

Abstract
The ability to remotely trigger CRISPR/Cas9 activity would enable new strategies to study cellular events with greater precision and complexity. We developed a method to photocage the activity of the guide RNA called ‘CRISPR-plus’ (CRISPR-precise light-mediated unveiling of sgRNAs). The photoactivatable capability of our CRISPR-plus method is compatible with simultaneous targeting of multiple DNA sequences and supports numerous modifications that can enable guide RNA labeling for use in imaging and mechanistic inquiries.
Keywords: Photochemistry, CRISPR, Nucleic acids, Gene technology, DNA Cleavage
Entry for the Table of Contents
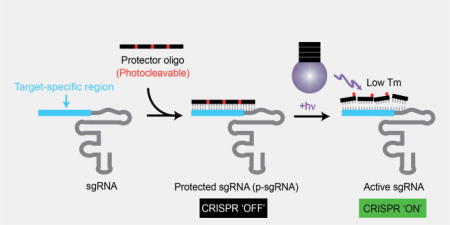
Turn ‘ON’ CRISPR with light: CRISPR can be brought under the control of light simply by hybridizing a single chimeric guide RNAs (sgRNA) with its complementary oligonucleotide containing photocleavable groups (protector oligonucleotide). These protected sgRNA (p-sgRNA) remain inactive, blocking CRISPR activity, until the protector oligonucleotides are cleaved by a remote light trigger.
The RNA-guided CRISPR/Cas9 system is a genome editing technology with broad biological and therapeutic applications.[1–2] The field’s enthusiasm for the potential of this platform has led to a rapidly-expanding toolbox[3], including site-specific single gene editing using photoactivatable CRISPR, via a modified Cas9 enzyme that incorporates light responsive domains or site-specific caging groups.[5–8]
The majority of light-activated approaches depend on modifications of the Cas9 enzyme, whereas recent efforts have modified the single chimeric guide RNA (sgRNA) as an alternative approach to genome editing.[9–10] In line with this shift, our method called ‘CRISPR-plus’ (CRISPR-precise light-mediated unveiling of sgRNAs) incorporates photocleavable oligonucleotides that complement target regions of sgRNA, in the absence of Cas9 modifications.
While photocleavable nucleotides have been used in other biological systems[11–15], we believe ours is the first use of photactivatable oligonucleotides in CRISPR activation. While other approaches genetically modify Cas9[5–8], our approach does not require any engineering of target cells. Also, these commercially available oligonucleotides are simple to design, chemically synthesize, modify, functionalize, purify, and characterize. Thus, the photoactivatable capability of CRISPR-plus affords simple and convenient control over editing within a genetic sequence, enables indirect and transient labeling of sgRNAs, and can multiplex different sgRNAs. We predict that these traits will permit greater mechanistic and causal testing of gene functions and roles in a wide range of cellular systems.
To establish CRISPR-plus, we designed complementary ssDNA oligonucleotides (commercially available from Gene Link, Inc.) or ‘protectors’, of varying lengths and positions, that contain photocleavable groups such that when they hybridize to the target region of an sgRNA, the resulting complex has a high melting temperature (Tm). The presence of the hybridized protector thus prevents the sgRNA from binding to the target DNA until the protector is photolyzed, releasing it from the sgRNA (Figure 1a, Supporting Information (SI) Figure S2 and Table 1c–d). Upon photolysis, the short fragments of the cleaved protector oligonucleotides will possess a reduced binding affinity for sgRNAs owing to their lower Tm, rendering the target DNA susceptible to Cas9-mediated cleavage. We tested a range of protectors designed to be complementary to the target regions (T) of sgRNA and that contain photocleavable (PC) groups (PCT1–PCT5) spaced 6 nucleotides (6-nt) apart, as well as corresponding non-photocleavable control protectors (T1–T5) that do not contain PC groups, using an in vitro DNA cleavage assay to determine the efficiency with which they block sgRNAs that target a GFP DNA sequence (Figure 1b, and Table 1c–d). In the absence of light, both the PCcontaining p-sgRNA (protected guide RNA) and the non-PC control protectors (T3–T5) eliminated virtually all Cas9-mediated cleavage of GFP target DNA, even at the lowest concentration tested (Figure 1b–d and SI Figure S3).
Figure 1.

