

Abstract
Mediator is a conserved, multi-subunit macromolecular machine divided structurally into head, middle, and tail modules, along with a transiently associating kinase module. Mediator functions as an integrator of transcriptional regulatory activity by interacting with DNA-bound transcription factors and with RNA polymerase II (Pol II) to both activate and repress gene expression. Mediator has been shown to affect multiple steps in transcription, including chromatin looping between enhancers and promoters, pre-initiation complex formation, transcriptional elongation, and mRNA splicing. Individual Mediator subunits participate in regulation of gene expression by the estrogen and androgen receptors and are altered in a number of endocrine cancers, including breast and prostate cancer. In addition to its role in genomic signaling, MED12 has been implicated in non-genomic signaling by interacting with and activating TGF-beta receptor 2 in the cytoplasm. Recent structural studies have revealed extensive inter-domain interactions and complex architecture of the Mediator-Pol II complex, suggesting that Mediator is capable of reorganizing its conformation and composition to fit cellular needs. We propose that alterations in Mediator subunit expression that occur in various cancers could impact the organization and function of Mediator, resulting in changes in gene expression that promote malignancy. A better understanding of the role of Mediator in cancer could reveal new approaches to the diagnosis and treatment of Mediator-dependent endocrine cancers, especially in settings of therapy resistance.
Keywords: Mediator, estrogen receptor, androgen receptor, cancer
1 Mediator: An Overview
The mediator complex, also known as Mediator, is a multi-subunit protein complex that is a critical regulator of RNA polymerase II (Pol II) activity [1, 2]. Mediator is conserved from yeast to human, with human Mediator comprising 33 subunits and associated factors (Table 1) [3]. Structural studies have divided Mediator into three main modules – head, middle, and tail – along with a dissociable kinase module [1, 3]. Mediator plays myriad roles in both positive and negative control of transcription, from regulating pre-initiation complex (PIC) formation and elongation to chromosome looping and mRNA splicing [2, 3]. Mediator also contributes to important physiological processes, and its dysregulation has been implicated in many disorders, including multiple cancers [2, 3].1
Table 1.
Mediator Subunits in Humans
| Module | Subunit |
|---|---|
| Head | MED6 |
| MED8 | |
| MED11 | |
| MED17 | |
| MED18 | |
| MED20 | |
| MED22 | |
| MED28 | |
| MED30 | |
| Middle | MED1 |
| MED4 | |
| MED7 | |
| MED9 | |
| MED10 | |
| MED19 | |
| MED21 | |
| MED26 | |
| MED31 | |
| Tail | MED15 |
| MED16 | |
| MED23 | |
| MED25 | |
| MED27 | |
| MED29 | |
| Kinase | MED12 |
| MED12L | |
| MED13 | |
| MED13L | |
| CDK8 | |
| CDK19 | |
| CCNC | |
| Backbone | MED14* |
Bold = human-specific subunits
Originally considered a middle subunit; re-classified as a backbone/scaffold subunit that contacts and holds together the head, middle, and tail modules
Mediator acts as a conduit between the promoter and enhancer by communicating regulatory signals from DNA-bound transcription factors to Pol II. The head and middle modules contact Pol II at the promoter, providing a scaffold to promote its recruitment and stability for contact with general transcription factors, while the tail module interacts with specific transcription factors occupying enhancers [1, 4, 5]. The kinase module, which has been implicated in both positive and negative regulation of gene transcription, was recently shown to occupy enhancers, with its dissociation from the rest of Mediator occurring with the formation of the PIC [6, 7].
Some Mediator subunits have been described as “essential” for general transcription (and hence viability in yeast cells), without which Mediator falls apart or is transcriptionally inactive. MED17, part of the head module, is essential for viability in yeast, with the head and middle modules unable to stably associate in its absence [8]. In yeast, MED14, which was originally proposed as part of the middle module, has in fact been found to act as a scaffold to bridge all three modules and enable conformational changes to support Pol II activity, making MED14 a key part of Mediator (Figure 1A) [1, 4].
Figure 1.
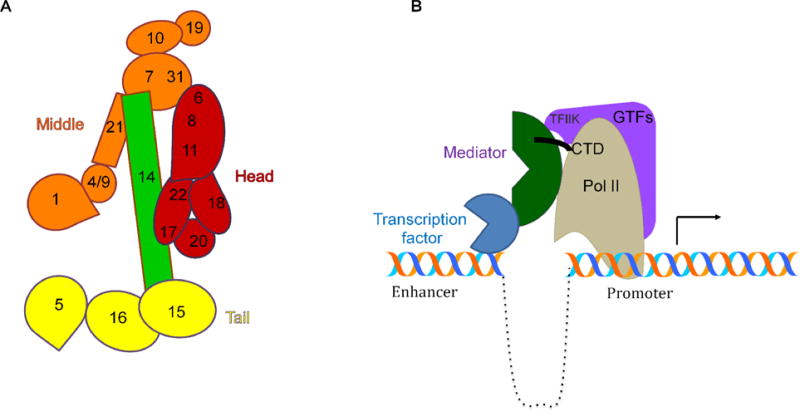
Schematic of Mediator. A. Model of individual subunits in Mediator based on the structures in S. pombe and in S. cerevisiae. B. Mediator bridges transcription factors at enhancers and Pol II at the promoter. Contacts with GTFs cover much of the surface of pol II. Contacts with Mediator dominate the surface of Rpb1, the largest subunit of pol II, from which emanate the RNA transcript and the CTD. Contacts between Mediator and the unphosphorylated CTD of Pol II bring Pol II to and stabilize it at the promoter.
Recent cryo-electron microscopy maps of Mediator from yeast (S. cerevisiae and S. pombe), in which Mediator subunits are individually resolved, revealed extensive interactions between the head and middle modules via MED14 (Figure 1A) [1, 9]. Mediator-Pol II holoenzyme complexes also revealed that MED14 facilitated large-scale Mediator rearrangement required for proper Mediator-Pol II interactions. This work also showed high-affinity binding of Mediator for the unphosphorylated C-terminal domain (CTD) tail of Pol II; it was proposed that the contacts between Mediator and the CTD of Pol II bring Pol II to the promoter, with phosphorylation of the CTD by TFIIK resulting in the release of Mediator and enabling Pol II promoter escape (Figure 1B) [1]. The structural data supports the idea that the Mediator-CTD-Pol II interactions are occluded by the kinase module. The implications of the elegant structural analysis of the Mediator-Pol II -PIC have been recently reviewed [10]. Consistent with such a key structural element, reconstituted human Mediator lacking MED14 cannot promote basal or transcription factor-induced transcription [11]. MED26 was also found to be essential for Pol II recruitment. However, it was proposed that MED26, rather than acting like MED14 to promote structural integrity, instead enabled transcription by counteracting negative co-factors, such as the transcriptional repressor DR1 [11].
