

Abstract
Cellular senescence is a fundamental biological process that has profound implications in cancer development and therapeutics, but the underlying mechanisms remain elusive. Here we show that carnitine palmitoyltransferase 1C (CPT1C), an enzyme that catalyzes carnitinylation of fatty acids for transport into mitochondria for β-oxidation, plays a major role in the regulation of cancer cell senescence through mitochondria-associated metabolic reprograming. Metabolomics analysis suggested alterations in mitochondria activity, as revealed by the marked decrease in acylcarnitines in senescent human pancreatic carcinoma PANC-1 cells, indicating low CPT1C activity. Direct analyses of mRNA and protein show that CPT1C is significantly reduced in senescent cells. Furthermore, abnormal mitochondrial function was observed in senescent PANC-1 cells, leading to lower cell survival under metabolic stress and suppressed tumorigenesis in a mouse xenograft model. Knock-down of CPT1C in PANC-1 cells induced mitochondrial dysfunction, caused senescence-like growth suppression and cellular senescence, suppressed cell survival under metabolic stress, and inhibited tumorigenesis in vivo. Further, CPT1C knock-down suppressed xenograft tumor growth in situ. Silencing of CPT1C in five other tumor cell lines also caused cellular senescence. On the contrary, gain-of-function of CPT1C reversed PANC-1 cell senescence and enhanced mitochondrial function. This study identifies CPT1C as a novel biomarker and key regulator of cancer cell senescence through mitochondria-associated metabolic reprograming, and suggests that inhibition of CPT1C may represent a new therapeutic strategy for cancer treatment through induction of tumor senescence.
Introduction
Cellular senescence is a fundamental biological process that has profound implications in cancer development and therapeutics, but the underlying mechanisms are still poorly understood. Cellular metabolomics can uncover information reflecting changes to metabolic pathways and processes occurring within cells [1]. In this study, metabolomics was performed on replicative PANC-1 cell senescence, revealing major alterations in mitochondrial metabolism as revealed by a decrease in acylcarnitines, indicating low CPT1C activity. Furthermore, low CPT1C expression caused abnormal energy metabolism and mitochondrial dysfunction of late passage PANC-1 cells, resulting in low cell survival under metabolic stress and suppression of tumorigenesis. Most importantly, knock-down of CPT1C can trigger mitochondrial dysfunction and consequently lead to impaired proliferation and cellular senescence, suppressed cell survival under metabolic stress and tumorigenesis. Further siRNA CPT1C treatment contributed to the suppression of xenograft tumor growth in situ. Silencing of CPT1C also induced mitochondrial dysfunction-mediated cellular senescence in five other tumor cell lines. On the contrary, gain-of-function of CPT1C reversed PANC-1 cell senescence and enhanced mitochondrial function. Taken together, CPT1C is a novel biomarker for mitochondrial dysfunction-associated cellular senescence, and suggests that inhibition of CPT1C may represent a new therapeutic strategy for cancer treatment through induction of tumor senescence.
Results
