS with 0.1% (v/v) Tween-20 (Sigma-Aldrich). Primary antibodies used were anti-cytochrome c mouse monoclonal 7H8.2C12 (BD Biosciences); anti-HA rat monoclonal 3F10 (Roche); anti-Mcl-1 rat monoclonal 19C4–15 (WEHI mAb Facility [56]), mouse monoclonal clone 22 (BD Transduction Laboratories) or rabbit polyclonal (Rockland Immunochemicals, Pottstown, PA, USA); anti-Bcl-x rabbit polyclonal (BD Biosciences) or mouse monoclonal clone 44 (BD Transduction Laboratories); anti-Bak rabbit polyclonal a23–38 (Sigma) or rat monoclonal 4B5 (WEHI mAb Facility [30]); anti-Bid rat monoclonal 2D1-3 (WEHI mAb Facility [57]); anti-FLAG rat monoclonal 11F3 (WEHI mAb Facility); and anti-Bim rat monoclonal 3C5 (WEHI mAb Facility [58]). Primary antibodies were detected with horseradish peroxidase-conjugated goat anti-mouse, anti-rabbit and anti-rat secondary antibodies (Southern Biotech, Birmingham, AL, USA).
Western blots were quantitated by densitometry using Image Lab 4.1 software (Bio-Rad, Hercules, CA, USA). Percentage cytochrome c release was calculated independently from the supernatant and the pellet as described in ref. [20]. Percentage Bak activation was calculated as percentage reduction in the band labelled ‘non-activated Bak’ on the ‘PK treated’ blots. Relative Bak:Mcl-1 or Bak:Bcl-xL IP was calculated relative to the brightest Bak:Mcl-1 band on the blot, and then normalised across blots by samples run on both blots.
DU145 cell experiments
N-terminally FLAG-tagged tBid and BimS were cloned into the Tetracycline Response Element (TRE)-tight vector, kindly provided by Marco Herold. TRE-linked tBid and BimS were then cloned into a modified version of the lentiviral vector FUGW [59]. Lentiviral particles were produced by transfecting HEK293T cells with lentiviral vector (10 μg), pMDL (5 μg), RSV-REV Eco (2.5 μg) and Eco (5 μg), using calcium phosphate co-precipitation [59]. After 48 h, each filtered (0.45 μm) viral supernatant was mixed with polybrene (4 μg/ml) and used to transduce DU145 cells by spin-infection (2000 rpm, 45 min, 21–26 °C). Fresh media was added to the remaining transfected HEK293T and after a further 24 h, viral supernatants were used to infect DU145 cells again. Transduced (mCherry positive) DU145 cells were selected by FACS. HEK293T and DU145 cells were maintained in Dulbecco’s modified Eagle's medium (Thermo-Fisher Scientific) supplemented with 10% fetal bovine serum (Thermo-Fisher Scientific).
DU145 cells (parental or cells transfected with tBid or BimS constructs) were pre-treated for at least 1 h with 50 μM Q-VD.oph (MP Biomedicals, Santa Ana, CA, USA) to block caspase activation, and then incubated with 5 μg/ml doxycycline (Sigma-Aldrich) for up to 8 h as indicated. Floating cells and trypsinised attached cells were collected, washed in PBS and pelleted by centrifugation. Cells were resuspended in MELB supplemented with protease inhibitor cocktail (Roche) and 4 mg/ml pepstatin A. To assess cytochrome c release, part of the cell sample was permeabilised by the addition of 0.025% digtonin for 10 min on ice, and the supernatant and pellet fractions tested as above. The remainder of the sample was solubilised by the addition of 1% digitonin and immunoprecipitated for Mcl-1, Bcl-xL and activated Bak as described above.
Electronic supplementary material
Acknowledgements
We thank Iris Tan and Marco Herold for the lentiviral constructs, Ray Bartolo for technical assistance, and Stephanie Grabow and Gemma Kelly for the Mcl-1flox/flox mice. This work was supported by NHMRC grants (1008434, 1016701, 1113133), Cancer Council Victoria (GNT1057949) and the Victorian State Government Operational Infrastructure Support and the Australian Government NHMRC IRIISS.