However, it is not completely clear which subunits are “essential” in human cells. In one study MED26 was proposed as a non-essential subunit, because its deletion in HeLa cells did not affect the structural integrity of Mediator [12]. Furthermore, although combined deletion of MED26 and MED19 specifically affected genes controlled by the transcription factor REST, deletion of MED26 by itself did not have an effect [12]. Interestingly, the combined deletion resulted in the de-repression of REST target genes, highlighting the role of mediator subunits in repression of gene expression, as well as activation [12]. Indeed, in one study the deletion of MED19 in yeast resulted in increased transcript levels, as did deletion of kinase module subunits. The deletion of other subunits, such as MED20 (head) and MED31 (middle), resulted in decreased transcript levels [13]. This suggests a potential negative transcriptional regulatory role for MED19, which may be responsible for transmitting repressive signals from the kinase module to the middle module [13].
In contrast to “essential” subunits like MED14, there are some subunits that have consistently been shown to be dispensable for general Mediator function. In yeast, for example, it was demonstrated that deletion of individual tail subunits, such as MED3 or MED15, did not affect viability. However, individual subunit deletions did result in moderate changes in the expression of SAGA-dependent genes [14]. This is consistent with the role of tail subunits in controlling the expression of particular subsets of genes through interaction with specific transcription factors. Interestingly, however, combinatorial deletion of MED3 and MED15 strongly affected SAGA-dependent gene expression and moderately reduced growth, suggesting at least some redundancy in subunit function [14].
To make for an even more complex picture, not only can some subunits compensate for others upon their deletion, but it also appears that Mediator can exist as heterogeneous “subcomplexes,” rather than as a strictly complete complex with every subunit present [2, 11, 15]. Some subunits can be added or taken out to regulate its function, and various subcomplexes that lack certain subunits have been isolated [11, 15, 16]. These subcomplexes are intact and deficient only in regulating transcription of specific genes, much like the MED3/15-less Mediator in the study by Ansari, et al [11, 14–16]. In that case, Mediator lacking MED3 or MED3 and MED15 specifically affected genes like MET2 and MET32, both involved in methionine biosynthesis [14].
Reconstitution of a minimal Mediator to determine the “core” mediator complex was found to contain head and middle subunits [11]. Consistent with studies on the role of MED14 as the backbone of Mediator, MED14 was found to be required for Mediator association with Pol II and transcriptional function; however, the middle subunits MED1 and MED19 were not determined to be necessary parts of the core Mediator [11]. This is consistent with other studies, where deletion of MED19 in yeast does not affect viability, and a functional MED19-less Mediator can be isolated [16]. Interestingly, however, Mediator lacking MED19 isolated under stringent conditions produced a head-tail subcomplex that lacks the middle module entirely and is structurally stable but defective in transcription [16]. It is worth noting that there is some controversy with regard to the dispensability of MED19 – in one study a MED19 deletion mutant was found to be inviable [3, 13].
Similarly, although MED1 knockout is embryonic lethal in mice, with MED1 playing a crucial role in development, MEFs taken from MED1-knockout mouse embryos contain intact Mediator that can be isolated, with the “core” subunits present in proportions similar to wild type Mediator, and drives gene expression [17]. However, Mediator lacking MED1 was deficient in transcription controlled by thyroid hormone receptor, suggesting a role for MED1 similar to tail subunits: interaction with specific transcription factors [17]. Indeed, MED1 has LXXLL motifs typical of co-regulators for nuclear receptors, and MED1 has been found to interact with the AF-2 domain of multiple nuclear receptors [2, 18]. Other mediator subunits have been implicated in nuclear receptor function as well, including the dispensable MED19, as well as the essential MED14 [19–21]. Of particular interest are their interactions with and regulation of the androgen receptor (AR) and estrogen receptor (ER) [3, 18].
2 Mediator and Nuclear Receptors: Estrogen Receptor alpha (ER) and Androgen Receptor (AR)
MED1 is the best characterized of the mediator subunits that act as a nuclear receptor co-factor. MED1 contains two LXXLL motifs, interacts with multiple nuclear receptors, and stimulates their transcriptional activity in a ligand-dependent manner [19, 22]. One of the nuclear receptors with which MED1 interacts with is ER, and MED1 also stimulates ER transcriptional activity [19, 23]. It was found that following estrogen treatment, ER binds to the promoter of target genes, and co-activators, including histone acetyltransferases and Mediator via MED1, are recruited, with Mediator in turn recruiting Pol II [24]. A recent study implicated Mediator in the early phase of ligand-dependent enhancer formation such that the co-activator p300 is recruited to ER by Mediator, followed by steroid receptor co-regulator (SRC) recruitment to ER for proper enhancer function [25].
Highlighting the importance of MED1 and Mediator in ER function, a later study demonstrated dysfunctional ER target gene expression and mammary development in MED1 LXXLL mutant knock-in mice [26]. However, these mice otherwise appeared healthy, indicating that other mediator subunits could act as co-regulators to compensate for MED1 dysfunction (discussed below). There also may be an alternative method of interaction between MED1 and nuclear receptors or co-factors, possibly mediated by the N-terminal domain of MED1 [18, 26]. There is some evidence for the latter possibility: MED1 was found to interact through its N-terminal domain with CCAR1, an ER co-activator, with depletion of CCAR1 disrupting Mediator and Pol II recruitment to ER target genes [27]. MED1 was also shown to cooperate with the protein ARGLU1, discovered as a MED1-interacting protein through mass spectrometry, to promote ER target gene expression [28].
MED1 also interacts with AR and is recruited along with Pol II to the promoter and enhancer of AR target genes, stimulating their expression [29–31]. Furthermore, MED1 was found to be essential for Pol II recruitment to and the expression of AR target genes [30]. Interestingly, MED1 was shown to participate in enhancer-promoter looping at the PSA locus mediated by androgen-induced PSA enhancer RNA, highlighting that Mediator subunits promote transcriptional activation through multiple mechanisms [32].
Although MED1 was initially found to interact with the ligand-binding domain of AR through the LXXLL motif, this interaction was weak [19]. A non-canonical binding motif on MED1 was later discovered that interacts with the N-terminal domain of AR, independently of the two LXXLL motifs [33]. This interaction is stabilized by phosphorylation of MED1, which also enhances promoter-enhancer looping, Pol II recruitment, and expression of AR target genes [33, 34]. Furthermore, phosphorylation of MED1 promotes recruitment of FOXA1, another AR co-regulator [34].
Several other Mediator subunits have been found to act as co-regulators for nuclear receptors, including ER and AR. MED14 was first found to interact with the glucocorticoid receptor (GR), with which MED1 also interacted [35, 36]. In contrast to MED1, which interacted with the AF-2 domain in the ligand-binding domain of GR, MED14 interacted with the AF-1 domain in the N-terminal domain. In addition, MED14 and MED1 appear to control distinct sets of GR target genes, with some overlap in genes that required both subunits for expression [35, 36]. This indicates that different mediator subunits have distinct functions in controlling nuclear receptor activity, though some overlap implies that there may be a degree of redundancy or some ability to compensate, as proposed in the study with LXXLL mutant knock-in mice [26, 36].