Culture-related replicative PANC-1 cell senescence
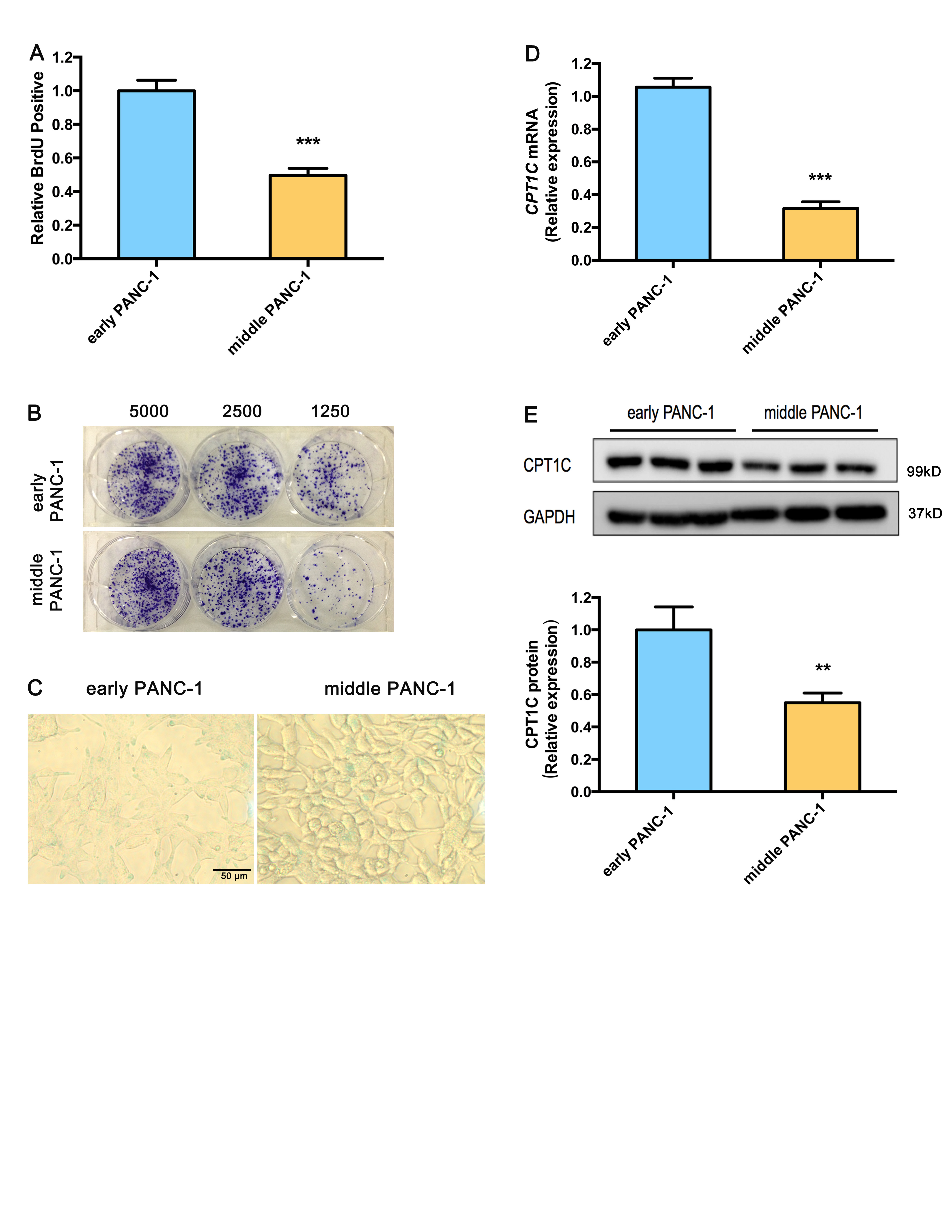
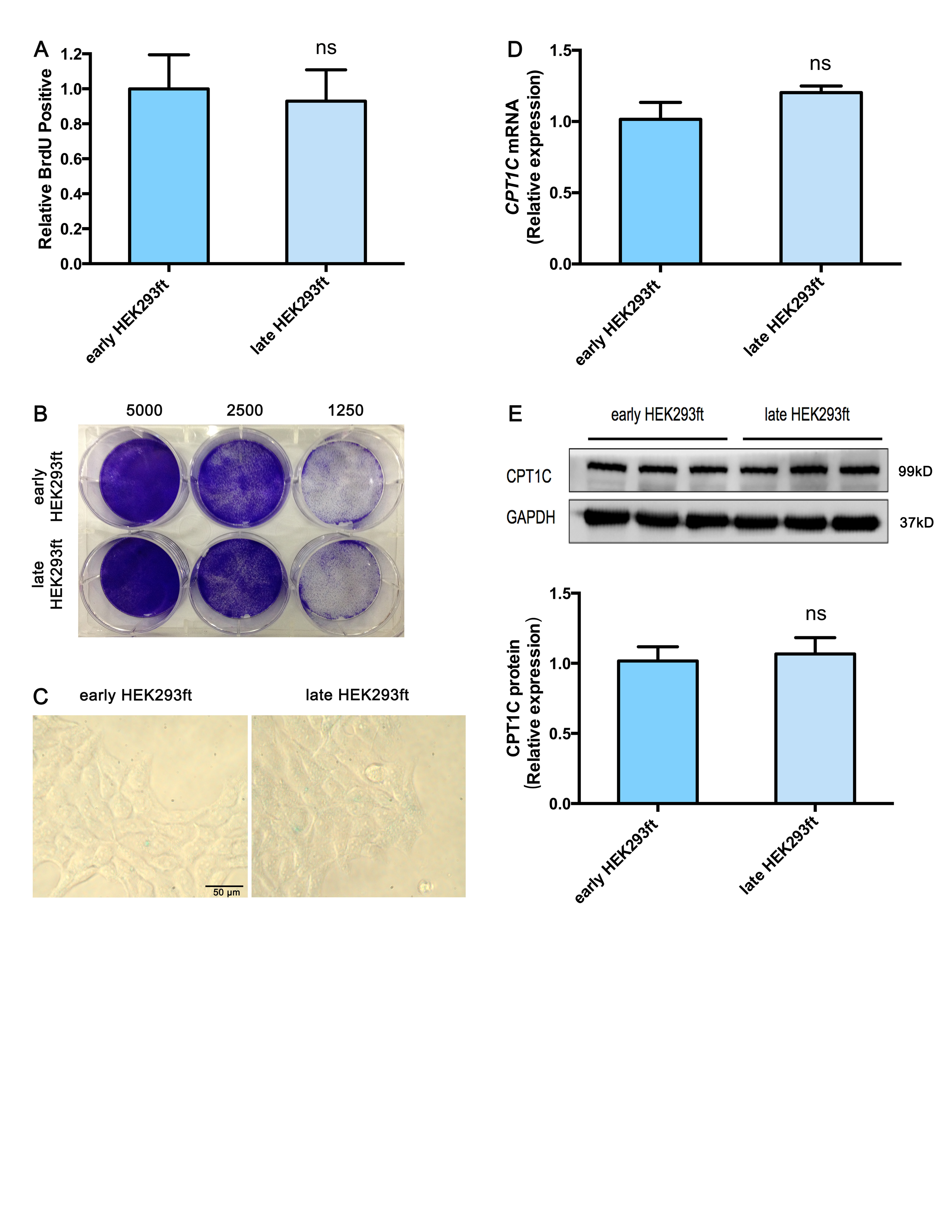
Extended culturing of human pancreatic epithelioid carcinoma PANC-1 cells led to growth arrest and severe cellular senescence. For growth arrest analysis, late PANC-1 (PANC-1 cells Passage/P 100) had lower proliferation than early PANC-1 (P 20) (Fig. 1a). Consistently, a weaker ability of late PANC-1 cells to form colonies was observed when seeded at the indicated limiting dilution (Fig. 1b, Supplementary Fig. S5A). The senescence-associated beta-galactosidase (SA-β-gal) staining showed early PANC-1 cells were nearly negative for β-gal, while the late PANC-1 cells had a significant increase in SA-β-gal activity and were positive for senescent signals (Fig. 1c, Supplementary Fig. S5B). Late PANC-1 cells were characterized by a flattened appearance arranged like flagstones, and increased granularities in the cytoplasm (Fig. 1c). Furthermore, DNA staining revealed a strikingly punctate pattern (positive DNA SAHF) in late PANC-1 cells, while early PANC-1 cells showed a completely different homogenous staining (Fig. 1d); degenerative changes of enlarged and irregular-shape nuclei were also triggered in late PANC-1 cells (Fig. 1d). Additionally, IL8 mRNA, encoding a key SASP factor [2], was increased in late PANC-1 cells (Fig. 1e). Finally, replication-triggered telomere shortening was approximately 2.7 kb in the late PANC-1 cells (Fig. 1f). Comparably, the middle PANC-1 (P 60) cells also displayed slight cellular senescence (Supplementary Fig. S2A–C, S5C, S5D). Whereas analysis of early HEK293ft (immortalized HEK293ft cells P 20) and late HEK293ft (P 100) cells, as a representative pair of negative controls for replicative senescence, were nearly negative for the senescence-associated phenotype after a large number of passages (Supplementary Fig. S3A–C). Taken together, these data indicated that extended cell culturing of PANC-1 cells triggered a strong senescence-like growth suppression and severe cellular senescence.
Fig. 1. Culture-related replicative cell senescence in late PANC-1 cells.

a BrdU incorporation during DNA synthesis in replicative late PANC-1 cells. Data are represented as mean ± S.E.M, n = 4 (***p < 0.001). b Ability of replicative late PANC-1 cells to form colonies when seeded at the indicated limiting dilutions. c SA-β-gal staining of replicative late PANC-1 cells. d Confocal fluorescent graphs of nuclei morphology to identify the positive DNA SAHF in replicative late PANC-1 cells. e Quantitative RT-PCR analysis of key SASP factor, IL8 mRNA in replicative late PANC-1 cells. Data are represented as mean ± S.E.M, n = 3 (**p < 0.01). f Telomere length was determined with the TRF length assay in replicative late PANC-1 cells. This experiment was repeated twice. See also Supplementary Figs. S2, S3 and S5
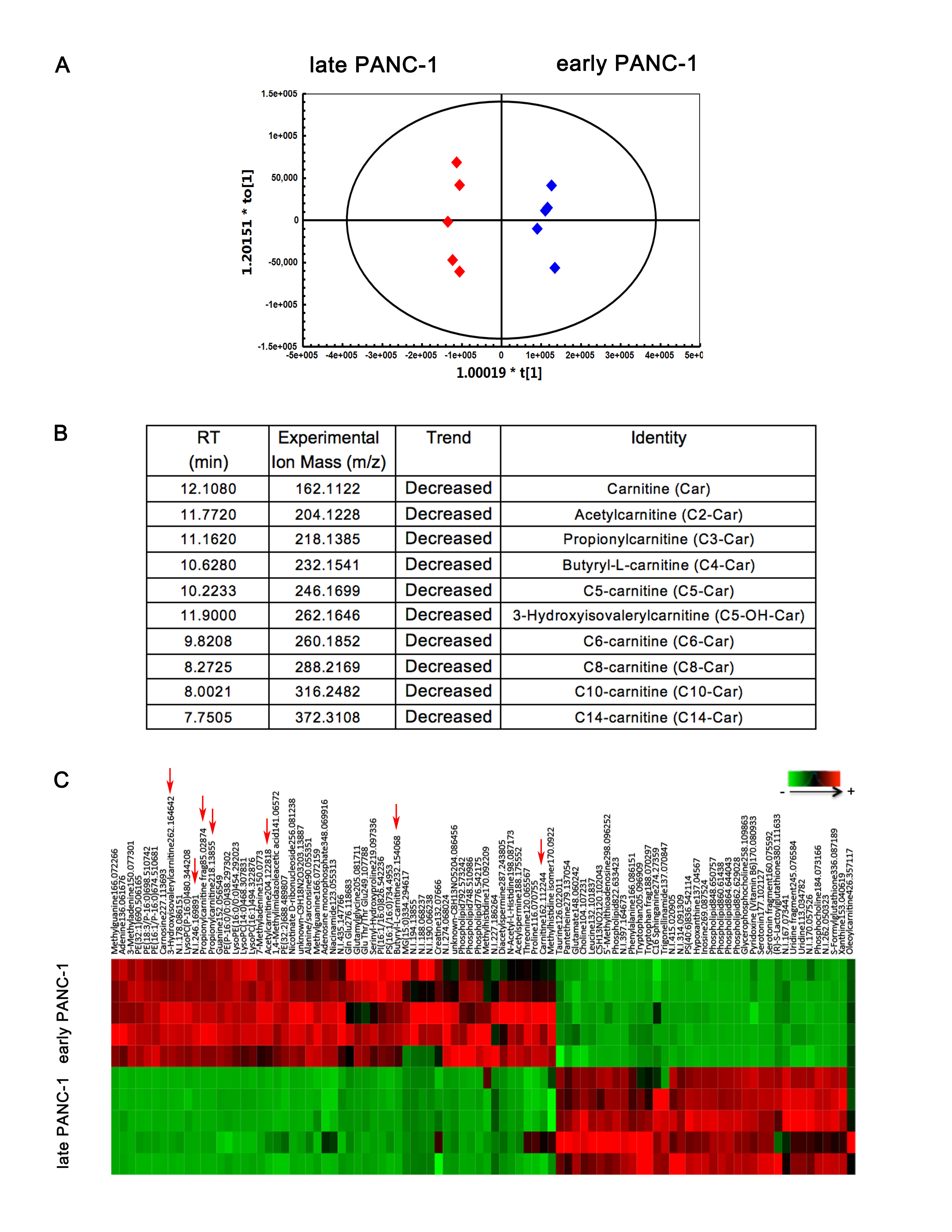
Metabolomics reveals much lower levels of acylcarnitines in senescent PANC-1 cells which is linked to a reduced CPT1C expression