Compliance with ethical standards
Conflict of interests
The authors declare that they have no competing interests.
Footnotes
Edited by C. Borner
Electronic supplementary material
The online version of this article (10.1038/s41418-017-0010-6) contains supplementary material, which is available to authorized users.
References
- 1.Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. doi: 10.1038/nrm3722. [DOI] [PubMed] [Google Scholar]
- 2.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- 4.Roberts AW, Stilgenbauer S, Seymour JF, Huang DC. Venetoclax in patients with previously treated chronic lymphocytic leukemia. Clin Cancer Res. 2017;23:4527–33. doi: 10.1158/1078-0432.CCR-16-0955. [DOI] [PubMed] [Google Scholar]
- 5.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–82. doi: 10.1038/nature19830. [DOI] [PubMed] [Google Scholar]
- 6.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–81. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 7.Shoemaker AR, Mitten MJ, Adickes J, Ackler S, Refici M, Ferguson D, et al. Activity of the Bcl-2 family inhibitor ABT-263 in a panel of small cell lung cancer xenograft models. Cli Cancer Res. 2008;14:3268–77. doi: 10.1158/1078-0432.CCR-07-4622. [DOI] [PubMed] [Google Scholar]
- 8.Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, et al. ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–8. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 9.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–99. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leber B, Lin J, Andrews DW. Embedded together: the life and death consequences of interaction of the Bcl-2 family with membranes. Apoptosis. 2007;12:897–911. doi: 10.1007/s10495-007-0746-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dewson G, Kluck RM. Mechanisms by which Bak and Bax permeabilise mitochondria during apoptosis. J Cell Sci. 2009;122(Pt 16):2801–8. doi: 10.1242/jcs.038166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strasser A, Cory S, Adams JM. Deciphering the rules of programmed cell death to improve therapy of cancer and other diseases. EMBO J. 2011;30:3667–83. doi: 10.1038/emboj.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Llambi F, Moldoveanu T, Tait SW, Bouchier-Hayes L, Temirov J, McCormick LL, et al. A unified model of mammalian BCL-2 protein family interactions at the mitochondria. Mol Cell. 2011;44:517–31. doi: 10.1016/j.molcel.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walensky LD, Gavathiotis E. BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem Sci. 2011;36:642–52. doi: 10.1016/j.tibs.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillies LA, Kuwana T. Apoptosis regulation at the mitochondrial outer membrane. J Cell Biochem. 2014;115:632–40. doi: 10.1002/jcb.24709. [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 17.Lopez J, Bessou M, Riley JS, Giampazolias E, Todt F, Rochegue T, et al. Mito-priming as a method to engineer Bcl-2 addiction. Nat Commun. 2016;7:10538. doi: 10.1038/ncomms10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, et al. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–35. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Oh KJ, Barbuto S, Pitter K, Morash J, Walensky LD, Korsmeyer SJ, et al. A membrane-targeted BID BCL-2 homology 3 peptide is sufficient for high potency activation of BAX in vitro. J Biol Chem. 2006;281:36999–7008. doi: 10.1074/jbc.M602341200. [DOI] [PubMed] [Google Scholar]
- 20.Hockings C, Anwari K, Ninnis RL, Brouwer J, O’Hely M, Evangelista M, et al. Bid chimeras indicate that most BH3-only proteins can directly activate Bak and Bax, and show no preference for Bak versus Bax. Cell Death Dis. 2015;6:e1735. doi: 10.1038/cddis.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Billen LP, Kokoski CL, Lovell JF, Leber B, Andrews DW. Bcl-XL inhibits membrane permeabilization by competing with Bax. PLoS Biol. 2008;6:e147. doi: 10.1371/journal.pbio.0060147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landeta O, Garcia Valero J, Flores-Romero H, Bustillo-Zabalbeitia I, Landajuela A, Garcia-Porras M, et al. Lipid-dependent bimodal MCL1 membrane activity. ACS Chem Biol. 2014;9:2852–63. doi: 10.1021/cb500592e. [DOI] [PubMed] [Google Scholar]
