

Abstract
Human blood plasma provides a highly accessible window to the proteome of any individual in health and disease. Since its inception in 2002, the Human Proteome Organization’s Human Plasma Proteome Project (HPPP) has been promoting advances in the study and understanding of the full protein complement of human plasma and on determining the abundance and modifications of its components. In 2017, we review the history of the HPPP and the advances of human plasma proteomics in general, including several recent achievements. We then present the latest 2017-04 build of Human Plasma PeptideAtlas, which yields ~43 million peptide-spectrum matches and 122,730 distinct peptide sequences from 178 individual experiments at a 1% protein-level FDR globally across all experiments. Applying the latest Human Proteome Project Data Interpretation Guidelines, we catalog 3509 proteins that have at least two non-nested uniquely-mapping peptides of 9 amino acids or more and >1300 additional proteins with ambiguous evidence. We apply the same two-peptide guideline to historical PeptideAtlas builds going back to 2006 and examine the progress made in the past ten years in plasma proteome coverage. We also compare the distribution of proteins in historical PeptideAtlas builds in various RNA-abundance and cellular localization categories. We then discuss advances in plasma proteomics based on targeted mass spectrometry as well as affinity assays, which during early 2017 target ~2000 proteins. Finally we describe considerations about sample handling and study design, concluding with an outlook for future advances in deciphering the human plasma proteome.
Keywords: Human Proteome Project, mass spectrometry, plasma, proteomics
Graphical Abstract
Introduction
Human blood is the most informative easily obtainable sample for assessing the wellness of an individual. Cells can be isolated to provide genomic, transcriptomic, proteomic, metabolomic, and epigenomic information, and the plasma can yield a wealth of systemic information based on the abundance of proteins, cell-free DNA and RNA, metabolites, lipids, and other small molecules. Blood is in contact with all internal organs and, therefore, contains signatures of normal function as well as the consequences of cellular damage and disease manifestation throughout the body. However, the abundances of proteins and other molecules vary across an enormous dynamic range (~1013 range), making the task of detecting and quantifying low abundance signatures a difficult challenge (1). The plasma proteome, therefore, is immensely important clinically and presents enormous analytical challenges.
As one of the very first initiatives of the Human Proteome Organization (2) (HUPO), the Human Plasma Proteome Project (HPPP) began in 2002 with the aim of establishing a comprehensive profile of the proteins present in human plasma in health and disease. The initial phase of the HPPP was a collaborative effort among 35 laboratories to catalog the human plasma proteome via mass spectrometry (MS) based on a set of defined reference samples of plasma and serum (3). The HPPP continues its efforts to the present day with MS as well as exploring orthogonal technologies.
The global Human Proteome Project (HPP) was announced in 2010 at the 10th HUPO World Congress in Sydney, Australia, with the aim of advancing our understanding of all proteins in the human proteome and making advanced protein measurement assays more accessible to the broader research community (4). The HPP comprises a chromosome-centric project (C-HPP; launched at HUPO 2011 in Geneva, Switzerland) (5) that aims to identify and characterize all proteins, annotated in an internationally distributed fashion; a biology/disease-focused project (B/D-HPP; launched at HUPO 2012 in Boston, USA) (6) that aims to understand better the complement of human proteins that are pertinent to a wide variety of human organ systems and biofluids in health and disease; and three resource pillars, including antibody profiling, mass spectrometry, and knowledge base/bioinformatics. The HPPP is one of the major initiatives of the B/D-HPP, focused on understanding the full complement of proteins that can be detected and measured in human plasma samples both in normal and diseased states.
In this article, we first provide a brief review of the advances of the HPPP and MS-based human plasma proteomics in general over the past 15 years. We present the latest and most comprehensive compendium of proteins that have been confidently detected in human plasma samples from the aggregated spectra generated in a large number of publicly available studies. We then discuss advances in MS, present other technologies beyond MS, and conclude with recommendations and an outlook for progress for the next five years.
Historical Perspective of Plasma Analysis with MS
The earliest large compendium of plasma proteins was presented in 2002 in the seminal article by Anderson & Anderson (1). The list of 289 proteins was collected from literature of the day. In a classic figure, a subset of proteins with abundances was depicted over 10 orders of magnitude in abundance in normal plasma and grouped broadly into the categories of classical plasma proteins, tissue leakage products, and cytokines and other very low abundance proteins. Most of the proteins were listed in a table with typical reported concentrations from ELISA (or similar technology) measurements.
In 2003 the HPPP obtained and distributed a series of serum and EDTA-, citrate-, and heparin-anticoagulated plasma samples from Caucasian, African-American, and Asian-American individuals to 35 different laboratories to run across a range of MS instruments and analysis of choice and report the resulting lists of peptides and proteins. There was tremendous variability in the reported results, with some groups providing a rather shallow analysis of one or two of the distributed samples, while other groups provided a far deeper analysis with substantial fractionation of many more samples. This heterogeneity made it particularly difficult to compare results among different groups. In the final HPPP report (3), the number of non-redundant proteins with one or more peptides reported by any group was over 9000. A more conservative list of proteins with at least two reported peptides amounted to 3020 proteins and those with three peptides to 1500 proteins. The danger of very high numbers of false positives from combining many such data lists was explicitly suspected. A subsequent, far more conservative analysis of the results by the same HPPP investigators concluded that there existed high confidence evidence for 889 proteins (7). This analysis uniquely used a Bonferroni-type adjustment for multiple comparisons. An independent analysis of the raw data from most (but not all) of the labs as well as some additional data sets yielded a similar estimate of 960 proteins (8).
Around the same time, several other labs made attempts at defining the MS-observable human plasma proteome. In retrospect, some of these attempts also suffered from substantially inflated numbers due to improperly controlled false positives. In 2008, a meticulous manually-validated effort using the latest instrumentation yielded only 697 proteins, albeit with 144 immunoglobulin entries removed (9). Although combining data sets from different investigators and platforms provides access to a higher number of legitimate detections, it also makes controlling the false discovery rate (FDR) far more challenging.
It was widely recognized that the most abundant proteins greatly complicated detection of much lower abundance proteins and their peptides. Antibody-based depletion of 1 (albumin), 6, 14, or 22 most abundant proteins (accounting for 50 to 99% of the protein mass) became common (10–12), with deeper in-house depletion methods also recently developed (13, 14).
Subsequently the PeptideAtlas resource collected many additional data sets from many different labs and provided substantial effort into controlling the FDR of the search results to provide a database of human plasma proteins with an approximate 1% FDR at the protein level across all experiments as an ensemble. In 2011 Farrah et al. reported 1929 proteins with a 1% protein-level FDR, along with estimated concentrations of the identified proteins based on PSM counting statistics across all datasets in the PeptideAtlas build (15). In 2013, 3553 non-redundant proteins were reported (16). In 2014, Farrah et al. extensively compared the proteomes of plasma, kidney, and urine, detecting proteins filtered from the plasma through the kidney to the urine, proteins appearing de novo in the urine, and proteins arising in the kidney (17).
More recent work has pushed the number of confidently-identified plasma proteome even higher. Keshishian et al. (18) reported on human plasma derived from four patients undergoing a planned myocardial infarction procedure for hypertrophic cardiomyopathy. Plasma was sampled at multiple time points, depleted of the ~64 most abundant proteins, cleaved with two proteases, labeled with iTRAQ reagents, fractionated by pH 10 RP-HPLC into 30 concatenated pools, and analyzed with modern high resolution Q Exactive Plus Orbitrap instrumentation. Whereas 3400 proteins were quantified in every sample, a total of 5300 distinct protein entries were claimed in at least one of the samples, with an estimated protein-level FDR <0.02% (1 decoy/5304). Alternative strategies attempt to minimize the total analysis time in an effort to make plasma proteomic analyses more clinically accessible. Recent efforts achieve high quantitative repeatability of ~1000 proteins using a single MS run per sample (19), in one case analyzing 1294 separate plasma proteomes (20).
The Human Plasma PeptideAtlas 2017
The PeptideAtlas project (21–23) has been collecting shotgun proteomics datasets from labs all around the world since 2004, processing the raw data through a uniform analysis pipeline based on the Trans-Proteomic Pipeline (24–26), and publicly releasing ensemble builds of all the processing results, for human and many other species for which substantial data were collected (27–34). For some species such as human, where samples from different tissue and biofluid type are available, PeptideAtlas has created special builds focusing only on a single sample type. The earliest such special build was for human plasma in 2005 (8); and this has been updated many times since. The Human Plasma PeptideAtlas has provided a publicly available, high stringency snapshot of the full complement of proteins detected in plasma in publicly available shotgun MS experiments over time since 2005.