MED14 also promotes ER and AR activity. MED14 was found to interact with and promote the transcriptional activation of ER, independently of LXXLL motifs [21]. MED14, along with MED24 and MED1, were found to stimulate the transcriptional activity of ligand-bound AR, when overexpressed in human cells [29]. MED24, like MED1, contains LXXLL motifs, but was initially found to have weak or negligible interactions with nuclear receptors [19]. However, it does stimulate the transcriptional activity of AR [29]. This indicates that some mediator subunits, like MED1, act as direct AR co-factors through physical interaction with AR, while others, like MED24, may act as indirect AR co-factors by stabilizing the interaction between AR and MED1 and transmitting signals to the rest of the mediator complex [29].
MED19 is a mediator subunit that has more recently been shown to act as an AR co-regulator. MED19 was identified in a genome-wide RNAi screen, with its depletion resulting in a reduction in AR target gene expression, suggesting that MED19 might function as an AR co-activator [20]. Indeed, depletion of MED19 by siRNA reduced the expression of AR target genes in LNCaP-abl cells (androgen-independent prostate cancer cell line), and reduced the proliferation in both LNCaP-abl cells and LNCaP cells (androgen-dependent prostate cancer cell line) [20]. MED19 has also been implicated in the growth of prostate cancer cells in other studies [37, 38]. Although no studies have examined MED19 as an ER co-regulator, it has been demonstrated to play a role in the growth of breast cancer cells, indicating that it may regulate ER signaling [39, 40].
3 Mediator and Endocrine Cancers
MED19, as well as MED1 and other mediator subunits, have been shown to play a role in multiple cancers, particularly prostate and breast cancer, where multiple Mediator subunits have altered expression (Figure 2) [20, 37–43]. MED1, in keeping with its detailed characterization as a nuclear receptor co-regulator, is one of the best-characterized Mediator subunits in cancer. MED1 has been shown to interact with and drive the activity of AR in prostate cancer cells [29]. Overexpression of MED1 drives AR target gene expression and growth in vitro and in vivo; conversely, depletion of MED1 reduces AR target gene expression, proliferation, and cell cycle progression, and increases apoptosis [9, 34, 43, 44]. In addition, MED1 is overexpressed in primary prostate cancer patient samples [9, 43].
Figure 2.
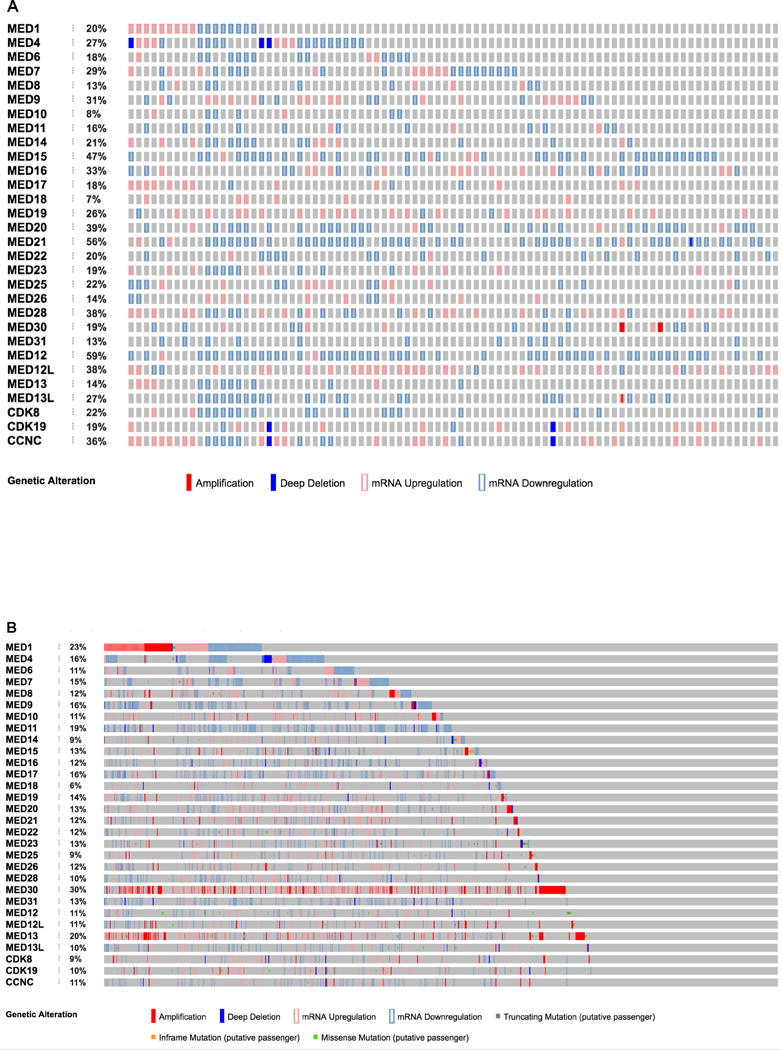
The expression of Mediator subunits is altered in prostate cancer and breast cancer. A. Expression of each Mediator subunit in 85 prostate adenocarcinoma complete tumor samples with sequencing, CNA, and mRNA data from Taylor, et al, 2010. Several Mediator subunits, such as MED19, show consistent mRNA upregulation (1.5-fold or greater). In the case of MED19 this is correlated with poor outcome. B. Expression of each Mediator subunit in 816 breast adenocarcinoma complete tumor samples with sequencing, CNA, and mRNA data from Ciriello, et al, 2015. Several Mediator subunits, such as MED1 or MED30, show mRNA upregulation (1.5-fold or greater) or amplification. In the case of MED1 this is correlated with poor outcome.
In castration-resistant prostate cancer (CRPC), AR is constitutively active in spite of low levels of androgens; this can occur through the formation of AR splice variants (AR-V) that lack the ligand-binding domain [45, 46]. Although MED1 was originally shown to interact with full length AR in a ligand-dependent manner, MED1 can also interact with AR-V and drive their activity, consistent with the discovery of the non-canonical binding motif of MED1 that can bind the N-terminal domain of AR [33, 34, 47]. Interestingly, it is the phosphorylated form of MED1 that interacts with AR-V [34, 47]. Phosphorylation of MED1 drives its interaction with AR-V and recruitment to the promoter and enhancer of UBE2C, an AR-V-specific target gene [34, 47]. Phosphorylated MED1, which is more stable than the non-phosphorylated form of MED1, facilitates recruitment of Pol II, TATA-binding protein (TBP), and FOXA1, resulting in promoter-enhancer looping and driving UBE2C expression and cell growth [33, 34, 43, 47]. Phosphorylation of MED1 can occur through AKT or ERK signaling, both of which are pathways that have been implicated in CRPC [34, 43, 45, 48, 49].
MED1 also plays an important role in breast cancer. Beyond its interaction with ER, MED1 was shown to be essential for the expression of ER target genes and proliferation in breast cancer cells [15, 50]. In line with other studies on the dispensability of MED1, MED1 was only present in about 20% of mediator complexes, with MED1-containing Mediator also enriched for MED19 and MED15, both of which have been implicated in breast cancer, as well as prostate cancer (discussed below) [15, 20, 38, 51, 52]. This indicates that MED19 and MED15 may cooperate with MED1 in breast cancer to promote ER activity. This may also apply to MED24, which when depleted in breast cancer cells, reduced growth [50].