- 23.Czabotar PE, Lee EF, Thompson GV, Wardak AZ, Fairlie WD, Colman PM, et al. Mutation to Bax beyond the BH3 domain disrupts interactions with pro-survival proteins and promotes apoptosis. J Biol Chem. 2011;286:7123–31. doi: 10.1074/jbc.M110.161281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai H, Ding H, Meng XW, Peterson KL, Schneider PA, Karp JE, et al. Constitutive BAK activation as a determinant of drug sensitivity in malignant lymphohematopoietic cells. Genes Dev. 2015;29:2140–52. doi: 10.1101/gad.267997.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fletcher JI, Meusburger S, Hawkins CJ, Riglar DT, Lee EF, Fairlie WD, et al. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc Natl Acad Sci USA. 2008;105:18081–7. doi: 10.1073/pnas.0808691105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ku B, Liang C, Jung JU, Oh BH. Evidence that inhibition of BAX activation by BCL-2 involves its tight and preferential interaction with the BH3 domain of BAX. Cell Res. 2011;21:627–41. doi: 10.1038/cr.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee EF, Grabow S, Chappaz S, Dewson G, Hockings C, Kluck RM, et al. Physiological restraint of Bak by Bcl-xL is essential for cell survival. Genes Dev. 2016;30:1240–50. doi: 10.1101/gad.279414.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uren RT, Dewson G, Chen L, Coyne SC, Huang DCS, Adams JM, et al. Mitochondrial permeabilization relies on BH3 ligands engaging multiple pro-survival Bcl-2 relatives, not Bak. J Cell Biol. 2007;177:277–87. doi: 10.1083/jcb.200606065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Renault TT, Floros KV, Chipuk JE. BAK/BAX activation and cytochrome c release assays using isolated mitochondria. Methods. 2013;61:146–55. doi: 10.1016/j.ymeth.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dewson G, Kratina T, Sim HW, Puthalakath H, Adams JM, Colman PM, et al. To trigger apoptosis Bak exposes its BH3 domain and homo-dimerizes via BH3:grooove interactions. Mol Cell. 2008;30:369–80. doi: 10.1016/j.molcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Pecot J, Maillet L, Le Pen J, Vuillier C, Trecesson SC, Fetiveau A, et al. Tight sequestration of BH3 proteins by BCL-xL at subcellular membranes contributes to apoptotic resistance. Cell Rep. 2016;17:3347–58. doi: 10.1016/j.celrep.2016.11.064. [DOI] [PubMed] [Google Scholar]
- 32.Okamoto T, Coultas L, Metcalf D, van Delft MF, Glaser SP, Takiguchi M, et al. Enhanced stability of Mcl1, a prosurvival Bcl2 relative, blunts stress-induced apoptosis, causes male sterility, and promotes tumorigenesis. Proc Natl Acad Sci USA. 2014;111:261–6. doi: 10.1073/pnas.1321259110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazarou M, Stojanovski D, Frazier AE, Kotevski A, Dewson G, Craigen WJ, et al. Inhibition of Bak activation by VDAC2 is dependent on the Bak transmembrane anchor. J Biol Chem. 2010;285:36876–83. doi: 10.1074/jbc.M110.159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilfling F, Weber A, Potthoff S, Vogtle FN, Meisinger C, Paschen SA, et al. BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death Differ. 2012;19:1328–36. doi: 10.1038/cdd.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Terrones O, Etxebarria A, Landajuela A, Landeta O, Antonsson B, Basanez G, et al. BIM and tBID are not mechanistically equivalent when assisting BAX to permeabilize bilayer membranes. J Biol Chem. 2008;283:7790–803. doi: 10.1074/jbc.M708814200. [DOI] [PubMed] [Google Scholar]
- 36.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–9. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 37.Gillissen B, Wendt J, Richter A, Muer A, Overkamp T, Gebhardt N, et al. Endogenous Bak inhibitors Mcl-1 and Bcl-xL: differential impact on TRAIL resistance in Bax-deficient carcinoma. J Cell Biol. 2010;188:851–62. doi: 10.1083/jcb.200912070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alsop AE, Fennell SC, Bartolo RC, Tan IK, Dewson G, Kluck RM, et al. Dissociation of Bak alpha1 helix from the core and latch domains is required for apoptosis. Nat Commun. 2015;6:6841. doi: 10.1038/ncomms7841. [DOI] [PubMed] [Google Scholar]