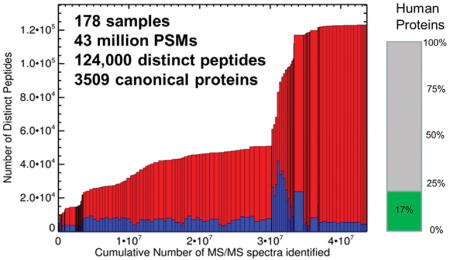
Since our last publication in 2014 (17), three additional builds have been released on-line in 2014 (unpublished), 2015 (unpublished), and 2017-04 (presented in this article). The latest build follows the same data analysis and FDR calculation procedures described in the 2014 publication, with the exception that the search database now used is the THISP database, which is based on neXtProt (Deutsch et al, JPR2016). Thresholds for all individual datasets are set so that when all combined, the combined protein-level FDR is 1%, with a corresponding combined PSM-level FDR of 0.0005 and a combined peptide-level FDR of 0.004. Further statistical details are displayed on the build summary page at the PeptideAtlas interface. Figure 1 depicts the total number of distinct peptides as a function of the total number of PSMs passing threshold added to the Human Plasma PeptideAtlas over time from the earliest datasets to the most recent. The dramatic increase in the number of distinct peptides heralds the advent of the collection and analysis of high resolution MS2 spectra with Orbitrap-based instruments.
Figure 1.

Number of distinct peptide sequences in the Human Plasma PeptideAtlas 2017-04 as a function of the total number of PSMs included (43 million). Each box represents one experiment. The width of the box corresponds to the total number of PSMs passing threshold. The total height of the boxes (blue+red) represents the cumulative number of distinct peptide sequences in the build (culminates at 122,730). The height of the blue boxes corresponds to the total number of distinct peptide sequences in each experiment.
Supplementary Table 1 lists all 178 experiments included in the latest build. Each experiment represents a distinct analyzed sample. Some samples are single MS runs, while in most experiments samples were fractionated to reduce complexity before each fraction was analyzed in a separate LC-MS run. In many cases, data sets that are submitted as a single ProteomeXchange dataset are separated into individual experiments when they represent different samples. For each experiment, attributes such as the total number of MS2 spectra, number of PSMs passing threshold, number of distinct peptides, etc. are listed. The 2017-04 build now contains 122,730 distinct peptides (including many semi-tryptic ones) derived from over 43 million PSMs that pass threshold, selected from a total of 172 million spectra searched. The number of proteins listed in Supplementary Table 1 is somewhat higher than the 3509 number listed in other parts of this article because the numbers in the supplementary table includes contaminants, alternative splice isoforms, variable region immunoglobulin sequences, and other TrEMBL entries with apparently unique peptides mapping to them, while the 3509 number is limited to just the ~20,000 neXtProt protein entries.
In order to gain a historical perspective on the total number of proteins gained for each of our consecutive builds in a consistent manner, we have remapped all the peptides from these builds to the 2017-01 version of neXtProt using the AtlasProphet algorithm (23). We have applied guideline # 15 of the HPP MS Dataset Interpretation Guidelines (35) such that for a protein to be listed as canonical in PeptideAtlas, it must have two distinct non-nested, uniquely mapping peptides of length ≥9 residues. In Figure 2, we show the progression of the number of these canonical proteins over historical builds, culminating in 3509 proteins in the 2017-04 build. Note that these numbers are smaller from those quoted in previous publications because new, stricter requirements have been imposed on the older data to reduce misidentifications. In addition, there are 1337 proteins in the ambiguous category; these include proteins with only one peptide, peptides of insufficient length, and cases where the detected peptides cannot distinguish between members of a group, although clearly at least one of the members of the group has been detected. A further 1700 entries are in a redundant category, meaning that although there are peptides that map to these entries, those peptides are better assigned to a canonical or ambiguous protein using rules of parsimony.
Figure 2.

Progression of the number of canonical proteins (using HPP Guidelines metrics) of historical Plasma PeptideAtlas builds. Builds since 2009 (in blue) do not contain N-glycosylation enriched datasets and were thresholded at a ~1% protein-level FDR prior to application of guideline 15 requirements. Builds prior to 2009 (in orange) contain N-glycosylation enriched datasets and were thresholded at a distinct peptide probability ≥ 0.9 prior to application of guideline 15 requirements.
In Figure 3 we compare the overlap between the lists of proteins in builds 2009 – 2017. A core set of 984 proteins are found in all of these builds, with 1153 added in 2013, 785 progressively added in 2015, and 573 progressively added in 2017. Only 14 proteins that were present in the 2009 and 2010 builds were lost in later builds. These correspond to identifications that were marginally above threshold in earlier builds, but below in later builds; thresholds must become more stringent with every build as more data are added in order to maintain a similar overall FDR. These 14 proteins are most likely false positives not borne out with far more data, implying a minimum FDR of 1.4% in the 2009 and 2010 builds. The 2013, 2014, 2015 builds have only a handful of protein entries not corroborated or carried through in later builds. There were 714 proteins with 2 peptides in the 2006 and 2007 builds that were not corroborated in any of the subsequent builds, suggesting very high FDRs of perhaps 20% in these earlier builds, although some of the discrepancy may be due to N-glycosylation enriched proteins as well as the requirement that the peptides be ≥ 9 aa.
Figure 3.

A non-proportional 5-way Venn diagram depicting the overlap of canonical proteins in Human Plasma PeptideAtlas builds 2009 – 2017; 2013 and 2014 builds are combined because they are very similar. A core set of 984 proteins are found in all of these builds, with 1153 proteins added in 2013, 785 progressively added in 2015, and 573 progressively added in 2017. The number 714 in the lower corner represents the numbers of proteins unique to the 2006 and 2007 builds, which are now known to have quite high FDRs.
We have also estimated average protein concentrations for canonical proteins in the latest 2017 build as well as older builds in order to compare the distributions of concentrations of added proteins as the Atlas expands. Concentrations were estimated based on the total number of PSMs for canonical proteins, corrected for the number of expected tryptic peptides of suitable attributes, and calibrated against a list of known protein concentrations, a method previously described in detail for the 2010 Human Plasma PeptideAtlas build by Farrah et al. (36). Figure 4 depicts the distribution of estimated protein concentrations from builds in 2010, 2013, 2015, and 2017. The mean of the distributions clearly moves to lower concentration from 2010 to 2013 to 2015. However, the shape of the distribution remains highly similar from 2015 to 2017, with very few proteins extending below 0.01 ng/ml estimated concentration. These concentrations are estimated based on a combination of all samples, which vary widely in instrumentation, quantification methods, degrees of fractionation, and sample preparation techniques. It would be useful to be able to estimate and provide plasma protein concentrations in specific physiological contexts or disease states, but data to provide that are not currently available; a highly uniform survey for plasma concentrations under carefully controlled specific physiological contexts or disease states would provide very valuable data.
Figure 4.

Relative distributions of estimated human plasma protein concentrations from four Human Plasma PeptideAtlas builds from 2010 to 2017. The mean of the distributions has moved markedly to include more proteins at concentrations below 1 ng/ml from 2010 to 2017.
All data associated with this build are made publicly available in the PeptideAtlas Data Repository and via links at http://www.peptideatlas.org/hupo/hppp/. This includes raw data and search results from each of the included experiments, listed with links at http://www.peptideatlas.org/hupo/hppp/repository/. The data are available packaged in BDBags that are uniquely identified with Minids (37). BDBags are compressed archives that contain embedded manifests and checksums that enable automated validation of completeness against the checksums. Minids provide a convenient and short minimal identifiers that can uniquely identify datasets and enable users to download and verify them using the associated toolset (http://minid.bd2k.org/).
Comparisons with RNA abundances
In order to compare the expansion of the proteome with overall RNA abundance, we group RNA abundance in tissues as compiled by the Human Protein Atlas (HPA; version 15) into four broad categories (high: FPKM > 50, medium: FPKM 10–50, low: FPKM 10-0.5; not detected: FPKM 0.5-0) (38, 39)). We note that correlation between RNA abundances and protein abundances is a subject of extensive discussions and generally low (e.g. (40)), but the completeness of RNA abundances is far higher and enables nearly complete comparisons with several protein sets. In Table 1 we show the relative distributions of proteins within these high, medium, low and not detected categories. While 42% of the population of all protein-coding genes from Ensembl (release 88.38) falls into the high RNA-abundance category in highest-expressed tissues, a somewhat higher percentage (50%) of proteins detected in all human samples are in this high abundance category, and in the Plasma PeptideAtlas an even higher percentage (77%) are in this high RNA abundance category. The total number of plasma proteins differs slightly from the figure of 3509 in the abstract and other totals in this article because there is a small number of proteins that could not be mapped to the RNA expression tables.