Analogous to its role in AR activity and prostate cancer, phosphorylated MED1 promotes constitutive activity of ER and treatment resistance in breast cancer. Phosphorylation of MED1 promotes its stability, interaction with ER, and recruitment to ER target genes in tamoxifen-resistant breast cancer cells [53, 54]. MED1 is likewise overexpressed in breast cancer patient tissue, and its depletion reverses tamoxifen resistance [53]. ERK and AKT signaling induce MED1 phosphorylation, which in HER2+ breast cancer cells, is driven by HER2 overexpression [54].
MED19 is another middle module subunit that has been implicated in prostate cancer and breast cancer progression, although it is not as well characterized as MED1. MED19, which was identified from a genome-wide RNAi screen for novel AR co-regulators, reduced the proliferation and AR activity of both androgen-dependent and androgen-independent prostate cancer cells, when depleted [20]. Alteration of MED19 mRNA, which was upregulated in a subset of prostate cancer patients, correlated with poor outcome [20]. A later study demonstrated overexpression of MED19 protein in primary prostate tumor compared to benign tissue [38]. Depletion of MED19 in LNCaP cells reduced proliferation, invasion, and xenograft growth, and it was suggested that MED19 may promote tumor growth and metastasis through the regulation of expression of pro-apoptotic, cell cycle, and EMT genes, such as Bid, p27, and N-cadherin [37, 38]. However, the mechanism by which MED19 regulates these genes, the specific signaling pathways it activates in prostate cancer, and how this intersects with its role as an AR co-regulator, have yet to be elucidated.
Similarly to prostate cancer, MED19 appears to be an important driver of breast cancer cell growth. MED19 is overexpressed in breast cancer tissue, and depletion of MED19 in breast cancer cells greatly reduced proliferation and cell cycle progression [40]. MED19 appears to promote growth through inhibition of CBFA2T3, a potential tumor suppressor that negatively regulates the E-box gene HEB. HEB is upregulated with MED19 overexpression and downregulated with MED19 depletion [39]. However, the mechanism by which this contributes to MED19-regulated breast cancer cell growth is unclear, as is the intersection of MED19 with ER function. No studies have yet directly linked MED19 to regulation of ER activity in breast cancer, unlike AR in prostate cancer. Emerging evidence points to a role for MED19 in many types of cancer, and future studies will likely shed light onto its mechanism of action in endocrine and other cancers [42, 55, 56].
The tail module MED15, like MED19, has recently been implicated in both prostate and breast cancer. MED15 was found to promote breast cancer metastasis through TGF-beta signaling. MED15 interacted with SMAD3, promoting SMAD2/3 phosphorylation and nuclear translocation. MED15 silencing reduced SMAD2/3 phosphorylation, TGF-beta signaling, and EMT marker expression, as well as lung metastases in breast cancer xenografts [47]. Similarly, in prostate cancer, MED15 is overexpressed in metastases, and correlates with TGF-beta signaling and proliferative markers, as well as AR overexpression and poor clinical outcome [57]. MED15 silencing in prostate cancer cells also reduces SMAD2/3 phosphorylation and nuclear translocation, as well as proliferation [57].
MED15 was also linked to PI3K signaling: in clinical samples, MED15 correlates to phospho-AKT, as well as phospho-SMAD3, and both PI3K/mTOR and TGF-beta receptor inhibition reduced MED15 expression in prostate cancer cells. Interestingly, MED15 silencing not only reduced the growth of prostate cancer cells, but also reduced the protein expression of AR [57, 58]. This is in line with the observation in clinical samples that MED15 overexpression correlates with AR overexpression [57]. This indicates that MED15 may act in an AR-dependent and an AR-independent manner, especially given that PI3K signaling intersects with AR activity but can promote CRPC independently of AR [48, 58]. Emphasizing the relevance of MED15 to CRPC, its expression increased in prostate cancer cells after androgen deprivation and in patient samples after androgen deprivation therapy [58].
Finally, subunits in the kinase module, particularly MED12, have been found to play a role in endocrine cancers, as well as non-endocrine cancers [3, 38, 52]. Several studies have shown that it is mutated or overexpressed in prostate cancer [52, 59, 60]. However, there is no evidence that MED12 acts as a co-factor for AR, and it does not seem to affect AR transcriptional activity in prostate cancer cells [20]. This indicates MED12 may promote prostate cancer progression through alternative signaling. Indeed, MED12 interacts with beta-catenin to promote WNT signaling, which contributes to CRPC progression [48, 57, 61]. MED12, like MED15, also activates TGF-beta signaling in prostate cancer [52].
Although MED12 has not been directly linked to AR function, it has been shown to play a potential role in regulating ER function in the context of breast cancer. For example, prevalent MED12 mutations occur in breast fibroadenomas and phyllodes tumors, and are associated with dysfunction in ER signaling and extracellular matrix proteins; furthermore, MED12 expression correlates with ER expression [62–64]. However, the mechanism by which MED12 affects ER signaling is unclear. One study found that silencing of MED12 reduced ER transcriptional activity; however, rather than regulating the function of ER through direct interaction, like MED1, MED12 appears to control the expression level of ER [65]. MED12 silencing reduced Pol II occupancy at the ESR1 promoter, as well as ESR1 expression. MED12 and cohesin were found to co-occupy the ESR1 gene promoter, indicating that MED12 cooperates with cohesin to promote the expression of ER [65].
However, control of ER expression may not be the only mechanism by which MED12 promotes growth in the context of ER-driven malignancies. Similarly to breast cancer, MED12 mutations have been linked to uterine cancer [66]. MED12 not only controls the expression of ER in uterine cancer cells, but also promotes growth through activation of WNT/beta-catenin signaling, as well as TGF-beta signaling [66, 67]. This indicates that MED12 in this context may function similarly to prostate cancer, promoting growth through regulation of proliferative pathways involved in treatment resistance [47, 68, 69].
4 Mediator and Non-Genomic Signaling
Although Mediator subunits have been shown to control growth through regulation of transcriptional processes, there is evidence that some Mediator subunits can regulate signaling pathways directly in the cytoplasm [70]. For example, MED12 in lung cancer cells was present in both the nucleus and the cytoplasm; in the latter compartment, MED12 regulated TGF-beta signaling by direct interaction with TGF-beta receptor 2, affecting its glycosylation and cell surface expression [70]. This indicates that MED12 can control TGF-beta signaling through both canonical regulation of transcription and non-genomic activity. Although the non-genomic activity of MED12, as well as other mediator subunits, in different types of cancer remains to be characterized, this provides an intriguing new insight into the many functions of Mediator.