- 39.Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev. 2011;21:12–20. doi: 10.1016/j.gde.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ, et al. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–92. doi: 10.1016/S1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- 41.Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak [see comment] Science. 2007;315:856–9. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 42.Meng XW, Lee SH, Dai H, Loegering D, Yu C, Flatten K, et al. Mcl-1 as a buffer for proapoptotic Bcl-2 family members during TRAIL-induced apoptosis: a mechanistic basis for sorafenib (Bay 43-9006)-induced TRAIL sensitization. J Biol Chem. 2007;282:29831–46. doi: 10.1074/jbc.M706110200. [DOI] [PubMed] [Google Scholar]
- 43.Certo M, Moore Vdel G, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–65. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 44.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and charactierization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–82. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 45.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–63. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 46.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.LeBlanc H, Lawrence D, Varfolomeev E, Totpal K, Morlan J, Schow P, et al. Tumor-cell resistance to death receptor-induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat Med. 2002;8:274–81. doi: 10.1038/nm0302-274. [DOI] [PubMed] [Google Scholar]
- 48.Wang C, Youle RJ. Predominant requirement of Bax for apoptosis in HCT116 cells is determined by Mcl-1’s inhibitory effect on Bak. Oncogene. 2012;31:3177–89. doi: 10.1038/onc.2011.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, et al. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell. 2013;51:751–65. doi: 10.1016/j.molcel.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Du H, Wolf J, Schafer B, Moldoveanu T, Chipuk JE, Kuwana T, et al. BH3 domains other than Bim and Bid can directly activate Bax/Bak. J Biol Chem. 2011;286:491–501. doi: 10.1074/jbc.M110.167148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–88. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 52.Chen HC, Kanai M, Inoue-Yamauchi A, Tu HC, Huang Y, Ren D, et al. An interconnected hierarchical model of cell death regulation by the BCL-2 family. Nat Cell Biol. 2015;17:1270–81. doi: 10.1038/ncb3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Senft D, Weber A, Saathoff F, Berking C, Heppt MV, Kammerbauer C, et al. In non-transformed cells Bak activates upon loss of anti-apoptotic Bcl-XL and Mcl-1 but in the absence of active BH3-only proteins. Cell Death Dis. 2015;6:e1996. doi: 10.1038/cddis.2015.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Banerjee A, Grumont R, Gugasyan R, White C, Strasser A, Gerondakis S, et al. NF-κB1 and c-Rel cooperate to promote the survival of TLR4-activated B cells by neutralizing Bim via distinct mechanisms. Blood. 2008;112:5063–73. doi: 10.1182/blood-2007-10-120832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tran VH, Bartolo R, Westphal D, Alsop A, Dewson G, Kluck RM, et al. Bak apoptotic function is not directly regulated by phosphorylation. Cell Death Dis. 2013;4:e452. doi: 10.1038/cddis.2012.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee EF, Czabotar PE, van Delft MF, Michalak E, Boyle M, Willis SN, et al. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. J Cell Biol. 2008;180:341–55. doi: 10.1083/jcb.200708096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaufmann T, Tai L, Ekert PG, Huang DC, Norris F, Lindemann RK, et al. The BH3-only protein Bid is dispensable for DNA damage- and replicative stress-induced apoptosis or cell-cycle arrest. Cell. 2007;129:423–33. doi: 10.1016/j.cell.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 58.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA. 2004;101:6164–9. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Herold MJ, van den Brandt J, Seibler J, Reichardt HM. Inducible and reversible gene silencing by stable integration of an shRNA-encoding lentivirus in transgenic rats. Proc Natl Acad Sci USA. 2008;105:18507–12. doi: 10.1073/pnas.0806213105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.