Table 1.
Distribution of several sets of proteins as a function of RNA abundance categories from the Human Protein Atlas. Various sources were used to collect immunoassays for plasma analysis.
| Source | Data type | Sample | Total | Annotated RNA expression | |||
|---|---|---|---|---|---|---|---|
| High > 50 FPKM |
Medium 50-10 FPKM |
Low 10-0.5 FPKM |
Not detected 0.5-0 FPKM |
||||
| Ensembl | Protein encoding genes | Tissues | 19,613 | 8,283 (42%) | 7,687 (39%) | 2,539 (13%) | 1,104 (6%) |
| PeptideAtlas | Peptides | Tissues | 15,013 | 7,555 (50%) | 6,212 (41%) | 1,114 (7%) | 132 (1%) |
| PeptideAtlas | Peptides | Plasma | 3,429 | 2,624 (77%) | 676 (20%) | 103 (3%) | 26 (1%) |
| Various | Immunoassays | Plasma | 1,918 | 1,281 (67%) | 476 (25%) | 126 (7%) | 35 (2%) |
The union of a broad range of immunoassays publicly available for plasma analysis during spring of 2017 was collected from five providers. The corresponding protein-coding genes for these targets were annotated with regards to their predicted location and RNA abundance using HPA: Somalogic (N = 1278), Olink (N = 978), R&D Systems (N = 540), Myriad RBM (N = 302) and Abcam (N = 238). For immunoassays, a greater number of targets are represented in the low RNA-abundance category than for the Plasma PeptideAtlas. The union of the 1918 proteins targeted by immunoassays and the 3429 Plasma PeptideAtlas proteins overlap by 1087 proteins.
In Table 2, we show the distribution of proteins detected in the Human Plasma PeptideAtlas over time as a function of RNA abundance categories from the Human Protein Atlas. In 2009, only ~10% of the proteins in the Plasma PeptideAtlas were found in the medium and low RNA-abundance categories, while as of 2017, nearly a quarter of all high-confidence detections are proteins with corresponding medium and low RNA-abundance. The distributions of plasma protein concentrations in relation to these RNA expression categories can be found in Supplementary Figure 1.
Table 2.
Distribution of proteins detected in the Human Plasma PeptideAtlas over time as a function of RNA abundance categories from the Human Protein Atlas.
| Build | Total | Annotated RNA expression | |||
|---|---|---|---|---|---|
| High > 50 FPKM |
Medium 50-10 FPKM |
Low 10-0.5 FPKM |
Not detected 0.5-0 FPKM |
||
| 2009 | 1,011 | 891 (88%) | 89 (9%) | 22 (2%) | 9 (1%) |
| 2010 | 1,013 | 893 (88%) | 89 (9%) | 22 (2%) | 9 (1%) |
| 2013 | 2,109 | 1698 (81%) | 330 (16%) | 60 (3%) | 21 (1%) |
| 2015 | 2,880 | 2211 (77%) | 550 (19%) | 95 (3%) | 24 (1%) |
| 2017 | 3,429 | 2624 (77%) | 676 (20%) | 103 (3%) | 26 (1%) |
In Table 3, we used the protein abundance from the 2017 Human Plasma PeptideAtlas to show the concentration distribution and increase in coverage from previous builds. A 33-fold increase in coverage was seen for low abundant proteins (< 0.1 ng/ml).
Table 3.
Distribution of the estimated protein concentration by abundance categories from the 2017 Human Plasma PeptideAtlas and previous builds adjusted to same criteria.
| Build | Total N (%) |
Protein Concentration [ng/ml] | |||
|---|---|---|---|---|---|
|
| |||||
| > 1000 | 1000-10 | 10-0.1 | < 0.1 | ||
| 2009 | 1008 (100%) | 237 (24%) | 495 (49%) | 259 (26%) | 17 (2%) |
|
| |||||
| 2010 | 1010 (100%) | 237 (23%) | 495 (49%) | 261 (26%) | 17 (2%) |
|
| |||||
| 2013 | 2139 (100%) | 257 (12%) | 791 (37%) | 991 (46%) | 100 (5%) |
|
| |||||
| 2015 | 2944 (100%) | 288 (10%) | 844 (29%) | 1458 (50%) | 354 (12%) |
| 2017 | 3521 (100%) | 309 (9%) | 913 (26%) | 1726 (49%) | 573 (16%) |
In Table 4, we show the numbers and percentages of proteins in the Plasma PeptideAtlas builds since 2009 in relation to their localizations predicted from protein sequences (39, 41). Between 2015 and 2017, the coverage for secreted proteins hardly changed, accompanied by a marked increase in the percentage of membrane and intracellular proteins.
Table 4.
Distribution of proteins detected in the Human Plasma PeptideAtlas over time as a function of localization categories from the Human Protein Atlas. The first row shows the distribution of annotations for all protein-encoding genes; the last row lists corresponding values for the immunoassays described in Table 1.
| Secreted-Membrane-Intracellular | Secreted | Secreted-Membrane | Intracellular-Secreted | Membrane | Intracellular-Membrane | Intracellular | |
|---|---|---|---|---|---|---|---|
| ALL | 298 (100%) | 1229 (100%) | 386 (100%) | 1020 (100%) | 3103 (100%) | 1693 (100%) | 11884 (100%) |
| 2017 | 127 (43%) | 421 (34%) | 133 (34%) | 422 (41%) | 320 (10%) | 300 (18%) | 1706 (14%) |
| 2015 | 124 (42%) | 412 (34%) | 128 (33%) | 407 (40%) | 243 (8%) | 233 (14%) | 1333 (11%) |
| 2013 | 99 (33%) | 331 (27%) | 92 (24%) | 299 (29%) | 176 (6%) | 148 (9%) | 964 (8%) |
| 2009 | 28 (9%) | 200 (16%) | 28 (7%) | 175 (17%) | 51 (2%) | 47 (3%) | 482 (4%) |
| Imm | 100 (34%) | 403 (33%) | 126 (33%) | 268 (26%) | 205 (7%) | 179 (11%) | 637 (5%) |
For the 2017 build, the distributions of concentrations in relation to the predicated protein localization (categorized in intracellular, secreted and membrane only) are illustrated in Supplementary Figure 2.
Targeted MS assays for the plasma proteome
While most of the efforts to expand the known plasma proteome have used shotgun proteomics techniques, the selected reaction monitoring (SRM) technique has been widely used to achieve more consistent, quantitative, and sensitive measurements of specific proteins of interest in plasma. SRM workflows are more difficult to deploy due to the requirement for predetermining effective signatures for each targeted protein, the need for spiking in heavy-labeled reference peptides for accurate quantitation, and the limited number (~200 – 1000) of those signatures that can be monitored per MS run. However, this extra effort required to carry out measurements is offset by improved sensitivity and specificity, high degree of quantitative accuracy and reproducibility, and the determination of upper limits of abundance for each targeted peptide. The earliest work by Anderson & Hunter (42) developed targeted assays (also known as multiple reaction monitoring (MRM) targeting 53 high and medium abundance proteins over 4.5 logs in abundance with a coefficient of variation (CV) of < 10% for most of them. This was followed by many other studies that have defined and demonstrated the effectiveness of signatures for certain proteins of interest (43–46). Of particular interest was the deployment of SRM assays over several MS runs for over 1000 cancer associated proteins and the detection of 182 of these cancer associated proteins in plasma covering 5 orders of magnitude (47).
To provide assistance in deploying targeted SRM assays, the SRMAtlas resource (48) was developed using highly characterized synthetic peptides representing the most proteotypic candidates of each protein, their variants, including spliced and non-synonymous variants, and some post-translational modifications. The human SRMAtlas has now determined recommended signatures for multiple peptides per protein for over 99% of all human proteins, enabling researchers to target any protein of interest without needing to undergo a lengthy signature selection process, fragment ion selection and SRM assay construction.
To further advance targeted quantitative proteomics, multiplexed fragment ion monitoring based assays have been developed into a technique, broadly termed data-independent acquisition (DIA), including SWATH-MS (49). SWATH-MS promises to provide similar sensitivity and specificity as SRM, without the problems associated with selecting targets in advance, by utilizing a well characterized spectral library developed a priori or on the fly (50, 51). In essence, high-resolution fragment ion data are collected for all peptides across chromatographic peaks above the detection threshold in a sample, but at the cost of substantially more complex informatics analysis after acquisition, which is needed to disentangle the high level of multiplexing in the data. DIA techniques perform well in comparison with SRM (52). Large scale-studies of over 1000 plasma proteins in many plasma samples are now feasible (53, 54), and also enable the study of unexpected post-translational modifications within the comprehensive multiplexed datasets (55, 56).