The potential cytoplasmic functions of Mediator may be particularly relevant in the context of breast cancer and prostate cancer, given that ER and AR themselves can function in a non-genomic manner. For example, cytoplasmic AR can activate several pathways, including PI3K and MAPK signaling, and cytoplasmic AR splice variants have been detected in metastatic prostate cancer [71, 72]. Furthermore, cytoplasmic AR interacts with Src, either directly or through LXXLL motifs on the scaffold protein PELP1, to activate MAPK signaling and promote growth, survival, and invasion, and Src activity correlates with resistance in prostate cancer patients [73–75]. ER also interacts with Src, either directly or through PELP1 to promote invasion and metastasis in breast cancer [76, 77]. Non-nuclear ER can activate rapid estrogen signaling, as well as PI3K and MAPK signaling, promote proliferation and survival, and is associated with tamoxifen and chemotherapy resistance in breast cancer [78, 79].
Although it is particularly interesting that both mediator subunits and Src via PELP1 may bind to steroid receptors through LXXLL motifs, no interactions between steroid receptors and mediator subunits outside of the nucleus, or between Src and mediator subunits, have been documented. Given the emerging non-genomic roles of steroid receptors and mediator subunits, however, this may be intriguing to study.
4 Conclusion and Therapeutic Implications
The role of Mediator and the functions of individual Mediator subunits in regulating transcription, nuclear receptor signaling, and proliferative pathways continue to be characterized. The dysregulation of Mediator in cancer and other disorders has strong therapeutic implications. Given that specific subunits act as AR and ER co-regulators and can contribute to treatment resistance and tumor growth in prostate and breast cancer, they may provide novel targets to overcome treatment resistance [20, 34, 55]. For example, a member of the kinase module, CDK19, is overexpressed in prostate cancer and is associated with aggressive growth and decreased survival [80]. Inhibitors against CDK19 and CDK8, another member of the kinase module that activates WNT signaling, reduced prostate cancer cell proliferation and invasion [80, 81]. In another study, compounds that specifically inhibit CDK8 and CDK19 were identified and shown to inhibit in vitro cell growth in multiple cancer types, as well as in patient-derived xenografts, showing as a proof-of-principle that mediator subunits can be targeted to inhibit tumor growth [81]. However, the toxic side effects of the inhibitors halted their use in pre-clinical studies and pointed to the challenges of targeting Mediator [81].
Another possibility would be to identify and target the pathways activated downstream of individual mediator subunits that are upregulated in different cancers. For example, the overexpression of MED15 in CRPC and its association with activation of TGF-beta and PI3K signaling indicate that MED15 may serve as a potential biomarker for resistance and as a predictive marker for TGF-beta or PI3K/mTOR inhibitors [52, 58]. MED1 overexpression in prostate cancer and the association of phosphorylated MED1 with constitutive AR activity and MAPK and PI3K signaling imply that these pathways may be potential targets in CRPC with high expression of MED1 [43, 44]. Finally, the overexpression of MED12 in prostate cancer, and its association with WNT and TGF-beta signaling, indicate that these pathways could potentially be targeted for tumors with high MED12 expression [52]. Given that MED12 may directly interact with TGF-beta-receptor 2 in the cytoplasm, there is potential for drugs that could target this interaction [70].
Furthermore, some drugs that have shown efficacy in pre-clinical models of cancer and are being evaluated for clinical trials may work in part through disruption of Mediator interactions. For example, BRD4, a BET bromodomain protein, is an AR co-factor that is upregulated in prostate cancer [82]. JQ1, a BET bromodomain inhibitor, has been shown in prostate cancer to disrupt the interaction between BRD4 and AR [82]. Interestingly, in acute myeloid leukemia and multiple myeloma cells, JQ1 inhibited cell growth and disrupted the interaction between BRD4 and Mediator, preventing super-enhancer formation and inhibiting the expression of pro-growth and survival genes [83, 84]. A similar mechanism may also contribute to the efficacy of BET inhibition in prostate cancer and other cancers, especially given that several Mediator subunits are AR co-regulators [82]. In line with this, BET inhibition is effective in breast cancer cells, where there is a strong association between BRD4 and MED1 [85]. Overall, continuing characterization of Mediator in cancer and other disorders will provide important insight for the development of novel and effective therapies.
Acknowledgments
This work was supported by the National Institutes of Health (grant numbers 5T32GM066704-13/14, T32GM007308). We thank Dr. Elina Shrestha and Jeffrey Schneider of the Garabedian and Logan laboratories for helpful comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations: ARGLU1 - Arginine And Glutamate Rich 1; CBFA2T3 -CBFA2/RUNX1 Translocation Partner 3; CCAR1 - Cell Division Cycle And Apoptosis Regulator 1; DR1 - Down-Regulator Of Transcription 1; HEB - Transcription Factor 12 (TCF 12); PELP1 - Proline, Glutamate And Leucine Rich Protein 1; MED - Mediator; REST - RE1 Silencing Transcription Factor; SAGA - Spt-Ada-Gcn5 Acetyltransferase; UBE2C - Ubiquitin Conjugating Enzyme E2 C
References
- 1.Robinson P, Trnka M, Bushnell D, Davis R, Mattei P, Burlingame A, et al. Structure of a Complete Mediator-RNA Polymerase II Pre-Initiation Complex. Cell. 2016;166:1411–22. doi: 10.1016/j.cell.2016.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen B, Taatjes D. The Mediator complex: a central integrator of transcription. Molecular Cell Biology. 2015;16:155–66. doi: 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.CJeronimo Robert F. The Mediator Complex: At the Nexus of RNA Polymerase II Transcription. Trends in Cell Biology. 2017 doi: 10.1016/j.tcb.2017.07.001. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 4.Tsai K-L, Yu X, Gopalan S, Chao T-C, Zhang Y, Florens L, et al. Mediator structure and rearrangements required for holoenzyme formation. Nature. 2017;544:196–203. doi: 10.1038/nature21393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsai K-L, Tomomori-Sato C, Sato S, Conaway R, Conaway H, Asturias F. Subunit Architecture and Functional Modular Rearrangements of the Transcriptional Mediator Complex. Cell. 2014;157:1430–44. doi: 10.1016/j.cell.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jeronimo C, Langelier M-F, Bataille A, Pascal J, Pugh B, Robert F. Tail and Kinase Modules Differently Regulate Core Mediator Recruitment and Function In Vivo. Molecular Cell. 2016;64:455–66. doi: 10.1016/j.molcel.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petrenko N, Jin Y, Wong K, Struhl K. Mediator Undergoes a Compositional Change during Transcriptional Activation. Molecular Cell. 2016;64:443–54. doi: 10.1016/j.molcel.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Linder T, Zhu X, Baraznenok V, Gustafsson C. The classical srb4-138 mutant allele causes dissociation of yeast Mediator. Biochem Biophys Res Commun. 2006;349:948–53. doi: 10.1016/j.bbrc.2006.08.099. [DOI] [PubMed] [Google Scholar]