To further investigate metabolism in culture-induced senescent PANC-1 cells, metabolomics was performed to identify potential biomarkers or regulators underlying replicative cellular senescence. Multiple notable differences of intensity at the same retention time clearly revealed significant differences in metabolite features between the two groups (Fig. 2a). Unsupervised PCA of the metabolomic profiles obtained from HILIC-ESI+-MS (Fig. 2b) and HILIC-ESI−-MS (Supplementary Fig. S1A) modes showed clear separation between early and late PANC-1 cells, indicating significant differences in the metabolome between the two groups. Supervised multivariate analysis and subsequent loadings S-plots from OPLS-DA models that showed many features were highly dysregulated between the early and late PANC-1 cells. These ions (P1 to P10) were further identified by comparison of retention times and MS/MS fragmentation patterns with authentic standards or with those annotated in the HMDB and METLIN databases (Supplementary Fig. S1B). A heatmap revealed major alterations in cellular metabolism and the relative increase or decrease of each metabolite (Supplementary Fig. S1C). Specifically, acyl-carnitines were markedly decreased in late PANC-1 cells (Fig. 2c).
Fig. 2. Metabolomics reveals lower acylcarnitines in senescent PANC-1 cells that result from the reduction of CPT1C.

a Comparison of total ion chromatograms (TICs) from early and late PANC-1 cell extracts displayed as a mirror plot, n = 5/group. b PCA score plots of HILIC-ESI+-MS metabolomics profiles obtained from HILIC-ESI+-MS, n = 5/group. c Analysis of the relative response of acylcarnitine ions in senescent late PANC-1 cells. Data are represented as mean ± S.E.M, n = 5 (**p < 0.01, ***p < 0.001). d Quantitative RT-PCR analysis of genes related to acylcarnitines. Data are represented as mean ± S.E.M, n = 3 (ns means no significance, *p < 0.05, **p < 0.01, ***p < 0.001). e Images and densitometric analysis of CPT1C protein bands of senescent late PANC-1 cells. Data are represented as mean ± S.E.M, n = 3 (**p < 0.01). See also Supplementary Figures S1, S2 and S3
To delineate the functional regulators behind such dramatic change in acylcarnitine levels, the mRNA expression of genes related to acylcarnitine biosynthesis, transfer and transport were determined. The results (Fig. 2d) showed that CPT1B, CPT2, carnitine octanoyltransferase (COT), carnitine/acylcarnitine translocase (CACT), carnitine/organic cation transporter 2 (OCTN2), and carnitine transporter 2 mRNA levels were significantly decreased in late PANC-1 cells, while CPT1A (CT2) mRNA levels slightly increased and the carnitine O-acetyltransferase (CRAT) mRNA expression remained unchanged. Most strikingly, CPT1C mRNA levels were decreased by >99% in late PANC-1 cells (Fig. 2d); reduced protein levels were further verified by immunoblots (Fig. 2e). In addition, compared to early PANC-1, a significant decrease in CPT1C mRNA (Supplementary Fig. S2D) and CPT1C protein (Supplementary Fig. S2E) levels were observed in middle PANC-1, suggesting that low CPT1C might play a crucial role in replicative senescence. Interestingly, there was no significant change in CPT1C mRNA (Supplementary Fig. S3D) and CPT1C protein levels (Supplementary Fig. S3E) between the early and late HEK293ft cells, consistent with no culture-related cellular senescence of this cell line. Together, these results suggest that CPT1C could be the most important contributor to the decrease in acylcarnitines and a potential driver behind replicative senescence in PANC-1 cells.