Affinity assays for the plasma proteome
As described above, the main driving force for defining new components of the plasma proteome has been mass spectrometric. An alternative for determining the constituent proteins in plasma is the use of affinity reagents to capture, enrich, and detect their target proteins. The use of such assays has provided general as well as clinically applied insights into the protein composition of body fluids, starting from albumin down to low abundance molecules, such as cytokines.
Affinity-based approaches require knowledge about which proteins are to be measured, and hence are by definition targeted assays. A primary bottleneck for expansion to a wider range of proteins has been linked to the availability of affinity reagents (57). According to Antibodypedia (www.antibodypedia.org; access date 2017-05-11; (58)) there are more than 2.8 million antibody reagents commercially available. Even though these cover 94% of the protein encoding genes, enormous effort is still required to validate and annotate their utility for the intended assay type and technology platform, as well as the sample type, preparation and formulation. For highly multiplexed assays, compatibility between different assay components also needs further attention. Recent discussions have indeed raised concerns about the reliability of affinity-based assays due to lack of reproducibility and specificity (59, 60). The community has therefore brought forward suggestions for how to apply communal standards on how to validate these reagents (61). The performance of reagents is indeed dependent on the application and sample type and needs to be assessed for every binder and every assay. A systematic approach using MS to qualify antibodies for their use in plasma proteomics assays has just recently been described (62). While the generation of natural or synthetic affinity reagents is discussed in detail elsewhere (63), and an increasing diversity of reagent types is making them attractive across the different fields of life science, they must be assessed separately for their use in plasma assays and the applied technologies.
Assay platforms
Today there are a variety of multiplexed assay technologies available to detect proteins in plasma (64). Whereas affinity reagents are also used in combination with MS (65, 66), the primary applications for plasma are driven towards high throughput assays in terms of sample and proteins. Here, multiplexing is achieved by different concepts such as coding the features either via their location (e.g. on a planar support or in channels) or with color or DNA barcode. Detection is then most often achieved via fluorescently labelled reporters or barcodes of oligonucleotides for amplification, hybridization or sequencing. A general feature of multiplexed affinity assays is that small quantities of samples (< 50 μl) suffice for a single highly multiplexed analysis or sets of medium-sized multiplexed analyses. Pre-fractionation is generally not required. In addition, larger sets of samples (N > 100) can be analyzed within a single lab day, since most assays are performed in microtiter plates for which automation solutions exist. Technological advances have further led to the availability of assays with very high sensitivity (fg/ml) (67). Nevertheless, the number of protein assays and degree of multiplexing remains limited in most applications, in particular when compared to MS. Challenges for immunoassays relate to interference, incompatibility of assay components causing cross-reactivity, different sample dilutions needed for reaching the optimal analytical window, assay specificity, and analytical selectivity (68).
In the long run, technologies that are capable of measuring proteins in multiplex and in a large number of samples will be beneficial for rapid analysis of plasma (69). One attractive solution is described for coupling multiplexed protein detection with read-out by DNA and to profile more than 90 proteins in a single assay (70, 71); they have been used and applied in extensive sample sets (72, 73). While these proximity extension assays use two binders per target protein, other large scale techniques employ single affinity reagents for high multiplexing capabilities. Those exist on planar arrays (74) as well as on color-coded beads (75), but both come at the expense of sensitivity and specificity, hence require complex validation schemes to assure the identity of the captured protein (76). Another single binder concept that has seen broad usage is based on modified aptamers that have been engineered to both enrich proteins from plasma and serve as the detection agent (77). The signatures of the obtained plasma profiles were associated with the protein quantitative trait loci (pQTLs) for validation (78). Recently, the system has been expanded in the number of targets per assay to 3000 proteins (79), and assays with 5000 proteins are under development although have not been released as of September 2017 (personal communication). The utilized selection technology and introduction of additional modifications may be one approach to enable the generation of even larger libraries of affinity binders, possibly reaching a possible proteome-wide coverage (80).
To provide insights into the current portfolio of immunoassays with data from MS, we collected protein assays offered by five commercial immunoassays providers, as introduced above (see Comparisons with RNA abundances). Cumulatively, these five resources constituted 1918 unique targets as of spring 2017, and provide an alternative starting point for a description of the protein assays with plasma. Out of these 1918 proteins, a set of 831 proteins were only detectable by immunoassays when compared to the 2017 build of the Human Plasma PeptideAtlas, leaving 1087 proteins detected in both. The 2017 Human Plasma PeptideAtlas contained a further 2422 proteins that remained addressed only by MS-based methods. We investigated the functional differences between the proteins covered by the two technologies by assessing their Gene Ontology (GO) term distribution using ToppCluster (81). This revealed that terms for cytokine related proteins were enriched for immunoassays (GO:0005126, GO:0005125, GO:0008009, GO:0042379). For MS, terms related to structural elements (GO:0005198, GO:0008092, GO:0003779, GO:0051015) were enriched (see also Supplementary Table 2). This indicates that (i) many low abundant proteins are still difficult to detect in plasma when using MS and that (ii) cellular proteins are found in plasma but are underrepresented in the portfolio of immunoassays. Lastly, our investigation revealed 1087 proteins found to be detectable with both technologies. GO analysis for these proteins revealed terms such as protein complex binding (GO:0032403; N =172), extracellular space (GO:0005615; N =466), cell mobility (GO:0048870; N =304), localization (GO:0051674; N =304) and cell migration (GO:0016477; N =295). These ~1000 proteins can provide a starting point for comparing the two technologies with the purpose of validating each other in increasing numbers of samples, studies and diseases. Eventually, databases such as PeptideAtlas should also be created for the widely used immunoassays to collect data and the concentrations of proteins found in plasma. These initiatives could also follow examples of the NCI assay portal (82) that currently hosts 1,517 quantitative mass spectrometry-based proteomic assays targeted for 820 proteins. In this portal, 374 assays listed for plasma or serum derived samples target 192 unique proteins.
Affinity assays will assist in our understanding of the plasma proteome once validated and sensitive assays exist for even more protein targets and for those currently not detectable by MS. The immunoassays are attractive for studies with larger sample numbers, especially if the amount of material is limited, but still less for de novo discovery purposes. Quantification remains a challenge for today’s immunoassay, since different standards are often used by different assay providers, and their detectability may differ from that of the protein found in-vivo. The emerging systems that offer either highly multiplexed or highly sensitive assays will accelerate our knowledge about the plasma proteome and move beyond the well-studied markers of inflammation. Specificity also remains a major challenge for affinity assays, as reflected in the goals of the International Working Group on Antibody Validation (61).
Considerations about samples in plasma proteomics
Studies aiming to determine the plasma proteome have largely been limited either by the number of targets or in terms of sample numbers. It is therefore likely that, if only a few phenotypes are used, the initial indications cannot be confirmed in an independent set of samples. Hence, the sample number of plasma proteomic studies must increase. Furthermore, plasma is a specimen that is attractive because it is accessible from a large number of subjects and frequently hosted by biobanks, although many are populated mainly with blood sera (83). Since the recommendations of the Human Proteome Project in 2006 (3), most plasma samples have used EDTA as anticoagulant; however, some specimens are still anticoagulated with heparin or citrate. Further, multitudes of dried blood spot cards known as Guthrie cards are available since the early 1960’s for all newborns almost worldwide providing a large resource for blood-based proteomics (84–86). This provides opportunities for new discoveries and is assisted by expanded metadata about each donor (health status and history) as well as the pre-analytical conditions related to the sample. However, both must be acknowledged and considered more carefully. The community is indeed becoming more aware that these sample related parameters may contribute to the detectability of a protein and thereby influence the outcome and reproducibility of a study (87–89). Hence, guidelines for the parameters related to the sample, such as blood draw (time point of collection, location), collection procedure (tube type, handling conditions), storage (aliquoting, container, temperature), use (thawing procedure, freeze-thawing cycles) as well as alternative formulation (dried plasma or blood spots), should therefore be considered and included in the experimental planning and downstream data analysis. A categorization of sample-related parameters can already be obtained (90) and a wider use of such codes will further enhance the validity of studies in plasma.
Conclusion
The advances in MS technologies continue to push our ability to detect and quantify a large number of proteins with high specificity and quantitative accuracy. Yet, affinity capture reagent technologies outperform MS in terms of sensitivity and speed. Ongoing efforts to better characterize affinity capture reagents in a given sample holds the potential to improve the specificity and reliability of the assays in which these are used. The combination of these two technologies is probably underutilized today, having the potential to combine the sensitivity of affinity capture and specificity of MS, albeit still with the lower speed of MS.