CRISPR-plus achieves photoactivatable blockade of Cas9-mediated DNA targeting. (a) CRISPR-plus concept. (b) Fold increase in DNA cleavage following light exposure (365 nm, 6.0 J/cm2) with complementary sgGFP1 protectors of different lengths and position that contain photocleavable (PC) groups spaced 6 nucleotides apart (PCT1–PCT5), or protectors without PC groups (T1–T5), tested using an in vitro cleavage assay (n=2) (c) Representative gel electrophoresis data from the in vitro cleavage assay, for a 2:1 ratio of protector PCT5:sgRNA. The 702 bp target GFP DNA is cut to yield 563 bp and 139 bp fragments in the presence of sgRNA, only after p-sgRNA photolysis (365 nm, 6.0 J/cm2). (d) Quantification of replicate in vitro assays from (c) (n=3), where data is expressed as a fraction of uncleaved DNA, as calculated based on band intensities. Data is normalized to control untreated DNA, and mean values with standard deviation are plotted (n=3). Unpaired student t-test was performed for sgRNA with PCT5 in the presence or absence of light exposure groups and p values are represented with asterisks ***p < 0.001.
However, after only 2–5 seconds of light exposure (equivalent to 0.4–1.0 J/cm2, using an OmniCure S2000, 365 nm filter, 200 mW/cm2), significant photolysis-mediated cleavage of target DNA was recovered using several of the PC-containing protectors, whereas the non-PC protectors retained complete blocking efficiency even in the presence of light (SI Figure S4). As an important control, we also confirmed both the purity and photolability of the protectors alone (without sgRNA) using denaturing PAGE gels and/or HPLC (SI Figure S5). Based on the positioning and length of the most efficient CRISPR-plus protectors for the GFP target sequence (Figures 1b–d and SI Figure S3), we designed and tested protectors for additional GFP target regions, as well as two endogenous genes, CD71 and CD33 (Figure 2 and SI Figure S6). Consistent with our initial findings, all 6 PC-protectors afforded protection from DNA cleavage that was lost after exposure to light. These results suggest applicability to other genomic targets.
Figure 2.

Validation of optimized 24-nt target-specific protectors with other sgRNAs and targets in an in vitro DNA cleavage assay. (a–c) PC-containing (PCT6–PCT11) or non-PC (T6–T11) oligonucleotides complementary to 6 additional sgRNAs targeting three different DNA targets (a) GFP (green), (b) CD71 (magenta), and (c) CD33 (blue), in the absence or presence of light (365 nm, 6.0 J/cm2). % uncleaved DNA was calculated from the band intensities of the gels (SI Figure S6). Data is normalized to the cleavage of control, untreated DNA. Mean values +/− s.d. are plotted for multiple repeat experiments (sgGFP-2, sgGFP-3 and sgCD71-1, n=4; sgCD71-2 and sgCD33-1, n=2; sgCD33-3, n=2 with T11, n=1). Unpaired student’s t-test was performed between irradiated and non-irradiated samples, as described in the SI data analysis section, and p values are represented with asterisks *p < 0.05, **p < 0.01, ****p < 0.0001.
The CRISPR-plus platform intentionally targets sgRNA, in order to provide distinct advantages over other light inducible Cas9 methods, yet in doing so, it is important to confirm that CRISPR-plus retains the capacity to target multiple guide RNAs simultaneously.[5–8] To assay for potential multiplex capacity, we performed a series of in vitro cleavage assays in which we combined 1, 2 or all 3 PC-protectors in the presence of GFP, CD33, and CD71 target DNA and their corresponding sgRNAs (Figure 3, SI Figures S7 and S8). We observed that the light-activated target DNA cleavage response was specific to the presence or absence of the photoactivatable protector-sgRNA complexes, even in the context of mixed targets and mixed sgRNA sequences (Figure 3, SI Figures S7 and S8). Notably, we only observed target DNA cleavage after its cognate sgRNA was unveiled with light-mediated disruption of the protectors. These results suggest that, by virtue of its photoactivatable capacity, CRIPSR-plus can enable simultaneous, synchronized gene editing of multiple targets.
Figure 3.