Mitochondrial function, survival, and tumorigenicity in low-CPT1C-expressing senescent PANC-1 cells
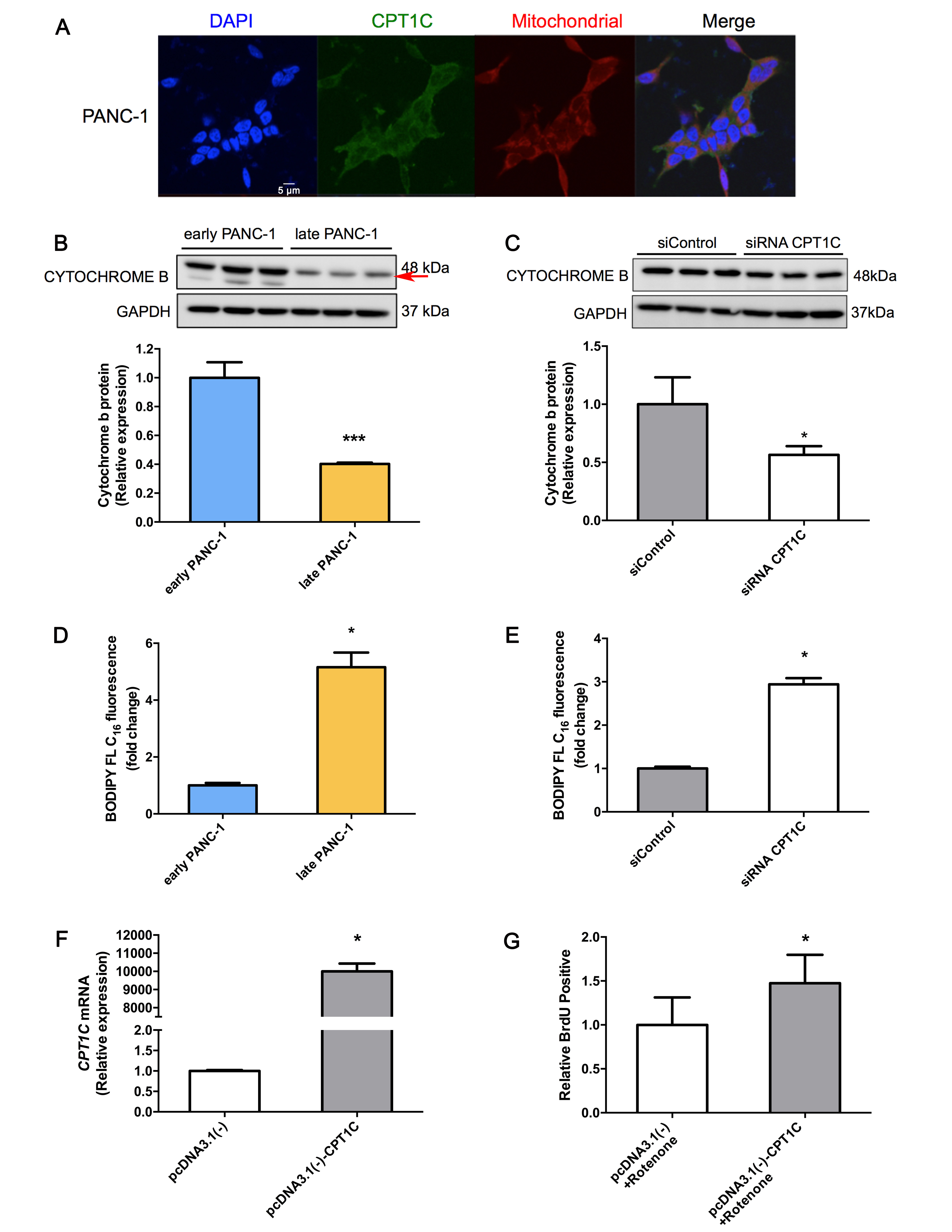
CPT1C, a mitochondrial enzyme, catalyzes the initiating step of fatty acid oxidation through by transporting fatty acids from the cytoplasm to the mitochondrial matrix for β-oxidation. Co-localization of CPT1C with mitochondria in PANC-1 cells was observed (Supplementary Fig. S7A). To determine the impact of low CPT1C expression on replicative senescence, ATP production and mitochondrial function were determined in senescent PANC-1 cells. Notably, under conditions of culture medium deprivation, late PANC-1 cells produced significantly less ATP than early PANC-1 cells (Fig. 3a). On the contrary, reactive oxygen species (ROS) accumulation was more severe in late PANC-1 cells (Fig. 3b). The intensity of absorbed rh123 dye decreased in late PANC-1 cells, indicating the loss of mitochondrial transmembrane potential upon low CPT1C expression (Fig. 3c).
Fig. 3. Abnormal mitochondrial function, attenuated survival and tumorigenesis in senescent late PANC-1 cells.

a ATP production in senescent late PANC-1 cells. Data are represented as mean ± S.E.M, n = 4 (**p < 0.01). b Increased accumulation of ROS in senescent late PANC-1. Data are represented as mean ± S.E.M, n = 4 (**p < 0.01, ***p < 0.001). c Loss of mitochondrial transmembrane potential measured by the rh123 dequenching method in senescent late PANC-1 cells. Data are represented as mean ± S.E.M, n = 4 (**p < 0.01). d Mitochondrial integrity (left panel), maximal respirations (250 ± 22 vs. 348 ± 146 pmol O2/min, middle panel) and spare respiratory capacity (10 ± 7 vs. 173 ± 21 pmol O2/min, right panel) in the forms of OCRs in senescent late PANC-1. OCRs was normalized to the total protein after harvesting and lysing cells used for the seahorse experiment. Data are represented as mean ± S.E.M, n = 4 (***p < 0.001). e Mitochondriogenesis analysis in senescent late PANC-1. Data are represented as mean ± S.E.M, n = 3 (**p < 0.01, ***p < 0.001). f Electron microscopy photomicrographs of mitochondrial morphology of senescent late PANC-1 cells. Sensitivity to metabolic stress from g glucose deprivation (0.5 mM glucose), h glycolytic inhibition (20 mM 2-deoxyglucose), and i rapamycin of senescent late PANC-1 cells. Data are represented as mean ± S.E.M, n = 5 (*p < 0.05, **p < 0.01, ***p < 0.001). j Nude mice were implanted with early or late PANC-1 cells, respectively. The body weights of mice in both groups increased with no significant difference between the two groups. Tumor sizes were presented as mean ± S.E.M over time, (n = 5) (*p < 0.05, **p < 0.01, ***p < 0.001). k Images of tumors after excision on day 42 post-implantation. (l) Comparison of dissected tumor weights (mean ± S.E.M, ***p < 0.001)
With inhibition of oxygen consumption rate (OCRs), late PANC-1 exhibited reduced FCCP-uncoupled maximal respiration but without statistical significance. More importantly, spare respiratory capacity as a parameter of the cell’s ability to respond to increased energy demand was significantly decreased in late PANC-1 (Fig. 3d), which was indicative of partially impaired mitochondrial integrity upon low CPT1C expression. Intriguingly, late PANC-1 exhibited higher basal respiratory rates and ATP-dependent oxygen consumption compared to early PANC-1, suggesting that these cells might adjust their mitochondrial respiration to supplement energy supply and maintain homeostasis status under a long-term progressive senescence program [3].
Mitochondrial biogenesis may be impaired as mitochondrial DNA (mtDNA) is a key target of oxidants produced during ageing [4, 5]. In late PANC-1 cells, PGC1A mRNA and its downstream gene mRNAs, NRF1 and TFAM, were decreased. Subsequently, CYTB mRNA was also lowered (Fig. 3e), consistent with the observation of decreased CYTB protein level and reduced mitochondrial mass (Supplementary Fig. S7B). There was strong evidence that low CPT1C induced lower renewal of the mitochondrial genome that led to defects in mitochondrial function. Electron microscopy also revealed that damage to the morphology of the mitochondria occurred upon replicative senescence in low-CPT1C-expressing late PANC-1 cells. This damage included mitochondrial swelling and disorganization, membrane breaks, loss of internal cristae, and increased lucency of the matrix and surrounding structures (Fig. 3f).
In order to further examine the consequences of mitochondrial dysfunction in late senescent PANC-1 cells with low CPT1C expression, cell survival under metabolic stress and tumorigenesis were studied. Late PANC-1 cells showed higher sensitivity to 0.5 mM glucose (Fig. 3g) or 20 mM 2-deoxyglucose (2-DG, a glycolysis inhibitor) (Fig. 3h) compared to early PANC-1 cells, and the magnitude of difference increased in a time-dependent manner. More importantly late PANC-1 cells showed a marked decrease in cell survival with increasing rapamycin (also as a source of stress and cytotoxicity that hinders cell growth) concentrations (Fig. 3i). These data suggest that low CPT1C expression might hinder cell growth under various forms of metabolic stress.