- 9.Vijayvargia R, May M, Fondell J. A Coregulatory Role for the Mediator Complex in Prostate Cancer Cell Proliferation and Gene Expression. Cancer Research. 2007;67:4034–41. doi: 10.1158/0008-5472.CAN-06-3039. [DOI] [PubMed] [Google Scholar]
- 10.Harper TM, Taatjes DJ. The complex structure and function of Mediator. J Biol Chem. 2017 doi: 10.1074/jbc.R117.794438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cevher M, Shi Y, Li D, Chait B, Malik S, Roeder R. Reconstitution of active human core Mediator complex reveals a critical role of the MED14 subunit. Nature Structural & Molecular Biology. 2014;21:1028–36. doi: 10.1038/nsmb.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ding N, Tomomori-Satto C, Sato S, Conaway R, Conaway J, Boyer T. MED19 and MED26 are synergistic functional targets of the RE1 silencing transcription factor in epigenetic silencing of neuronal gene expression. Journal of Biological Chemistry. 2009;284:2648–56. doi: 10.1074/jbc.M806514200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peppel Jvd, Kettelarij N, Bakel Hv, Kockelkorn T, Leenen Dv, Holstege F. Mediator Expression Profiling Epistasis Reveals a Signal Transduction Pathway with Antagonistic Submodules and Highly Specific Downstream Targets. Molecular Cell. 2005;19:511–22. doi: 10.1016/j.molcel.2005.06.033. [DOI] [PubMed] [Google Scholar]
- 14.Ansari S, Ganapathi M, Benschop J, Holstege F, Wade J, Morse R. Distinct role of Mediator tail module in regulation of SAGA-dependent, TATA-containing genes in yeast. EMBO Journal. 2012;31:44–57. doi: 10.1038/emboj.2011.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang X, Krutchinsky A, Fukuda A, Chen W, Yamamura S, Chait B, et al. MED1/TRAP220 Exists Predominantly in a TRAP/Mediator Subpopulation Enriched in RNA Polymerase II and Is Required for ER-Mediated Transcription. Molecular Cell. 2005;19:89–100. doi: 10.1016/j.molcel.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 16.Baidoobonso S, Guidi B, Myers L. Med19(Rox3) Regulates Intermodule Interactions in the Saccharomyces cerevisiae Mediator Complex. Journal of Biological Chemistry. 2007;282:5551–9. doi: 10.1074/jbc.M609484200. [DOI] [PubMed] [Google Scholar]
- 17.Malik S, Guermah M, Yuan C-X, Wu W, Yamamura S, Roeder R. Structural and Functional Organization of TRAP220, the TRAP/Mediator Subunit That Is Targeted by Nuclear Receptors. Molecular and Cellular Biochemistry. 2004;24:8244–54. doi: 10.1128/MCB.24.18.8244-8254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W, Roeder R. Mediator-dependent nuclear receptor function. Seminars in Cell & Developmental Biology. 2011;22:749–58. doi: 10.1016/j.semcdb.2011.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan C-X, Ito M, Fondell J, Fu Z-Y, Roeder R. The TRAP220 component of a thyroid hormone receptor-associated protein (TRAP) coactivator complex interacts directly with nuclear receptors in a ligand-dependent fashion. Proceedings of the National Academy of Sciences. 1998;95:7939–44. doi: 10.1073/pnas.95.14.7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imberg-Kazdan K, Ha S, Greenfield A, Poultney C, Bonneau R, Logan S, et al. A genome-wide RNA interference screen identifies new regulators of androgen receptor function in prostate cancer cells. Genome Research. 2013;23:581–91. doi: 10.1101/gr.144774.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J, Kim K, Sacchettini J, Smith C, Safe S. DRIP150 Coactivation of Estrogen Receptor in ZR-75 Breast Cancer Cells Is Independent of LXXLL Motifs. Journal of Biological Chemistry. 2005;280:8819–30. doi: 10.1074/jbc.M413184200. [DOI] [PubMed] [Google Scholar]
- 22.Hittelman AB, Burakov D, Iniguez-Lluhi JA, Freedman LP, Garabedian MJ. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J. 1999;18:5380–8. doi: 10.1093/emboj/18.19.5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang Y, Guermah M, Yuan C-X, Roeder R. The TRAP/Mediator coactivator complex interacts directly with estrogen receptors and through the TRAP220 subunit and directly enhances estrogen receptor function in vitro. Proceedings of the National Academy of Sciences. 2002;99:2642–7. doi: 10.1073/pnas.261715899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shang Y, Hu X, DiRenzo J, Lazar M, Brown M. Cofactor Dynamics and Sufficiency in Estrogen Receptor–Regulated Transcription. Cell. 2000;103 doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 25.Murakami S, Nagari A, Kraus WL. Dynamic assembly and activation of estrogen receptor alpha enhancers through coregulator switching. Genes Dev. 2017;31:1535–48. doi: 10.1101/gad.302182.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang P, Hua Q, Itoc M, Meyer S, Waltz S, Khan S, et al. Key roles for MED1 LxxLL motifs in pubertal mammary gland development and luminal-cell differentiation. Proceedings of the National Academy of Sciences. 2010;107:6765–70. doi: 10.1073/pnas.1001814107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim J, Yang C, Heo K, Roeder R, An W, Stallcup M. CCAR1, a key regulator of Mediator complex recruitment to nuclear receptor transcription complexes. Molecular Cell. 2008;31:510–9. doi: 10.1016/j.molcel.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang D, Jiang P, Xu Q, Zhang X. Arginine and Glutamate-rich 1 (ARGLU1) Interacts with Mediator Subunit 1 (MED1) and Is Required for Estrogen Receptor-mediated Gene Transcription and Breast Cancer Cell Growth. Journal of Biological Chemistry. 2011;286:17746–54. doi: 10.1074/jbc.M110.206029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Q, Sharma D, Ren Y, Fondell J. A coregulatory role for the TRAP-mediator complex in androgen receptor-mediated gene expression. Journal of Biological Chemistry. 2002;277:42852–8. doi: 10.1074/jbc.M206061200. [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, Carroll J, Brown M. Spatial and Temporal Recruitment of Androgen Receptor and Its Coactivators Involves Chromosomal Looping and Polymerase Tracking. Molecular Cell. 2005;19:631–42. doi: 10.1016/j.molcel.