CRISPR-plus enables simultaneous targeting of multiple genes. (a) Schematic representation of combining three different sgRNAs with respective protectors and their activation with light. (b) Simultaneous in vitro targeting of three genes (GFP, CD71 and CD33) by their corresponding sgRNAs (sgGFP (green), sgCD71 (magenta), and sgCD33 (blue)) with (p-sgRNA) or without (sgRNA) their cognate photolabile protectors in the absence (solid bars) or presence (open bars) of light (365 nm, 6.0 J/cm2). The proportion of uncleaved DNA is expressed relative to untargeted DNA, averaged over two individual experiments and sgCon bars represent non-targeting scrambled sgGFP-1. Error bars represent standard deviation (n=2). DNA cleavage was blocked only in the presence of the corresponding protector, and all the p-sgRNA-treated conditions regained CRISPR activity in the presence of light.
After establishing that photolabile protection mediated by PC-containing protectors’ functions as anticipated in an in vitro cleavage assay, we sought to determine whether this capacity is maintained in a more complex cellular environment. To this end, we generated a Cas9 / destabilized GFP (Cas9/d2eGFP) co-expressing reporter line using HeLa cells and tested the efficiency of previously screened ssDNA protectors designed to be complementary to GFP-targeting guide RNA (sgGFP1). To quantify CRISPR activity, we performed FACS analysis of Cas9/d2eGFP HeLa cells to measure GFP protein expression and, as anticipated, observed an increase in the frequency of GFP-negative cells upon addition of sgGFP RNA. We observed a reduced efficiency of CRISPR-blocking activity in cells with shorter protectors (12-nt and 18-nt) relative to their inhibition of Cas9-mediated DNA cleavage in the in vitro cleavage assay, possibly due to easier dissociation of shorter protectors inside cells (data not shown). However, when longer ssDNA protectors (24-nt) containing PC groups were used in the cell-based assay, the CRISPR-plus system yielded a population change that was conditionally blocked in the presence of PCT5 p-sgGFP, prior to exposure to UV light (Figure 4c–d and SI Figure S9). To further confirm that the light-mediated loss of p-sgRNA-dependent protection of target DNA sequences is actually mediated by genomic DNA cleavage, we performed a SURVEYOR nuclease assay on intact, Cas9/d2eGFP HeLa cells and measured % indels (insertion/deletion mutations) according to published protocols.[16] We observed significant, light-insensitive indel formation with the transfection of GFP- or CD71-targeted sgRNA, whereas the inclusion of the appropriate PC protector RNA for either target significantly blocked indel formation. Furthermore, this protection from Cas9-mediated cleavage was recoverable after exposure to UV light (4.0 J/cm2 at 365 nm, generated using a CL-1000 UV Cross-linker UVP light source with power density of 4.45 mW/cm2, as measured by an OAI 306 UV power meter) for both GFP and CD71 (Figure 4a–b). It has been previously confirmed in multiple studies that a single exposure of up to 5.0 J/cm2 365 nm UV light is non-photogenotoxic in the HaCaT (human keratinocyte) cell line[17] and has also been used in different tumor models in vivo[14, 18–19], consistent with the lack of overt, acute photocytotoxicity observed in our platform (SI Figure S10).
Figure 4.

Validation of CRISPR-plus in cells. (a, b) SURVEYOR nuclease assay was performed 72 hours post-transfection using Cas9/d2eGFP-expressing HeLa cells transfected with sgGFP-1 (a), or sgCD71-1 (b), with PCT5 or PCT8, respectively, in the absence or presence of light. Uncleaved GFP DNA (green, 500 bp) cut to a shorter fragment (black, 408 bp), and uncleaved CD71 DNA (magenta, 434 bp) with its 350 bp & 84 bp cleavage fragments are indicated. (c) Representative flow cytometry histograms of Cas9/d2eGFP-expressing HeLa cells transfected with sgGFP-1, with or without PCT5. The presence or absence of light (365 nm, 4.0 J/cm2) is indicated for each condition and control traces (top) represent untreated cells. Additional controls included in SI Figure S9. (d) Quantification of the fraction of GFP negative cells observed in (c), where mean values are plotted with error bars indicating standard deviation (n=3). Unpaired student’s t-test was performed for sgRNA with PCT5 in the presence or absence of light exposure groups and p values are represented with asterisks ***p < 0.001.