The contribution of low CPT1C expression to PANC-1 cell tumorigenicity was further characterized in a mouse xenograft model. Tumors arising from late PANC-1 cells grew much more slowly (Fig. 3j–l).
Taken together, in addition to morphological damage, low CPT1C expression might be involved in replicative senescence by decreasing mitochondrial function, thus, eventually enhancing cellular senescence. Low CPT1C expression also impaired metabolic adaptation of late PANC-1 cells in vitro, which further hindered the ability of the transformed cells to form tumors in vivo.
Silencing of CPT1C confers mitochondrial dysfunction in PANC-1 cells
Since low CPT1C expression triggered replicative senescence in late PANC-1 cells as a result of lower mitochondrial function, further studies were carried out on the putative role of low CPT1C in PANC-1 cell proliferation. An optimal CPT1C reduction (Supplementary Fig. S4A, B) was determined in PANC-1 cells transfected with siRNA CPT1C-3. Upon culture medium deprivation and nutrient deficiency, ATP synthesis was markedly attenuated after depletion of CPT1C (Fig. 4a). On the contrary, ROS accumulation was enhanced after CPT1C depletion (Fig. 4b). Lower CPT1C also caused rapid rh123 dequenching and loss of the mitochondrial electrochemical gradient (Fig. 4c), which destabilizes the mitochondrial membrane. Furthermore, depletion of CPT1C significantly affected mitochondrial respiration (Fig. 4d), with inhibition of OCRs in basal respiration. However, adjusted mitochondrial respiration was not observed in these transient siCPT1C cells, indicating different respiratory profiles of early and late PANC-1, and siContrl and siRNA CPT1C models. Depletion of CPT1C limited PANC-1 cells to a lower FCCP-uncoupled maximal respiratory capacity. In addition, CPT1C depletion attenuated nonphosphorylating proton leak and depressed ATP-related OCRs, while spare respiratory capacity did not fluctuate significantly. More importantly, after depletion of CPT1C, PGC1α and its down-steam NRF1 and TFAM mRNAs were reduced (Fig. 4e). Subsequently, mtDNA subunit CYTB was lowered, in accordance with reduced quantification of mitochondrial mass by examining the decreased abundance of CYTB protein (Supplementary Fig. S7C). Finally, lowering the CPT1C caused an increase in the presence of swollen mitochondria exhibiting loss of internal cristae density and abnormal membrane structures (Fig. 4f). These data revealed a causal role for CPT1C depletion in a decreasing mitochondrial and cellular function.
Fig. 4. CPT1C depletion confers to mitochondrial dysfunction in PANC-1 cells.

a ATP synthesis in PANC-1 cells upon knockdown of CPT1C. Data are represented as mean ± S.E.M, n = 4 (*p < 0.05). b Depletion of CPT1C induced increased accumulation of ROS. Data are represented as mean ± S.E.M, n = 4 (**p < 0.01, ***p < 0.001). c Loss of mitochondrial transmembrane potential measured by the rh123 dequenching method upon siRNA knockdown of CPT1C. Data are represented as mean ± S.E.M, n = 4 (**p < 0.01). d Mitochondrial integrity (left panel), basal respirations (middle panel, 593 ± 45 pmol O2/min in controls and 384 ± 41 pmol O2/min in CPT1C-depleted PANC-1 cells) and maximal respiratory capacity (right panel, 1120 ± 15 pmol O2/min in controls and 929 ± 43 pmol O2/min in CPT1C-depleted PANC-1 cells) in the forms of OCRs in PANC-1 cells depleted of CPT1C. In addition, CPT1C depletion attenuated nonphosphorylating proton leak and depressed ATP-related OCRs, while spare respiratory capacity did not fluctuate significantly. Data are represented as mean ± S.E.M, n = 4 (*p < 0.05, ***p < 0.001). e Mitochondriogenesis analysis in PANC-1 cells after siRNA knockdown of CPT1C. Data are represented as mean ± S.E.M, n = 4 (**p < 0.01, ***p < 0.001). f Electron microscopy photomicrographs of mitochondrial morphology of PANC-1 cells after CPT1C depletion. See also Supplementary Figure S4, S7
CPT1C depletion leads to cancer cell senescence, and suppresses the survival and tumorigenesis of PANC-1 cells
The decrease in cellular function as an indicator of senescence driven by CPT1C knockdown is likely a direct consequence of disruption of mitochondrial function. Upon CPT1C loss, the number of PANC-1 cells actively replicating DNA decreased (Fig. 5a), consistent with effective inhibition of colony formation in a cell concentration-dependent manner (Fig. 5b, Supplementary Fig. S5E), indicating that PANC-1 cells lacking expression of CPT1C have reduced proliferation. In addition, strong SA-β-gal activity indicated that CPT1C depletion led to the severe senescence state of PANC-1 cells (Fig. 5c, Supplementary Fig. S5F). As expected, loss of CPT1C caused PANC-1 cells to change shape and increase cytoplasmic granularities (Fig. 5c). DNA SAHF was specifically enriched in CPT1C-depleted PANC-1 cells, while controls were excluded from SAHF (Fig. 5d). CPT1C-depleted PANC-1 cells also had abnormal nuclear size, when compared to control cells (Fig. 5d). Furthermore, a significant increase in IL8 mRNA levels (Fig. 5e) confirmed a sharp induction of the SA phenotypic changes observed by the SA-β-gal assay. Finally, the TRF length assay revealed that CPT1C depletion inhibited the elongation of telomeres by 0.8 kb in PANC-1 cells (Fig. 5f).
Fig. 5. Knock-down of CPT1C triggers cellular senescence and reduction of PANC-1 cells survival and tumorigenesis.

a The number of PANC-1 cells actively replicating DNA measured by BrdU incorporation upon knocking down of CPT1C. Data are represented as mean ± S.E.M, n = 4 (*p < 0.05). b Colony formation of PANC-1 cells after CPT1C depletion. c SA-β-gal staining of PANC-1 cells upon silencing of CPT1C. This experiment was repeated three times. d Confocal fluorescent graphs of nuclei morphology to identify the positive DNA SAHF in PANC-1 cells after CPT1C depletion. e Quantitative RT-PCR analysis of IL8 mRNA in PANC-1 cells after CPT1C depletion. Data are represented as mean ± S.E.M, n = 3 (**p < 0.01). f Determination of telomere length in PANC-1 cells after CPT1C silencing. This experiment was repeated two times. Sensitivity to metabolic stress from g glucose withdrawal (0.5 mM glucose), h glycolytic inhibition (20 mM 2-deoxyglucose), and i rapamycin stimuli in PANC-1 cells depleted of CPT1C. Data are represented as mean ± S.E.M, n = 5 (*p < 0.05, ***p < 0.001). j Images of xenograft tumor mice derived from PANC-1 cells that were infected with retroviruses expressing pRS-CPT1C shRNA or pRS shRNA (n = 8), tumor growth was monitored for 6 weeks. No significant difference of the increased body weights of mice was observed between these two groups. k Comparison of tumor formation incidence. l Comparison of dissected tumor weights (mean ± S.E.M, ***p < 0.001). m Images of tumors after excision at 1 week post-delivery of 2′OMe + 5′Chol siRNA CPT1C or siControl (n = 10). Subcutaneous xenograft tumors arising from PANC-1 cells were monitored for 4.5 weeks. There was no significant difference in tumor sizes between siRNA CPT1C and siControl group before intratumoral delivery of 2′OMe + 5′Chol siRNA CPT1C. After intratumoral injection of 2′OMe + 5′Chol siRNA CPT1C twice for a week, no significant difference of body weight changes was observed between these two groups. n Tumor growth curve was presented as mean tumor volume ± S.E.M on the indicated days (*p < 0.05). See also Supplementary Figure S4, S5 and S6
Notably, senescence activation was clearly observed in PANC-1 cells transfected with siRNA CPT1C-2 (Supplementary Fig. S6A–C). Similar senescence phenotypes (Supplementary Fig. S9A–C, S10A, S10B) were also observed in other cells transfected with CPT1C siRNA (Supplementary Fig. S8A–S8J), suggesting that depletion of CPT1C can induce cellular senescence in most tumor cell lines. Taken together, these findings demonstrated that depletion of CPT1C induces a potent decline of mitochondrial function, which further renders tumor cells more vulnerable to proliferation arrest and cellular senescence. Furthermore, depletion of CPT1C also induced a decrease of mitochondrial function in other cancer cell lines (Supplementary Fig. S11A–L).