We have presented the 2017-04 latest build of the Human Plasma PeptideAtlas, with 3509 core neXtProt proteins meeting guideline # 15 of the HPP MS Data Interpretation Guidelines. In comparison with older plasma builds, the addition of datasets with high resolution MS2 spectra have dramatically increased the number of distinct peptides observed and the sequence coverage, although the increase in the number of proteins was less pronounced. More datasets will be added as they are deposited into the public ProteomeXchange repositories. Datasets that use new instrumentation, additional proteases beyond trypsin, and additional enrichment strategies are most likely to continue increasing the total number of proteins successfully detected in human plasma samples via MS. Challenges remain—when is the project considered closed? Human variability is enormous and so individual differences in the plasma proteome will be apparent and cloud the analyses and interpretation. For example, tissue leakage markers from growing juveniles need to be distinguished from similar or identical markers present in adults undergoing tissue healing and repair after pathology or injury. Moreover, nutritional status, diurnal variation and hormonal status will all need to be considered and hallmark proteins subclassified accordingly. Nonetheless, annotating the Human Plasma Proteome Project heralds an important achievement necessary to define and form baselines for disease biomarker discovery and monitoring before and during treatment.
Supplementary Material
Supplementary Table 1: Listing of all experiments included in the Human Plasma PeptideAtlas 2017-04 along with selected attributes.
Supplementary Table 2: GO analysis of contrasting the proteins detectable with the proteomic technologies.
Supplementary Figure 1. Relative distributions of estimated human plasma protein concentrations from the 2017 Human Plasma PeptideAtlas build in relation to RNA abundance.
Supplementary Figure 2. Relative distributions of estimated human plasma protein concentrations from the 2017 Human Plasma PeptideAtlas build in relation to annotated protein location.
Acknowledgments
This work was funded in part by the National Institutes of Health grants R01GM087221 (EWD/RLM), R24GM127667 (EWD), U54EB020406 (EWD), P50GM076547 (RLM), U54ES017885 (GSO), and U24CA210967-01 (GSO). JMS acknowledges the Knut and Alice Wallenberg Foundation for funding to Human Protein Atlas, and the Erling-Persson Family foundation for support to the KTH Center for Applied Precision Medicine (KCAP).
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Molecular & cellular proteomics: MCP. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 2.Hanash S, Celis JE. The Human Proteome Organization: a mission to advance proteome knowledge. Molecular & cellular proteomics: MCP. 2002;1:413–414. doi: 10.1074/mcp.r200002-mcp200. [DOI] [PubMed] [Google Scholar]
- 3.Omenn GS, States DJ, Adamski M, Blackwell TW, Menon R, Hermjakob H, Apweiler R, Haab BB, Simpson RJ, Eddes JS, Kapp EA, Moritz RL, Chan DW, Rai AJ, Admon A, Aebersold R, Eng J, Hancock WS, Hefta SA, Meyer H, Paik YK, Yoo JS, Ping P, Pounds J, Adkins J, Qian X, Wang R, Wasinger V, Wu CY, Zhao X, Zeng R, Archakov A, Tsugita A, Beer I, Pandey A, Pisano M, Andrews P, Tammen H, Speicher DW, Hanash SM. Overview of the HUPO Plasma Proteome Project: results from the pilot phase with 35 collaborating laboratories and multiple analytical groups, generating a core dataset of 3020 proteins and a publicly-available database. Proteomics. 2005;5:3226–3245. doi: 10.1002/pmic.200500358. [DOI] [PubMed] [Google Scholar]
- 4.Legrain P, Aebersold R, Archakov A, Bairoch A, Bala K, Beretta L, Bergeron J, Borchers CH, Corthals GL, Costello CE, Deutsch EW, Domon B, Hancock W, He F, Hochstrasser D, Marko-Varga G, Salekdeh GH, Sechi S, Snyder M, Srivastava S, Uhlén M, Wu CH, Yamamoto T, Paik Y-K, Omenn GS. The human proteome project: current state and future direction. Molecular & cellular proteomics: MCP. 2011;10:M111.009993. doi: 10.1074/mcp.M111.009993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paik YK, Omenn GS, Overall CM, Deutsch EW, Hancock WS. Recent Advances in the Chromosome-Centric Human Proteome Project: Missing Proteins in the Spot Light. Journal of Proteome Research. 2015;14:3409–3414. doi: 10.1021/acs.jproteome.5b00785. [DOI] [PubMed] [Google Scholar]
- 6.Van Eyk JE, Corrales FJ, Aebersold R, Cerciello F, Deutsch EW, Roncada P, Sanchez JC, Yamamoto T, Yang P, Zhang H, Omenn GS. Highlights of the Biology and Disease-driven Human Proteome Project, 2015–2016. Journal of Proteome Research. 2016;15:3979–3987. doi: 10.1021/acs.jproteome.6b00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.States DJ, Omenn GS, Blackwell TW, Fermin D, Eng J, Speicher DW, Hanash SM. Challenges in deriving high-confidence protein identifications from data gathered by a HUPO plasma proteome collaborative study. Nature Biotechnology. 2006;24:333–338. doi: 10.1038/nbt1183. [DOI] [PubMed] [Google Scholar]
- 8.Deutsch EW, Eng JK, Zhang H, King NL, Nesvizhskii AI, Lin B, Lee H, Yi EC, Ossola R, Aebersold R. Human Plasma PeptideAtlas. Proteomics. 2005;5:3497–3500. doi: 10.1002/pmic.200500160. [DOI] [PubMed] [Google Scholar]
- 9.Schenk S, Schoenhals GJ, de Souza G, Mann M. A high confidence, manually validated human blood plasma protein reference set. BMC medical genomics. 2008;1:41. doi: 10.1186/1755-8794-1-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gianazza E, Miller I, Palazzolo L, Parravicini C, Eberini I. With or without you - Proteomics with or without major plasma/serum proteins. J Proteomics. 2016;140:62–80. doi: 10.1016/j.jprot.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Roche S, Tiers L, Provansal M, Seveno M, Piva MT, Jouin P, Lehmann S. Depletion of one, six, twelve or twenty major blood proteins before proteomic analysis: the more the better? J Proteomics. 2009;72(6):945–51. doi: 10.1016/j.jprot.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 12.Smith MP, Wood SL, Zougman A, Ho JT, Peng J, Jackson D, Cairns DA, Lewington AJ, Selby PJ, Banks RE. A systematic analysis of the effects of increasing degrees of serum immunodepletion in terms of depth of coverage and other key aspects in top-down and bottom-up proteomic analyses. Proteomics. 2011;11(11):2222–35. doi: 10.1002/pmic.201100005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan SH, Mohamedali A, Kapur A, Baker MS. Ultradepletion of human plasma using chicken antibodies: a proof of concept study. J Proteome Res. 2013;12(6):2399–413. doi: 10.1021/pr3007182. [DOI] [PubMed] [Google Scholar]
- 14.Tan SH, Kapur A, Baker MS. Chicken immune responses to variations in human plasma protein ratios: a rationale for polyclonal IgY ultraimmunodepletion. J Proteome Res. 2012;11(12):6291–4. doi: 10.1021/pr300717b. [DOI] [PubMed] [Google Scholar]
- 15.Farrah T, Deutsch EW, Aebersold R. Using the Human Plasma PeptideAtlas to study human plasma proteins. Methods Mol Biol. 2011;728:349–74. doi: 10.1007/978-1-61779-068-3_23. [DOI] [PubMed] [Google Scholar]
- 16.Farrah T, Deutsch EW, Hoopmann MR, Hallows JL, Sun Z, Huang CY, Moritz RL. The state of the human proteome in 2012 as viewed through PeptideAtlas. Journal of Proteome Research. 2013;12:162–171. doi: 10.1021/pr301012j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Farrah T, Deutsch EW, Omenn GS, Sun Z, Watts JD, Yamamoto T, Shteynberg D, Harris MM, Moritz RL. State of the human proteome in 2013 as viewed through PeptideAtlas: comparing the kidney, urine, and plasma proteomes for the biology- and disease-driven Human Proteome Project. Journal of Proteome Research. 2014;13:60–75. doi: 10.1021/pr4010037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Keshishian H, Burgess MW, Gillette MA, Mertins P, Clauser KR, Mani DR, Kuhn EW, Farrell LA, Gerszten RE, Carr SA. Multiplexed, Quantitative Workflow for Sensitive Biomarker Discovery in Plasma Yields Novel Candidates for Early Myocardial Injury. Molecular & cellular proteomics: MCP. 2015;14:2375–2393. doi: 10.1074/mcp.M114.046813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geyer PE, Kulak NA, Pichler G, Holdt LM, Teupser D, Mann M. Plasma Proteome Profiling to Assess Human Health and Disease. Cell Systems. 2016;2:185–195. doi: 10.1016/j.cels.2016.02.015. [DOI] [PubMed] [Google Scholar]