We developed CRISPR-plus as a modular approach using a photocleavable complement oligo against the target region of sgRNAs to achieve inducible, target-specific editing of any gene(s) of interest. This light-dependent approach permits simultaneous targeting of multiple sequences and offers the possibility of achieving temporal precision in the activation of sgRNAs. Using the CRISPR-plus methodology, we performed targeted cleavage of the PCR products of three genomic sequences, including two genes relevant to multiple myeloma and acute myeloid leukemia development.[20–21] This removable protector approach can be immediately extended to numerous Cas nuclease and sgRNA variants with other effector functions.[9,23–29]
Recently, Dieters et al., reported a caged Cas9 approach that achieved a robust off/on switch for multiple sgRNAs in cells, minimal leakage of Cas9 activity in the absence of light, and with robust recovery of Cas9 activity in a subset of sgRNAs after light exposure.[7] In comparison, our system also showed a robust off/on switch in an in vitro cleavage assay, using multiple guide RNAs individually and in combination, but our dynamic range was lower when tested in cells. We did observe some light-independent cleavage activity after day 5 (data not shown), possibly due to complex dissociation inside cells. Despite this time-dependent leakage of activity, an important distinguishing factor is that our approach is based on sgRNA modifications, and thus it opens up new possibilities of modifying, controlling and improving CRISPR activity by non-genetic methods.
In an attempt to improve the dynamic range, and to test whether protectors can tolerate modifications that may enable future functionalization, we 1) changed the backbone of the protectors to RNA or 2’-OMe RNA, 2) decreased the number of PC groups on a ssDNA protector, and 3) modified the 3’ end of ssDNA protectors to sterically block the 5’ end of sgRNA. Each of these modifications was tested in cells, with or without pre-irradiation (SI Figure S11). In the first case, despite achieving a more stable RNA/sgRNA duplex, the RNA protectors showed very poor blocking of CRISPR activity (SI Figure S11), while 2’-OMe RNA protectors performed similar to DNA protectors. Secondly, decreasing the number of PC groups in a 24-nt protector resulted in a comparable blocking of activity in the absence of light, yet activation of CRISPR activity in the presence of light was not as robust (slightly reduced), likely due to higher residual binding of the 8-nt protector fragments, compared to 6-nt spaced PC groups. Finally, the addition of a FAM dye to the 3’ end of a ssDNA protector yielded strong inhibition of CRISPR activity in the absence of light and resulted in robust CRIPSR-plus activity, similar to that observed using an unmodified ssDNA protector. This finding emphasizes that protectors tolerate modifications near the 5’ end of sgRNA, and highlight that they are thus amenable for use in indirect labeling of sgRNAs while maintaining their utility as a CRISPR-plus switch.
While we acknowledge that our first generation CRISPR-plus method lacks a perfect off/on switch, we believe that it still offers an attractive, simple platform to many researchers to adapt for their own applications by further modifying protectors, or by conjugating the protector to the sgRNA. Notably, Doudna and coworkers have shown that 10-nt ssDNA can stabilize the Cas9/sgRNA complex and hence is required for target binding with Cas9/sgRNA.[30] Our efforts extend these findings to highlight that the position of ssDNA binding to the sgRNA impacts Cas9-mediated cleavage (Figure 1b), and may also influence enzyme binding to the complex.
Further studies may provide additional mechanistic insights, improved control and specificity. While our current demonstrations of genome editing via CRISPR-plus activation of sgRNAs are irreversible and dependent on activation with UV light, activation duration has the potential to be controlled by modulating Cas9/sgRNA persistence, and orthogonal photocleavable groups can be employed to achieve multiplexed activation of protectors in a spatiotemporal-controlled manner. Future generations of CRISPR-plus could also incorporate photocleavable oligonucleotides across the sgRNA backbone region to yield universal, off-target sequence-agnostic protectors, or other modified sgRNAs designed to mediate selective blockade and thereby prevent the recruitment of other effector domains.[9] Overall, our CRISPR-plus approach provides a rapid and simple approach for light-mediated control of genome editing.
Supplementary Material
Acknowledgments
The authors thank Dr. Phillip A. Sharp for providing HeLa cells expressing destabilized GFP (HeLa-d2eGFP cells) and Dr. Feng Zhang and his lab members for helpful discussions. This study was supported in part by the Ludwig Center for Molecular Oncology, the Marie D. & Pierre Casimir-Lambert Fund, and a Koch Institute Support Grant P30-CA14051 from the National Cancer Institute (Swanson Biotechnology Center). S.N.B is a Howard Hughes Medical Institute Investigator.