CPT1C-depleted PANC-1 cells grew significantly slower than control cells under 0.5 mM glucose (Fig. 5g) or 20 mM 2-DG (Fig. 5h) in a time-dependent manner. In addition, CPT1C-depleted PANC-1 cells showed a significant increase in rapamycin sensitivity, while control cells were partially resistant to rapamycin stimuli (Fig. 5i). Thus, CPT1C depletion may attenuate survival of PANC-1 cells subjected to conditions of metabolic stress.
Reduction in CPT1C levels was found in PANC-1 cells transduced with CPT1C shRNA-4 (Supplementary Fig. S4C, D). When these cells were inoculated into nude mice, tumor formation incidence of controls was 100% (Fig. 5j, k), whereas there was minimal or no tumors from PANC-1 cells expressing CPT1C shRNA-4 (tumor formation was 25%), while sustained depletion of CPT1C reduced tumor growth and sizes (Fig. 5l). These data revealed that knocking down CPT1C reduced the malignancy of senescent PANC-1 cells in vitro, and further suppressed the tumorigenicity of PANC-1 cells in vivo.
siCPT1C treatment contributed to suppression of xenograft tumor growth in situ
Xenograft tumor-bearing nude mice were used to evaluate the antitumor effect of siCPT1C injection in situ. After intratumoral injection of 2′OMe + 5′Chol siRNA CPT1C twice a week, tumors grew much slower than controls (Fig. 5m). Sustained depletion of CPT1C by methyl and cholesterol-conjugated siRNA CPT1C delivery correlated with reduce tumor sizes in xenograft models (Fig. 5n), although no significant difference in tumor weights was observed. These data suggest that CPT1C siRNA treatment induced tumor growth suppression in situ, which might be a consequence of tumor cell senescence.
Gain-of-function of CPT1C reverses PANC-1 cell senescence
Strikingly higher expression of CPT1C was found in pcDNA3.1(-)-CPT1C transfected late PANC-1 cells (Fig. 6a, b). Upon gain of CPT1C, the cell number of late PANC-1 actively replicating DNA increased (Fig. 6c), consistent with effective enhancement of colony formation ability (Fig. 6d), indicating that late PANC-1 expressing CPT1C showed increased proliferation. In addition, late PANC-1 over-expressing CPT1C showed very weak SA-β-gal activity, which indicated that gain-of-function of CPT1C was sufficient to reverse the replicative PANC-1 cell senescence (Fig. 6e). Late PANC-1 over-expressing CPT1C mutant plasmids showed the same strong SA-β-gal activity as controls (Fig. 6f), indicating that inactive CPT1C overexpression could not rescue the senescence phenotype in “late” PANC-1. Furthermore, enhanced cellular bioenergetics (Fig. 6g) and recovered mitochondrial transmembrane potential (Fig. 6h) were observed upon CPT1C gain-of-function in late PANC-1. In addition, after incubation with rotenone, cell proliferation increased in PANC-1 cells with CPT1C overexpressing (Supplementary Fig. S7F, G), indicating that CPT1C gain-of-function led to a faster cellular proliferation if mitochondrial function was inhibited.
Fig. 6. Gain-of-function of CPT1C reverses cellular senescence in late PANC-1.

a A human CPT1C plasmid was designed to up-regulate CPT1C expression in senescent late PANC-1 (PANC-1 P 100). Quantitative RT-PCR analysis of CPT1C mRNA expression was observed post-transfection with CPT1C plasmid in senescent late PANC-1. Data are represented as mean ± S.E.M, n = 3 (***p < 0.001). b Western blot analysis of CPT1C protein levels in senescent late PANC-1 after transfection of CPT1C expression plasmid (upper panel of B) images and (bottom panel of B) densitometric analysis of CPT1C protein bands. Data are represented as mean ± S.E.M, n = 3 (***p < 0.001). c The number of senescent late PANC-1 actively replicating DNA measured by BrdU incorporation upon CPT1C gain-of-function. Data are represented as mean ± S.E.M, n = 4 (*p < 0.05). d Colony formation capacity of senescent late PANC-1 after CPT1C gain-of-function. This experiment was independently repeated two times. e SA-β-gal staining of senescent late PANC-1 upon gain of CPT1C function. f SA-β-gal assay of senescent late PANC-1 gaining of inactive CPT1C function (**p < 0.01, ***p < 0.001). g Mitochondriogenesis analysis in late PANC-1 gaining of CPT1C function. Data are represented as mean ± S.E.M, n = 4 (ns means no significance, *p < 0.05). h Recovered mitochondrial transmembrane potential measured with rh123 dequenching method in late PANC-1 gaining of CPT1C function. Data are represented as mean ± S.E.M, n = 4 (*p < 0.05)
Discussion
In this study, a culture-related and passage-dependent replicative cellular senescence phenotype was observed in PANC-1 cells. Furthermore, replicative senescence is the consequence of a DNA damage response triggered by short telomeres [6,7]. Similarly, shortened telomeres were also observed in senescent PANC-1 cells. However, the underlying mechanisms involved in cellular senescence are largely not known. Senescence results in dramatic changes in metabolism due to metabolic reprograming [8,9]. Metabolomic profiling indicated a significant change in the endogenous metabolome suggesting striking alterations in cellular metabolism and metabolic reprograming during the onset of senescence in PANC-1 cells. Levels of nucleic acids, amino acids and phospholipid derivatives involved in lipid and energy metabolism were markably altered in late PANC-1 cells. Most interestingly, carnitine and short-chain acyl-carnitines were markedly decreased in senescent PANC-1 cells and are known growth factors [10]. Free carnitine and acylcarnitines are essential for cell energy metabolism as carriers of long-chain fatty acids for β-oxidation or as reservoir pools of acyl-CoA [11]. Thus, a dramatic decrease in carnitine and acylcarnitines may reflect a significant decrease in energy metabolism as well as mitochondrial dysfunction in senescent PANC-1 cells. It was reported that as a result of gradual deterioration of cellular function characteristic, impaired oxidation of 13C1-palmitate nonesterified fatty acids was observed in elderly humans [12], which further suggested a mitochondrial defect upon aging. ATP and ROS production and mitochondrial function assays consistently indicated collapse of mitochondrial function in late PANC-1 cells. Slightly morphological aberrations of endoplasmic reticulum were also observed in late PANC-1. Consequently, the senescent PANC-1 cells with mitochondrial dysfunction had low survival under metabolic stress and suppressed tumorigenesis.