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 31.Louie M, Yang H, Ma A-H, Xu W, Zou J, Kung H-J, et al. Androgen-induced recruitment of RNA polymerase II to a nuclear receptor–p160 coactivator complex. Proceedings of the National Academy of Sciences. 2003;100:2226–30. doi: 10.1073/pnas.0437824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsieh C-L, Feia T, Brown M, Kantoff P. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proceedings of the National Academy of Science. 2014;111:7319–24. doi: 10.1073/pnas.1324151111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin F, Claessens F, Fondell J. Regulation of Androgen Receptor-dependent Transcription by Coactivator MED1 Is Mediated through a Newly Discovered Noncanonical Binding Motif. Journal of Biological Chemistry. 2012;287:858–70. doi: 10.1074/jbc.M111.304519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Z, Zhang C, Wu D, Chen H, Rorick A, Zhang X, et al. Phospho-MED1-enhanced UBE2C locus looping drives castration-resistant prostate cancer growth. EMBO Journal. 2011;30:2405–19. doi: 10.1038/emboj.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hittelman A, Burakov D, Iniguez-Lluhi J, Freedman L, Garabedian M. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO Journal. 1999;18:5380–8. doi: 10.1093/emboj/18.19.5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen W, Rogatsky I, Garabedian M. MED14 and MED1 Differentially Regulate Target-Specific Gene Activation by the Glucocorticoid Receptor. Molecular Endocrinology. 2006;20:560–72. doi: 10.1210/me.2005-0318. [DOI] [PubMed] [Google Scholar]
- 37.Cui X, Xu D, Yao Y, Yu H. Suppression of MED19 expression by shRNA induces inhibition of cell proliferation and tumorigenesis in human prostate cancer cells. BMB reports. 2011;44:547–52. doi: 10.5483/bmbrep.2011.44.8.547. [DOI] [PubMed] [Google Scholar]
- 38.Yu S, Wang Y, Yuan H, Zhao H, Lv W, Chen J, et al. Knockdown of Mediator Complex Subunit 19 Suppresses the Growth and Invasion of Prostate Cancer Cells. PLoS ONE. 2017;12:1–14. doi: 10.1371/journal.pone.0171134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Fan Y, Liu B, Qi X, Guo Z, Li L. Med19 promotes breast cancer cell proliferation by regulating CBFA2T3/HEB expression. Breast Cancer. 2017;24:433–41. doi: 10.1007/s12282-016-0722-3. [DOI] [PubMed] [Google Scholar]
- 40.Li L-H, He J, Hua D, Guo Z, Gao Q. Lentivirus-mediated inhibition of Med19 suppresses growth of breast cancer cells in vitro. Cancer Chemotherapy and Pharmacology. 2011;68:207–15. doi: 10.1007/s00280-010-1468-9. [DOI] [PubMed] [Google Scholar]
- 41.Cui J, Germer K, Wu T, Wang J, Luo J, Wang Q, et al. Cross-talk between HER2 and MED1 regulates tamoxifen resistance of human breast cancer cells. Cancer Research. 2012;72:5625–34. doi: 10.1158/0008-5472.CAN-12-1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang LCJ, Leonard M, Nephew K, Li Y, Zhang X. Silencing MED1 sensitizes breast cancer cells to pure anti-estrogen fulvestrant in vitro and in vivo. PLoS ONE. 2013;8:1–11. doi: 10.1371/journal.pone.0070641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin F, Irshad S, Yu W, Belakavadi M, Chekmareva M, Ittmann M, et al. ERK and AKT Signaling Drive MED1 Overexpression in Prostate Cancer in Association with Elevated Proliferation and Tumorigenicity. Molecular Cancer Research. 2013;11:736–47. doi: 10.1158/1541-7786.MCR-12-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu G, Sprenger C, Wu P-J, Sun S, Uo T, KvHaugk, et al. MED1 mediates androgen receptor splice variant induced gene expression in the absence of ligand. Oncotarget. 2014;6:288–304. doi: 10.18632/oncotarget.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watson P, Arora V, Sawyers C. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nature Reviews Cancer. 2015;15:701–11. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu J, Steen TVd, Tindall D. Are androgen receptor variants a substitute for the full-length receptor? Nature Reviews Urology. 2015;12:137–44. doi: 10.1038/nrurol.2015.13. [DOI] [PubMed] [Google Scholar]
- 47.Wang Z, Zhang H, Hou J, Niu J, Ma Z, Zhao H, et al. Clinical implications of β-catenin protein expression in breast cancer. Int J Clin Exp Pathol. 2015;8:14989–94. [PMC free article] [PubMed] [Google Scholar]
- 48.Robinson D, Allen EV, Sawyers C, Chinnaiyan A. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell. 2015;161:1215–122. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kato M, Banuelos C, Imamura Y, Leung J, Caley D, Wang J, et al. Co-targeting androgen receptor splice variants and mTOR signaling pathway for the treatment of castration-resistant prostate cancer. Clinical Cancer Research. 2016;22:2744–54. doi: 10.1158/1078-0432.CCR-15-2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hasegawa N, Sumitomo A, Fujita A, Aritome N, Mizuta S, Matsui K, et al. Mediator Subunits MED1 and MED24 Cooperatively Contribute to Pubertal Mammary Gland Development and Growth of Breast Carcinoma Cells. Molecular and Cellular Biology. 2012;32:1483–95. doi: 10.1128/MCB.05245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao M, Yang X, Fu Y, Wang H, Ning Y, Yan J, et al. Mediator MED15 modulates transforming growth factor beta (TGFb)/Smad signaling and breast cancer cell metastasis. Journal of Molecular Cell Biology. 2013;5:57–60. doi: 10.1093/jmcb/mjs054. [DOI] [PubMed] [Google Scholar]
- 52.Shaikhibrahim Z, Offermann A, Kirfel J, Perner S. MED12 overexpression is a frequent event in castration-resistant prostate cancer. Endocrine-Related Cancer. 2014;21:663–75. doi: 10.1530/ERC-14-0171. [DOI] [PubMed] [Google Scholar]
- 53.Nagalingam A, Tighiouart M, Ryden L, Joseph L, Landberg G, Saxena N, et al. Med1 plays a critical role in the development of tamoxifen resistance. Carcinogenesis. 2012;33:918–30. doi: 10.1093/carcin/bgs105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mansouria S, Naghavi-al-Hosseinia F, Farahmanda L, Majidzadeh-Ab K. MED1 may explain the interaction between receptor tyrosine kinases and ERα66 in the complicated network of Tamoxifen resistance. European Journal of Pharmacology. 2017;804:78–81. doi: 10.1016/j.ejphar.2017.03.026. [DOI] [PubMed] [Google Scholar]
- 55.He G, Hu J, Zhou L, Zhu X, Xin S, Zhang D, et al. The FOXD3/miR-214/MED19 axis suppresses tumour growth and metastasis in human colorectal cancer. Br J Cancer. 2016;115:1367–78. doi: 10.1038/bjc.2016.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wei L, Wang X, Sun J, Lv L, Xie L, Song X. Knockdown of Med19 suppresses proliferation and enhances chemo-sensitivity to cisplatin in non-small cell lung cancer cells. Asian Pac J Cancer Prev. 2015;16:875–80. doi: 10.7314/apjcp.2015.16.3.875. [DOI] [PubMed] [Google Scholar]
- 57.Shaikhibrahim Z, Menon R, Biskup S, Perner S. MED15, encoding a subunit of the mediator complex, is overexpressed at high frequency in castration-resistant prostate cancer. International Journal of Cancer. 2014;135:19–26. doi: 10.1002/ijc.28647. [DOI] [PubMed] [Google Scholar]