References
- 1.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doudna JA, Charpentier E. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- 3.Sander JD, Joung JK. Nat. Biotechnol. 2014;32:347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang F. Hum. Gene Ther. 2015;26:409–410. doi: 10.1089/hum.2015.29002.fzh. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nihongaki Y, Yamamoto S, Kawano F, Suzuki H, Sato M. Chem. Biol. 2015;22:169–174. doi: 10.1016/j.chembiol.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Nihongaki Y, Kawano F, Nakajima T, Sato M. Nat. Biotechnol. 2015;33:755–760. doi: 10.1038/nbt.3245. [DOI] [PubMed] [Google Scholar]
- 7.Hemphill J, Borchardt EK, Brown K, Asokan A, Deiters A. J. Am. Chem. Soc. 2015;137:5642–5645. doi: 10.1021/ja512664v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polstein LR, Gersbach CA. Nat. Chem. Biol. 2015;11:198–200. doi: 10.1038/nchembio.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hendel A, Bak RO, Clark JT, Kennedy AB, Ryan DE, Roy S, Steinfeld I, Lunstad BD, Kaiser RJ, Wilkens AB, et al. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah S, Rangarajan S, Friedman SH. Angew. Chem. Int. Ed. 2005;44:1328–1332. doi: 10.1002/anie.200461458. [DOI] [PubMed] [Google Scholar]
- 12.Tang X, Dmochowski IJ. Mol. Biosyst. 2007;3:100–110. doi: 10.1039/b614349k. [DOI] [PubMed] [Google Scholar]
- 13.Shestopalov IA, Sinha S, Chen JK. Nat. Chem. Biol. 2007;3:650–651. doi: 10.1038/nchembio.2007.30. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Tong R, Chu H, Wang W, Langer R, Kohane DS. Proc. Natl. Acad. Sci. USA. 2014;111:17099–17103. doi: 10.1073/pnas.1420105111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hemphill J, Liu Q, Uprety R, Samanta S, Tsang M, Juliano RL, Deiters A. J. Am. Chem. Soc. 2015;137:3656–3662. doi: 10.1021/jacs.5b00580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Toyooka T, Ishihama M, Ibuki Y. J. Invest. Dermatol. 2011;131:1313–1321. doi: 10.1038/jid.2011.28. [DOI] [PubMed] [Google Scholar]
- 18.Tong R, Chiang HH, Kohane DS. Proc. Natl. Acad. Sci. USA. 2013;110:19048–19053. doi: 10.1073/pnas.1315336110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dudani JS, Jain PK, Kwong GA, Stevens KR, Bhatia SN. ACS Nano. 2015;9:11708–11717. doi: 10.1021/acsnano.5b05946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ruiz-Arguelles GJ, San Miguel JF. Mayo. Clin. Proc. 1994;69:684–690. doi: 10.1016/s0025-6196(12)61350-0. [DOI] [PubMed] [Google Scholar]
- 21.Liu Q, Wang M, Hu Y, Xing H, Chen X, Zhang Y, Zhu P. Leuk. Lymphoma. 2014;55:892–898. doi: 10.3109/10428194.2013.819100. [DOI] [PubMed] [Google Scholar]
- 22.Walter RB. Expert. Opin. Ther. Targets. 2014;18:715–718. doi: 10.1517/14728222.2014.909413. [DOI] [PubMed] [Google Scholar]
- 23.Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. Cell. 2014;159:647–661. doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Connell MR, Oakes BL, Sternberg SH, East-Seletsky A, Kaplan M, Doudna JA. Nature. 2014;516:263–266. doi: 10.1038/nature13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales AP, Li Z, Peterson RT, Yeh JR, et al. Nature. 2015;523:481–485. doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, et al. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zetsche B, Volz SE, Zhang F. Nat. Biotechnol. 2015;33:139–142. doi: 10.1038/nbt.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. Cell. 2015;163:759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang F, Zhou K, Ma L, Gressel S, Doudna JA. Science. 2015;348:1477–1481. doi: 10.1126/science.aab1452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.