Expressions of most functional regulators that cause significant metabolic reprograming of acylcarnitines were significantly lower in senescent PANC-1 cells. Strikingly, among these top candidate biomarkers, CPT1C decreased most dramatically in the senescent late PANC-1 cells. These data suggest that CPT1C may be the most crucial contributor to the decrease in acylcarnitines and triggering replicative senescence. In multiple cancer cell lines, CPT1C mRNA was increased in response to several different DNA-damaging stress stimuli, while no apparent change was observed in CPT1A, CPT1B, and CPT2 mRNAs [13]. Considering that tumor cells can be forced into senescence by epigenetic DNA-damaging stimuli factors [14], CPT1C may be the only CPT isoform to specifically protect cancer cells from cellular senescence. Notably, fatty acid uptake increased upon low CPT1C expression, which could be ruled out to explain reduced fatty acid oxidation and reduction in acylcarnitines, and might potentially contribute to lipid accumulation and lipotoxicity [15].
CPT1C, as a functional enzyme in the outer mitochondrial membrane, is linked to lipid metabolism and cellular energy supply [16,17]. Besides the regulation of fatty acid oxidation, CPT1C also plays an important role in other physiological and pathological processes [18–21]. The current study revealed that CPT1C depletion is sufficient to trigger a senescence phenotype in other cancer cell lines. Moreover, CPT1C may have a unique function in the regulation of cellular senescence via modulation of bioenergetics in mitochondria. Complete depletion of CPT1C directly drives abnormal energy metabolism and a decrease of mitochondrial function in PANC-1 cells.
Mounting evidence suggests a very close relationship between aberrant mitochondria and cellular senescence [22,23]. A recent study revealed that mitochondria dysfunction triggers cellular senescence [24]. Mitochondrial dysfunction was also observed in cardiomyocytes at late passage P39 compared to early passage P10 [25], implying that mitochondrial damage might be progressive over time and cell passage. One hypothesis is that impaired mitochondrial biogenesis may be induced during aging via increased ROS [26,27]. The current study showed that late PANC-1 cells with low CPT1C expression leads to the adjustment of mitochondrial respiration and an abnormal mitochondrial energy metabolism. On the other hand, transient depletion of CPT1C directly triggers a collapse of mitochondrial function without adjusted mitochondrial respiration. In addition, another report revealed that CPT1C depletion caused impaired cell ATP production after glucose deprivation [16]. Furthermore, CPT1C depletion in mouse ES cells led to reduced ATP production and activation of intrinsic mitochondrial apoptosis. The present data demonstrated that knockdown of CPT1C caused PANC-1 cells to undergo senescence-like growth suppression and cellular senescence. Consistent with this observation, long-term cultures of Cpt1cgt/gt ES cells showed decreased total cell numbers and enhanced apoptosis [16]. Depletion of CPT1C further delayed tumor growth in a murine neurofibromatosis type I tumor model [13]. Notably, similar mitochondrial dysfunction-mediated senescent phenotypes were also observed when CPT1C was efficiently depleted in other five tumor cell lines, suggesting that cellular senescence induced by CPT1C knockdown is independent of the specific tumor cell lines. On the contrary, when CPT1C was efficiently over-expressed in late PANC-1 cells, gain-of-function of CPT1C increased proliferation and reversed cell senescence and impaired-mitochondrial function of PANC-1 cells, suggesting the important role of CPT1C in cell proliferation and senescence.
CPT1C depletion contributed to the disruption of cell homeostasis, especially in cells experiencing metabolic stress. In addition, studies have shown that fewer CPT1C-depleted cells were detected on exposure to 2-DG, and cells deficient for CPT1C showed reduced growth in respond to hypoxia stress and increased rapamycin sensitivity [16]. Conversely, CPT1C over-expression promoted cells survival under limited glucose and hypoxia [16]. Ectopic CPT1C expression protected cells with high metabolic sensitivity from 2-DG-induced apoptosis [13]. A previous report demonstrated that sustained depletion of CPT1C reduced the growth of tumors arising from MDA-MB-468 cells [16]. Furthermore, CPT1C depletion significantly suppressed tumor transformation/sarcomas and metastasis in the murine neurofibromatosis type I tumor model [13]. However, these previously published data did not clarify the mechanism by which CPT1C depletion can trigger suppressed tumor cell tumorigenesis. On the other hand, the current study clearly showed that CPT1C depletion triggers mitochondrial dysfunction and then cellular senescence resulting in lower survival under metabolic stress along with suppressed tumorigenesis. Most recently, we reported that PPARα could regulate cellular senescence by its novel target gene CPT1C [28]. But how this novel CPT1C function was discovered and how CPT1C regulates cancer cell senescence remains unknown. In the current study, metabolomics revealed a novel function of CPT1C in the regulation cellular senescence.
In a summary, cellular senescence is a complex process limiting the proliferative lifespan of tumor cells, the mechanism of which needs to be clearly understood. For the first time, we found that low CPT1C expression is a potential biomarker for culture-related cellular senescence and CPT1C regulates cell senescence through mitochondria-associated metabolic reprogramming in tumor cell lines, suggesting novel therapeutic opportunities through the induction of cancer cell senescence. Inhibition of CPT1C may also be a therapeutic option for cancer therapy, since mice lacking the Cpt1c gene are viable [29] and thus inhibition of this transporter would not be expected to be toxic.
Materials and Methods
Cell culture
Information on cell lines and culture conditions is presented in the Supplemental Experimental Procedures
Senescence analysis
Senescence analysis is described in the Supplemental Experimental Procedures.
Metabolomics analysis
Protocols for metabolomics analysis are presented in the Supplemental Experimental Procedures.
Quantitative RT-PCR and Western blot
Procedures for genes analysis are described in the Supplemental Experimental Procedures.
Mitochondrial dysfunction analysis
Analyses of mitochondrial function and morphology are described in the Supplemental Experimental Procedures.
Metabolic Stress Stimuli
Protocols for metabolic Stress Stimuli are presented in the Supplemental Experimental Procedures.
Sulforhodamine B (SRB) assay
Procedures for SRB are described in the Supplemental Experimental Procedures.
Xenograft mice models
Xenograft mice model assays are described in the Supplemental Experimental Procedures. All procedures were in accordance with the Regulations of Experimental Animal Administration issued by the Ministry of Science and Technology of the People’s Republic of China, and all animal protocols were approved by the Ethics Committee on the Care and Use of Laboratory Animals of Sun Yat-sen University.
Intratumoral delivery of methyl and cholesterol-conjugated siRNA CPT1C
Delivery of 2′OMe + 5′Chol siRNA CPT1C or siControl are presented in the Supplemental Experimental Procedures.
Gene silencing by RNA interference
RNAi assays are presented in the Supplemental Experimental Procedures.
CPT1C plasmid transfection
Plasmid transient transfection was performed as described in the Supplemental Experimental Procedures.
Statistical Analysis
Statistical analysis is described in the Supplemental Experimental Procedures.