- 58.Offermann A, Vlasic I, Syring I, Vogel W, Ruiz C, Zellweger T, et al. MED15 overexpression in prostate cancer arises during androgen deprivation therapy via PI3K/mTOR signaling. Oncotarget. 2017;8:7964–76. doi: 10.18632/oncotarget.13860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barbieri C, Baca S, Rubin M, Garraway L. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nature Genetics. 2012;44:685–91. doi: 10.1038/ng.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kampjarvi K, Kim N, Boyer T, Vahteristo P. Somatic MED12 Mutations in Prostate Cancer and Uterine Leiomyomas Promote Tumorigenesis Through Distinct Mechanisms. The Prostate. 2016;76:22–31. doi: 10.1002/pros.23092. [DOI] [PubMed] [Google Scholar]
- 61.Kim S, Xu X, Hecht A, Boyer T. Mediator Is a Transducer of Wnt/B-Catenin Signaling. Journal of Biological Chemistry. 2006;281:14066–75. doi: 10.1074/jbc.M602696200. [DOI] [PubMed] [Google Scholar]
- 62.Yoshida M, Sekine S, Ogawa R, Yoshida H, Maeshima A, Kanai Y, et al. Frequent MED12 mutations in phyllodes tumours of the breast. Br J Cancer. 2015;112:1703–8. doi: 10.1038/bjc.2015.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lim W, Ong C, Tan P, Teh B. Exome sequencing identifies highly recurrent MED12 somatic mutations in breast fibroadenoma. Nature Genetics. 2014;46:877–80. doi: 10.1038/ng.3037. [DOI] [PubMed] [Google Scholar]
- 64.Tan W, Chan J, Teh B, Tan P. MED12 protein expression in breast fibroepithelial lesions: correlation with mutation status and oestrogen receptor expression. Journal of Clinical Pathology. 2016;69:858–65. doi: 10.1136/jclinpath-2015-203590. [DOI] [PubMed] [Google Scholar]
- 65.Prenzel T, Kramer F, Bedi U, Nagarajan S, Beissbarth T, Johnsen S. Cohesin is required for expression of the estrogen receptor-alpha (ESR1) gene. Epigenetics & Chromatin. 2012;5:1–13. doi: 10.1186/1756-8935-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kampjarvi K, Park M, Boyer T, Vahteristo P. Mutations in Exon 1 Highlight the Role of MED12 in Uterine Leiomyomas. Human Mutation. 2014;35:1136–41. doi: 10.1002/humu.22612. [DOI] [PubMed] [Google Scholar]
- 67.Al-Hendy A, Laknaur A, Diamond M, Ismail N, Boyer T, Halder S. Silencing Med12 Gene Reduces Proliferation of Human Leiomyoma Cells Mediated via Wnt/b-Catenin Signaling Pathway. Endocrinology. 2017;158:592–603. doi: 10.1210/en.2016-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Howe L, Brown A. Wnt Signaling and Breast Cancer. Cancer Biology & Therapy. 2004;3:36–41. doi: 10.4161/cbt.3.1.561. [DOI] [PubMed] [Google Scholar]
- 69.Hiemer S, Szymaniak A, Varelas X. The Transcriptional Regulators TAZ and YAP Direct Transforming Growth Factor -induced Tumorigenic Phenotypes in Breast Cancer Cells. Journal of Biological Chemistry. 2014;289:13461–74. doi: 10.1074/jbc.M113.529115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang S, Holzel M, Wessels L, Bernards R. MED12 controls the response to multiple cancer drugs through regulation of TGF-β receptor signaling. Cell. 2012;151:937–50. doi: 10.1016/j.cell.2012.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jagla M, Fève M, Kessler P, Lapouge G, Erdmann E, Serra S, et al. A Splicing Variant of the Androgen Receptor Detected in a Metastatic Prostate Cancer Exhibits Exclusively Cytoplasmic Actions. Endocrinology. 2007;148:4334–43. doi: 10.1210/en.2007-0446. [DOI] [PubMed] [Google Scholar]
- 72.Liao R, Ma S, Miao L, Li R, Yin Y, Raj G. Androgen receptor-mediated non-genomic regulation of prostate cancer cell proliferation. Translational Andrology and Urology. 2013;2:187–96. doi: 10.3978/j.issn.2223-4683.2013.09.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Migliaccio A, Castoria G, Auricchio F. Analysis of Androgen Receptor Rapid Actions in Cellular Signaling Pathways: Receptor/Src Association. Androgen Action, Methods in Molecular Biology (Methods and Protocols) 2011;776:361–70. doi: 10.1007/978-1-61779-243-4_21. [DOI] [PubMed] [Google Scholar]
- 74.Zarif J, Lamb L, Schulz V, Nollet E, Miranti C. Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget. 2015;6:6862–76. doi: 10.18632/oncotarget.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Migliaccio A, Castoria G, Abbondanza C, Auricchio F. Steroid-induced androgen receptor–oestradiol receptor β–Src complex triggers prostate cancer cell proliferation. EMBO Journal. 2000;19:5406–17. doi: 10.1093/emboj/19.20.5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chakravarty D, Nair S, Kumar R, Vadlamudi R. Extranuclear functions of ER impact invasive migration and metastases of breast cancer cells. Cancer Research. 2010;70:4092–101. doi: 10.1158/0008-5472.CAN-09-3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zarif J, Miranti C. The importance of non-nuclear AR signaling in prostate cancer progression and therapeutic resistance. Cellular Signalling. 2016;28:348–56. doi: 10.1016/j.cellsig.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chaudhri R, Schwartz N, Elbaradie K, Schwartz Z, Boyan B. Role of ERα36 in membrane-associated signaling by estrogen. Steroids. 2014;81:74–80. doi: 10.1016/j.steroids.2013.10.020. [DOI] [PubMed] [Google Scholar]
- 79.Wang Z-Y, Yin L. Estrogen receptor alpha-36 (ER-α36): A new player in human breast cancer. Molecular and Cellular Endocrinology. 2015;418:193–206. doi: 10.1016/j.mce.2015.04.017. [DOI] [PubMed] [Google Scholar]
- 80.Bragelmann J, Klumper N, Kirfel J, Perner S. Pan-cancer analysis of the Mediator complex transcriptome identifies CDK19 and CDK8 as therapeutic targets in advanced prostate cancer. Clinical Cancer Research. 2017;23:1829–40. doi: 10.1158/1078-0432.CCR-16-0094. [DOI] [PubMed] [Google Scholar]
- 81.Clarke P, Ortiz-Ruiz M-J, Workman P, Wienke D. Assessing the mechanism and therapeutic potential of modulators of the human Mediator complex-associated protein kinases. eLife. 2016;5:1–25. doi: 10.7554/eLife.20722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Asangani I, Dommeti V, Feng F, Chinnaiyan A. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature. 2014;510:278–85. doi: 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bhagwat A, Roe J-S, Mok B, Hohmann A, Shi J, Vakoc C. BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Reports. 2016;15:519–30. doi: 10.1016/j.celrep.2016.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Loven J, Hoke H, Lin C, Lau A, Orlando D, Vakoc C, et al. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell. 2013;153:320–34. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shu S, Lin C, Bradner J, Polyak K. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413–20. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]