- 20.Geyer PE, Wewer Albrechtsen NJ, Tyanova S, Grassl N, Iepsen EW, Lundgren J, Madsbad S, Holst JJ, Torekov SS, Mann M. Proteomics reveals the effects of sustained weight loss on the human plasma proteome. Molecular Systems Biology. 2016;12:901. doi: 10.15252/msb.20167357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desiere F, Deutsch EW, Nesvizhskii AI, Mallick P, King NL, Eng JK, Aderem A, Boyle R, Brunner E, Donohoe S, Fausto N, Hafen E, Hood L, Katze MG, Kennedy KA, Kregenow F, Lee H, Lin B, Martin D, Ranish JA, Rawlings DJ, Samelson LE, Shiio Y, Watts JD, Wollscheid B, Wright ME, Yan W, Yang L, Yi EC, Zhang H, Aebersold R. Integration with the human genome of peptide sequences obtained by high-throughput mass spectrometry. Genome Biology. 2005;6:R9. doi: 10.1186/gb-2004-6-1-r9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN, Aebersold R. The PeptideAtlas project. Nucleic Acids Research. 2006;34:D655–658. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deutsch EW, Sun Z, Campbell D, Kusebauch U, Chu CS, Mendoza L, Shteynberg D, Omenn GS, Moritz RL. State of the Human Proteome in 2014/2015 As Viewed through PeptideAtlas: Enhancing Accuracy and Coverage through the AtlasProphet. Journal of Proteome Research. 2015;14:3461–3473. doi: 10.1021/acs.jproteome.5b00500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller A, Eng J, Zhang N, Li X-j, Aebersold R. A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Molecular Systems Biology. 2005;1:2005.0017. doi: 10.1038/msb4100024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deutsch EW, Mendoza L, Shteynberg D, Farrah T, Lam H, Tasman N, Sun Z, Nilsson E, Pratt B, Prazen B, Eng JK, Martin DB, Nesvizhskii AI, Aebersold R. A guided tour of the Trans-Proteomic Pipeline. Proteomics. 2010;10:1150–1159. doi: 10.1002/pmic.200900375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deutsch EW, Mendoza L, Shteynberg D, Slagel J, Sun Z, Moritz RL. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clinical Applications. 2015;9:745–754. doi: 10.1002/prca.201400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bislev SL, Deutsch EW, Sun Z, Farrah T, Aebersold R, Moritz RL, Bendixen E, Codrea MC. A Bovine PeptideAtlas of milk and mammary gland proteomes. Proteomics. 2012;12:2895–2899. doi: 10.1002/pmic.201200057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bundgaard L, Jacobsen S, Sørensen MA, Sun Z, Deutsch EW, Moritz RL, Bendixen E. The Equine PeptideAtlas: a resource for developing proteomics-based veterinary research. Proteomics. 2014;14:763–773. doi: 10.1002/pmic.201300398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan QWT, Parker R, Sun Z, Deutsch EW, Foster LJ. A honey bee (Apis mellifera L.) PeptideAtlas crossing castes and tissues. BMC genomics. 2011;12:290. doi: 10.1186/1471-2164-12-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hesselager MO, Codrea MC, Sun Z, Deutsch EW, Bennike TB, Stensballe A, Bundgaard L, Moritz RL, Bendixen E. The Pig PeptideAtlas: A resource for systems biology in animal production and biomedicine. Proteomics. 2016;16:634–644. doi: 10.1002/pmic.201500195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.King NL, Deutsch EW, Ranish JA, Nesvizhskii AI, Eddes JS, Mallick P, Eng J, Desiere F, Flory M, Martin DB, Kim B, Lee H, Raught B, Aebersold R. Analysis of the Saccharomyces cerevisiae proteome with PeptideAtlas. Genome Biology. 2006;7:R106. doi: 10.1186/gb-2006-7-11-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loevenich SN, Brunner E, King NL, Deutsch EW, Stein SE, Aebersold R, Hafen E Consortium F. The Drosophila melanogaster PeptideAtlas facilitates the use of peptide data for improved fly proteomics and genome annotation. BMC bioinformatics. 2009;10:59. doi: 10.1186/1471-2105-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van PT, Schmid AK, King NL, Kaur A, Pan M, Whitehead K, Koide T, Facciotti MT, Goo YA, Deutsch EW, Reiss DJ, Mallick P, Baliga NS. Halobacterium salinarum NRC-1 PeptideAtlas: toward strategies for targeted proteomics and improved proteome coverage. Journal of Proteome Research. 2008;7:3755–3764. doi: 10.1021/pr800031f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vialas V, Sun Z, Loureiro y Penha CV, Carrascal M, Abián J, Monteoliva L, Deutsch EW, Aebersold R, Moritz RL, Gil C. A Candida albicans PeptideAtlas. Journal of Proteomics. 2014;97:62–68. doi: 10.1016/j.jprot.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deutsch EW, Overall CM, Van Eyk JE, Baker MS, Paik YK, Weintraub ST, Lane L, Martens L, Vandenbrouck Y, Kusebauch U, Hancock WS, Hermjakob H, Aebersold R, Moritz RL, Omenn GS. Human Proteome Project Mass Spectrometry Data Interpretation Guidelines 2. 1. J Proteome Res. 2016;15(11):3961–3970. doi: 10.1021/acs.jproteome.6b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Farrah T, Deutsch EW, Omenn GS, Campbell DS, Sun Z, Bletz JA, Mallick P, Katz JE, Malmstrom J, Ossola R, Watts JD, Lin B, Zhang H, Moritz RL, Aebersold R. A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol Cell Proteomics. 2011;10(9):M110 006353. doi: 10.1074/mcp.M110.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chard K, Arcy MD, Heavner B, Foster I, Kesselman C, Madduri R, Rodriguez A, Soiland-Reyes S, Goble C, Clark K, Deutsch EW, Dinov I, Price N, Toga A. I’ll take that to go: Big data bags and minimal identifiers for exchange of large, complex datasets. 2016 IEEE International Conference on Big Data (Big Data); 5–8 Dec 2016; 2016. pp. 319–328. [Google Scholar]
- 38.Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, Asplund A, Sjostedt E, Lundberg E, Szigyarto CA, Skogs M, Takanen JO, Berling H, Tegel H, Mulder J, Nilsson P, Schwenk JM, Lindskog C, Danielsson F, Mardinoglu A, Sivertsson A, von Feilitzen K, Forsberg M, Zwahlen M, Olsson I, Navani S, Huss M, Nielsen J, Ponten F, Uhlen M. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014;13(2):397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. Proteomics. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 40.Fortelny N, Overall CM, Pavlidis P, Freue GVC. Can we predict protein from mRNA levels? Nature. 2017;547(7664):E19–E20. doi: 10.1038/nature22293. [DOI] [PubMed] [Google Scholar]
- 41.Fagerberg L, Jonasson K, von Heijne G, Uhlen M, Berglund L. Prediction of the human membrane proteome. Proteomics. 2010;10(6):1141–9. doi: 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- 42.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5(4):573–88. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Kuzyk MA, Smith D, Yang J, Cross TJ, Jackson AM, Hardie DB, Anderson NL, Borchers CH. Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol Cell Proteomics. 2009;8(8):1860–77. doi: 10.1074/mcp.M800540-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whiteaker JR, Zhao L, Anderson L, Paulovich AG. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol Cell Proteomics. 2010;9(1):184–96. doi: 10.1074/mcp.M900254-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin S, Zhou Y, Lok AS, Tsodikov A, Yan X, Gray L, Yuan M, Moritz RL, Galas D, Omenn GS, Hood L. SRM targeted proteomics in search for biomarkers of HCV-induced progression of fibrosis to cirrhosis in HALT-C patients. Proteomics. 2012;12(8):1244–52. doi: 10.1002/pmic.201100601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qin S, Zhou Y, Gray L, Kusebauch U, McEvoy L, Antoine DJ, Hampson L, Park KB, Campbell D, Caballero J, Glusman G, Yan X, Kim TK, Yuan Y, Wang K, Rowen L, Moritz RL, Omenn GS, Pirmohamed M, Hood L. Identification of Organ-Enriched Protein Biomarkers of Acute Liver Injury by Targeted Quantitative Proteomics of Blood in Acetaminophen- and Carbon-Tetrachloride-Treated Mouse Models and Acetaminophen Overdose Patients. J Proteome Res. 2016;15(10):3724–3740. doi: 10.1021/acs.jproteome.6b00547. [DOI] [PubMed] [Google Scholar]