Electronic supplementary material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Supplemental Information on Methods and Figures
Acknowledgements
The authors are grateful to Drs. Jun Du, Xiaoyan Shen, and Jun Li from Sun Yat-sen University for generously providing various cell lines. This study was supported by the Natural Science Foundation of China (Grants: 81522047, 81573489, 81373470, 81320108027), the 111 Project (Grant: B16047), the Key Laboratory Foundation of Guangdong Province (Grant: 2011A060901014), and the Guangzhou Health Care Collaborative Innovation Program (Grant: 201508020250).
Author contributions
HB, MH, PH and YW designed the experiments, YW, YC, LG, HZ and YH conducted the experiments, YW and HB performed data analysis, YW, HB, FJG, CHJ, AY, and PH wrote the paper, ZW, SY, YW, PC and XF contributed to new reagents or analytical tools.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no competing interests.
Footnotes
Edited by G. Melino
Electronic supplementary material
The online version of this article (10.1038/s41418-017-0013-3) contains supplementary material, which is available to authorized users.
References
- 1.Idle JR, Gonzalez FJ. Metabolomics. Cell Metab. 2007;6(5):348–51. doi: 10.1016/j.cmet.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25(20):2125–36. doi: 10.1101/gad.17276711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nogueira V, Hay N. Molecular pathways: reactive oxygen species homeostasis in cancer cells and implications for cancer therapy. Clin Cancer Res. 2013;19(16):4309–14. doi: 10.1158/1078-0432.CCR-12-1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vina J, Gomez-Cabrera MC, Borras C, Froio T, Sanchis-Gomar F, Martinez-Bello VE, et al. Mitochondrial biogenesis in exercise and in ageing. Adv Drug Deliv Rev. 2009;61(14):1369–74. doi: 10.1016/j.addr.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88(5):529–35. doi: 10.1161/01.RES.88.5.529. [DOI] [PubMed] [Google Scholar]
- 6.Simonsen JL, Rosada C, Serakinci N, Justesen J, Stenderup K, Rattan SI, et al. Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat Biotechnol. 2002;20(6):592–6. doi: 10.1038/nbt0602-592. [DOI] [PubMed] [Google Scholar]
- 7.Xu D, Neville R, Finkel T. Homocysteine accelerates endothelial cell senescence. FEBS Lett. 2000;470(1):20–24. doi: 10.1016/S0014-5793(00)01278-3. [DOI] [PubMed] [Google Scholar]
- 8.Panopoulos AD, Yanes O, Ruiz S, Kida YS, Diep D, Tautenhahn R, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22(1):168–77. doi: 10.1038/cr.2011.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu LE, Gomes AP, Sinclair DA. Geroncogenesis: metabolic changes during aging as a driver of tumorigenesis. Cancer cell. 2014;25(1):12–19. doi: 10.1016/j.ccr.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bremer J. Carnitine—metabolism and functions. Physiol Rev. 1983;63(4):1420–80. doi: 10.1152/physrev.1983.63.4.1420. [DOI] [PubMed] [Google Scholar]
- 11.Wu R, Wu Z, Wang X, Yang P, Yu D, Zhao C, et al. Metabolomic analysis reveals that carnitines are key regulatory metabolites in phase transition of the locusts. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(9):3259–63. doi: 10.1073/pnas.1119155109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen D, Samson SL, Reddy VT, Gonzalez EV, Sekhar RV. Impaired mitochondrial fatty acid oxidation and insulin resistance in aging: novel protective role of glutathione. Aging cell. 2013;12(3):415–25. doi: 10.1111/acel.12073. [DOI] [PubMed] [Google Scholar]
- 13.Sanchez-Macedo N, Feng J, Faubert B, Chang N, Elia A, Rushing EJ, et al. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. 2013;20(4):659–68. doi: 10.1038/cdd.2012.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res. 2003;63(11):2705–15. [PubMed] [Google Scholar]
- 15.Zhang H, Gao Y, Sun J, Fan S, Yao X, Ran X et al. Optimization of lipid extraction and analytical protocols for UHPLC-ESI-HRMS-based lipidomic analysis of adherent mammalian cancer cells. Anal Bioanal Chem. 2017;409:5349-58. [DOI] [PubMed]
- 16.Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25(10):1041–51. doi: 10.1101/gad.1987211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinaldi C, Schmidt T, Situ AJ, Johnson JO, Lee PR, Chen KL, et al. Mutation in CPT1C associated with pure autosomal dominant spastic paraplegia. JAMA Neurol. 2015;72(5):561–70. doi: 10.1001/jamaneurol.2014.4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolfgang MJ, Kurama T, Dai Y, Suwa A, Asaumi M, Matsumoto S, et al. The brain-specific carnitine palmitoyltransferase-1c regulates energy homeostasis. Proc Natl Acad Sci USA. 2006;103(19):7282–7. doi: 10.1073/pnas.0602205103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrasco P, Sahun I, McDonald J, Ramirez S, Jacas J, Gratacos E, et al. Ceramide levels regulated by carnitine palmitoyltransferase 1C control dendritic spine maturation and cognition. J Biol Chem. 2012;287(25):21224–32. doi: 10.1074/jbc.M111.337493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimizu N, Chikahisa S, Nishi Y, Harada S, Iwaki Y, Fujihara H, et al. Maternal dietary restriction alters offspring’s sleep homeostasis. PloS One. 2013;8(5):e64263. doi: 10.1371/journal.pone.0064263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Virmani A, Pinto L, Bauermann O, Zerelli S, Diedenhofen A, Binienda ZK, et al. The carnitine palmitoyl transferase (CPT) system and possible relevance for neuropsychiatric and neurological conditions. Mol Neurobiol. 2015;52(2):826–36. doi: 10.1007/s12035-015-9238-7. [DOI] [PubMed] [Google Scholar]
- 22.Sun N, Youle RJ, Finkel T. The mitochondrial basis of aging. Mol Cell. 2016;61(5):654–66. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallage S, Gil J. Mitochondrial dysfunction meets senescence. Trends Biochem Sci. 2016;41(3):207–9. doi: 10.1016/j.tibs.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 24.Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23(2):303–14. doi: 10.1016/j.cmet.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu Y, Nguyen P, Baris TZ, Poirier MC. Molecular analysis of mitochondrial compromise in rodent cardiomyocytes exposed long term to nucleoside reverse transcriptase inhibitors (NRTIs) Cardiovasc Toxicol. 2012;12(2):123–34. doi: 10.1007/s12012-011-9148-5. [DOI] [PubMed] [Google Scholar]
- 26.Hiona A, Leeuwenburgh C. The role of mitochondrial DNA mutations in aging and sarcopenia: implications for the mitochondrial vicious cycle theory of aging. Exp Gerontol. 2008;43(1):24–33. doi: 10.1016/j.exger.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet (London, England) 1989;1(8639):642–5. doi: 10.1016/S0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Wang Y, Huang Y, Zeng H, Hu B, Guan L, et al. PPARalpha regulates tumor cell proliferation and senescence via a novel target gene carnitine palmitoyltransferase 1C. Carcinogenesis. 2017;38(4):474–83. doi: 10.1093/carcin/bgx023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao XF, Chen W, Kong XP, Xu AM, Wang ZG, Sweeney G, et al. Enhanced susceptibility of Cpt1c knockout mice to glucose intolerance induced by a high-fat diet involves elevated hepatic gluconeogenesis and decreased skeletal muscle glucose uptake. Diabetologia. 2009;52(5):912–20. doi: 10.1007/s00125-009-1284-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information on Methods and Figures