- 47.Huttenhain R, Soste M, Selevsek N, Rost H, Sethi A, Carapito C, Farrah T, Deutsch EW, Kusebauch U, Moritz RL, Nimeus-Malmstrom E, Rinner O, Aebersold R. Reproducible quantification of cancer-associated proteins in body fluids using targeted proteomics. Sci Transl Med. 2012;4(142):142ra94. doi: 10.1126/scitranslmed.3003989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kusebauch U, Campbell DS, Deutsch EW, Chu CS, Spicer DA, Brusniak MY, Slagel J, Sun Z, Stevens J, Grimes B, Shteynberg D, Hoopmann MR, Blattmann P, Ratushny AV, Rinner O, Picotti P, Carapito C, Huang CY, Kapousouz M, Lam H, Tran T, Demir E, Aitchison JD, Sander C, Hood L, Aebersold R, Moritz RL. Human SRMAtlas: A Resource of Targeted Assays to Quantify the Complete Human Proteome. Cell. 2016;166(3):766–78. doi: 10.1016/j.cell.2016.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics. 2012;11(6):O111 016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenberger G, Koh CC, Guo T, Rost HL, Kouvonen P, Collins BC, Heusel M, Liu Y, Caron E, Vichalkovski A, Faini M, Schubert OT, Faridi P, Ebhardt HA, Matondo M, Lam H, Bader SL, Campbell DS, Deutsch EW, Moritz RL, Tate S, Aebersold R. A repository of assays to quantify 10,000 human proteins by SWATH-MS. Sci Data. 2014;1:140031. doi: 10.1038/sdata.2014.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tsou CC, Avtonomov D, Larsen B, Tucholska M, Choi H, Gingras AC, Nesvizhskii AI. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods. 2015;12(3):258–64. doi: 10.1038/nmeth.3255. 7 p following 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Y, Huttenhain R, Surinova S, Gillet LC, Mouritsen J, Brunner R, Navarro P, Aebersold R. Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics. 2013;13(8):1247–56. doi: 10.1002/pmic.201200417. [DOI] [PubMed] [Google Scholar]
- 53.Liu Y, Buil A, Collins BC, Gillet LC, Blum LC, Cheng LY, Vitek O, Mouritsen J, Lachance G, Spector TD, Dermitzakis ET, Aebersold R. Quantitative variability of 342 plasma proteins in a human twin population. Mol Syst Biol. 2015;11(1):786. doi: 10.15252/msb.20145728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nigjeh EN, Chen R, Brand RE, Petersen GM, Chari ST, von Haller PD, Eng JK, Feng Z, Yan Q, Brentnall TA, Pan S. Quantitative Proteomics Based on Optimized Data-Independent Acquisition in Plasma Analysis. J Proteome Res. 2017;16(2):665–676. doi: 10.1021/acs.jproteome.6b00727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenberger G, Liu Y, Rost HL, Ludwig C, Buil A, Bensimon A, Soste M, Spector TD, Dermitzakis ET, Collins BC, Malmstrom L, Aebersold R. Inference and quantification of peptidoforms in large sample cohorts by SWATH-MS. Nat Biotechnol. 2017;35(8):781–788. doi: 10.1038/nbt.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keller A, Bader SL, Kusebauch U, Shteynberg D, Hood L, Moritz RL. Opening a SWATH Window on Posttranslational Modifications: Automated Pursuit of Modified Peptides. Mol Cell Proteomics. 2016;15(3):1151–63. doi: 10.1074/mcp.M115.054478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stoevesandt O, Taussig MJ. Affinity proteomics: the role of specific binding reagents in human proteome analysis. Expert Rev Proteomics. 2012;9(4):401–14. doi: 10.1586/epr.12.34. [DOI] [PubMed] [Google Scholar]
- 58.Bjorling E, Uhlen M. Antibodypedia, a portal for sharing antibody and antigen validation data. Molecular & cellular proteomics : MCP. 2008;7(10):2028–37. doi: 10.1074/mcp.M800264-MCP200. [DOI] [PubMed] [Google Scholar]
- 59.Baker M. Antibody anarchy: A call to order. Nature. 2015;527(7579):545–51. doi: 10.1038/527545a. [DOI] [PubMed] [Google Scholar]
- 60.Baker M. Reproducibility crisis: Blame it on the antibodies. Nature. 2015;521(7552):274–6. doi: 10.1038/521274a. [DOI] [PubMed] [Google Scholar]
- 61.Uhlen M, Bandrowski A, Carr S, Edwards A, Ellenberg J, Lundberg E, Rimm DL, Rodriguez H, Hiltke T, Snyder M, Yamamoto T. A proposal for validation of antibodies. Nat Methods. 2016;13(10):823–7. doi: 10.1038/nmeth.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fredolini C, Bystrom S, Sanchez-Rivera L, Ioannou M, Tamburro D, Branca RM, Lehtio J, Nilsson P, Schwenk JM. Mass spectrometry based qualification of antibodies for plasma proteomics. bioRxiv. 2017 doi: https://doi.org/10.1101/158022.
- 63.Taussig MJ, Stoevesandt O, Borrebaeck CA, Bradbury AR, Cahill D, Cambillau C, de Daruvar A, Dubel S, Eichler J, Frank R, Gibson TJ, Gloriam D, Gold L, Herberg FW, Hermjakob H, Hoheisel JD, Joos TO, Kallioniemi O, Koegl M, Konthur Z, Korn B, Kremmer E, Krobitsch S, Landegren U, van der Maarel S, McCafferty J, Muyldermans S, Nygren PA, Palcy S, Pluckthun A, Polic B, Przybylski M, Saviranta P, Sawyer A, Sherman DJ, Skerra A, Templin M, Ueffing M, Uhlen M. ProteomeBinders: planning a European resource of affinity reagents for analysis of the human proteome. Nat Methods. 2007;4(1):13–7. doi: 10.1038/nmeth0107-13. [DOI] [PubMed] [Google Scholar]
- 64.Ayoglu B, Haggmark A, Neiman M, Igel U, Uhlen M, Schwenk JM, Nilsson P. Systematic antibody and antigen-based proteomic profiling with microarrays. Expert review of molecular diagnostics. 2011;11(2):219–34. doi: 10.1586/erm.10.110. [DOI] [PubMed] [Google Scholar]
- 65.Fredolini C, Bystrom S, Pin E, Edfors F, Tamburro D, Iglesias MJ, Haggmark A, Hong MG, Uhlen M, Nilsson P, Schwenk JM. Immunocapture strategies in translational proteomics. Expert Rev Proteomics. 2016;13(1):83–98. doi: 10.1586/14789450.2016.1111141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anderson NL, Anderson NG, Haines LR, Hardie DB, Olafson RW, Pearson TW. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA) J Proteome Res. 2004;3(2):235–44. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- 67.Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, Piech T, Patel PP, Chang L, Rivnak AJ, Ferrell EP, Randall JD, Provuncher GK, Walt DR, Duffy DC. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat Biotechnol. 2010;28(6):595–9. doi: 10.1038/nbt.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jani D, Allinson J, Berisha F, Cowan KJ, Devanarayan V, Gleason C, Jeromin A, Keller S, Khan MU, Nowatzke B, Rhyne P, Stephen L. Recommendations for Use and Fit-for-Purpose Validation of Biomarker Multiplex Ligand Binding Assays in Drug Development. AAPS J. 2016;18(1):1–14. doi: 10.1208/s12248-015-9820-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith JG, Gerszten RE. Emerging Affinity-Based Proteomic Technologies for Large-Scale Plasma Profiling in Cardiovascular Disease. Circulation. 2017;135(17):1651–1664. doi: 10.1161/CIRCULATIONAHA.116.025446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gullberg M, Gustafsdottir SM, Schallmeiner E, Jarvius J, Bjarnegard M, Betsholtz C, Landegren U, Fredriksson S. Cytokine detection by antibody-based proximity ligation. Proc Natl Acad Sci U S A. 2004;101(22):8420–4. doi: 10.1073/pnas.0400552101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gustafsdottir SM, Ostman A, Landegren U. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20(5):473–7. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- 72.Folkersen L, Fauman E, Sabater-Lleal M, Strawbridge RJ, Franberg M, Sennblad B, Baldassarre D, Veglia F, Humphries SE, Rauramaa R, de Faire U, Smit AJ, Giral P, Kurl S, Mannarino E, Enroth S, Johansson A, Enroth SB, Gustafsson S, Lind L, Lindgren C, Morris AP, Giedraitis V, Silveira A, Franco-Cereceda A, Tremoli E, Gyllensten U, Ingelsson E, Brunak S, Eriksson P, Ziemek D, Hamsten A, Malarstig A Is group. Mapping of 79 loci for 83 plasma protein biomarkers in cardiovascular disease. PLoS Genet. 2017;13(4):e1006706. doi: 10.1371/journal.pgen.1006706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Enroth S, Enroth SB, Johansson A, Gyllensten U. Protein profiling reveals consequences of lifestyle choices on predicted biological aging. Sci Rep. 2015;5:17282. doi: 10.1038/srep17282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schroder C, Jacob A, Tonack S, Radon TP, Sill M, Zucknick M, Ruffer S, Costello E, Neoptolemos JP, Crnogorac-Jurcevic T, Bauer A, Fellenberg K, Hoheisel JD. Dual-color proteomic profiling of complex samples with a microarray of 810 cancer-related antibodies. Molecular & cellular proteomics : MCP. 2010;9(6):1271–80. doi: 10.1074/mcp.M900419-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bystrom S, Ayoglu B, Haggmark A, Mitsios N, Hong MG, Drobin K, Forsstrom B, Fredolini C, Khademi M, Amor S, Uhlen M, Olsson T, Mulder J, Nilsson P, Schwenk JM. Affinity proteomic profiling of plasma, cerebrospinal fluid, and brain tissue within multiple sclerosis. J Proteome Res. 2014;13(11):4607–19. doi: 10.1021/pr500609e. [DOI] [PubMed] [Google Scholar]
- 76.Neiman M, Fredolini C, Johansson H, Lehtio J, Nygren PA, Uhlen M, Nilsson P, Schwenk JM. Selectivity analysis of single binder assays used in plasma protein profiling. Proteomics. 2013;13(23–24):3406–10. doi: 10.1002/pmic.201300030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, Flather D, Forbes A, Foreman T, Fowler C, Gawande B, Goss M, Gunn M, Gupta S, Halladay D, Heil J, Heilig J, Hicke B, Husar G, Janjic N, Jarvis T, Jennings S, Katilius E, Keeney TR, Kim N, Koch TH, Kraemer S, Kroiss L, Le N, Levine D, Lindsey W, Lollo B, Mayfield W, Mehan M, Mehler R, Nelson SK, Nelson M, Nieuwlandt D, Nikrad M, Ochsner U, Ostroff RM, Otis M, Parker T, Pietrasiewicz S, Resnicow DI, Rohloff J, Sanders G, Sattin S, Schneider D, Singer B, Stanton M, Sterkel A, Stewart A, Stratford S, Vaught JD, Vrkljan M, Walker JJ, Watrobka M, Waugh S, Weiss A, Wilcox SK, Wolfson A, Wolk SK, Zhang C, Zichi D. Aptamer-based multiplexed proteomic technology for biomarker discovery. PloS one. 2010;5(12):e15004. doi: 10.1371/journal.pone.0015004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suhre K, Arnold M, Bhagwat AM, Cotton RJ, Engelke R, Raffler J, Sarwath H, Thareja G, Wahl A, DeLisle RK, Gold L, Pezer M, Lauc G, El-Din Selim MA, Mook-Kanamori DO, Al-Dous EK, Mohamoud YA, Malek J, Strauch K, Grallert H, Peters A, Kastenmuller G, Gieger C, Graumann J. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun. 2017;8:14357. doi: 10.1038/ncomms14357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, Oliver-Williams C, Kamat MA, Prins BP, Wilcox SK, Zimmerman ES, Chi A, Bansal N, Spain SL, Wood AM, Morrell NW, Bradley JR, Janjic N, Roberts DJ, Ouwehand WH, Todd JA, Soranzo N, Suhre K, Paul DS, Fox CS, Plenge RM, Danesh J, Runz H, Butterworth AS. Consequences Of Natural Perturbations In The Human Plasma Proteome. bioRxiv. 2017 doi: https://doi.org/10.1101/134551.
- 80.Gawande BN, Rohloff JC, Carter JD, von Carlowitz I, Zhang C, Schneider DJ, Janjic N. Selection of DNA aptamers with two modified bases. Proc Natl Acad Sci U S A. 2017;114(11):2898–2903. doi: 10.1073/pnas.1615475114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kaimal V, Bardes EE, Tabar SC, Jegga AG, Aronow BJ. ToppCluster: a multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 2010;38(Web Server issue):W96–102. doi: 10.1093/nar/gkq418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Whiteaker JR, Halusa GN, Hoofnagle AN, Sharma V, MacLean B, Yan P, Wrobel JA, Kennedy J, Mani DR, Zimmerman LJ, Meyer MR, Mesri M, Rodriguez H, Paulovich AG Clinical Proteomic Tumor Analysis C. CPTAC Assay Portal: a repository of targeted proteomic assays. Nat Methods. 2014;11(7):703–4. doi: 10.1038/nmeth.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wichmann HE, Kuhn KA, Waldenberger M, Schmelcher D, Schuffenhauer S, Meitinger T, Wurst SH, Lamla G, Fortier I, Burton PR, Peltonen L, Perola M, Metspalu A, Riegman P, Landegren U, Taussig MJ, Litton JE, Fransson MN, Eder J, Cambon-Thomsen A, Bovenberg J, Dagher G, van Ommen GJ, Griffith M, Yuille M, Zatloukal K. Comprehensive catalog of European biobanks. Nat Biotechnol. 2011;29(9):795–7. doi: 10.1038/nbt.1958. [DOI] [PubMed] [Google Scholar]
- 84.Guthrie R, Susi A. A Simple Phenylalanine Method for Detecting Phenylketonuria in Large Populations of Newborn Infants. Pediatrics. 1963;32:338–43. [PubMed] [Google Scholar]
- 85.Chambers AG, Percy AJ, Yang J, Borchers CH. Multiple Reaction Monitoring Enables Precise Quantification of 97 Proteins in Dried Blood Spots. Mol Cell Proteomics. 2015;14(11):3094–104. doi: 10.1074/mcp.O115.049957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bjorkesten J, Enroth S, Shen Q, Wik L, Hougaard D, Cohen A, Sorensen L, Giedraitis V, Ingelsson M, Larsson A, Kamali-Moghaddam M, Landegren U. Stability of Proteins in Dried Blood Spot Biobanks. Mol Cell Proteomics. 2017;16(7):1286–1296. doi: 10.1074/mcp.RA117.000015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zimmerman LJ, Li M, Yarbrough WG, Slebos RJ, Liebler DC. Global stability of plasma proteomes for mass spectrometry-based analyses. Molecular & cellular proteomics : MCP. 2012;11(6):M111 014340. doi: 10.1074/mcp.M111.014340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ostroff R, Foreman T, Keeney TR, Stratford S, Walker JJ, Zichi D. The stability of the circulating human proteome to variations in sample collection and handling procedures measured with an aptamer-based proteomics array. Journal of proteomics. 2010;73(3):649–66. doi: 10.1016/j.jprot.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 89.Qundos U, Hong MG, Tybring G, Divers M, Odeberg J, Uhlen M, Nilsson P, Schwenk JM. Profiling post-centrifugation delay of serum and plasma with antibody bead arrays. J Proteomics. 2013;95:46–54. doi: 10.1016/j.jprot.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 90.Lehmann S, Guadagni F, Moore H, Ashton G, Barnes M, Benson E, Clements J, Koppandi I, Coppola D, Demiroglu SY, DeSouza Y, De Wilde A, Duker J, Eliason J, Glazer B, Harding K, Jeon JP, Kessler J, Kokkat T, Nanni U, Shea K, Skubitz A, Somiari S, Tybring G, Gunter E, Betsou F International Society for B, Environmental Repositories Working Group on Biospecimen S. Standard preanalytical coding for biospecimens: review and implementation of the Sample PREanalytical Code (SPREC) Biopreserv Biobank. 2012;10(4):366–74. doi: 10.1089/bio.2012.0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Listing of all experiments included in the Human Plasma PeptideAtlas 2017-04 along with selected attributes.
Supplementary Table 2: GO analysis of contrasting the proteins detectable with the proteomic technologies.
Supplementary Figure 1. Relative distributions of estimated human plasma protein concentrations from the 2017 Human Plasma PeptideAtlas build in relation to RNA abundance.
Supplementary Figure 2. Relative distributions of estimated human plasma protein concentrations from the 2017 Human Plasma PeptideAtlas build in relation to annotated protein location.