

Abstract
The Curtius rearrangement is a versatile reaction in which a carboxylic acid can be converted to an isocyanate through acyl azide intermediate under mild conditions. The resulting stable isocyanate can then be readily transformed into a variety of amines and amine derivatives including urethanes and ureas. There have been wide-ranging applications of Curtius rearrangement in the synthesis of natural products and their derivatives. Also, this reaction has been extensively utilized in the synthesis and application of a variety of biomolecules. In this review, we present mechanistic studies, chemical methodologies and reagents for the synthesis of isocyanates from carboxylic acids, conversion of isocyanates to amines and amine derivatives, and their applications in the synthesis of bioactive natural products and their congeners.
Graphical Abstract
An extensive review of the Curtius reaction and its recent applications in the synthesis of bioactive natural products are reported
1. Introduction
In 1885, at Heidelberg University, Julius Wilhelm Theodor Curtius discovered that acyl azides derived from carboxylic acids undergo thermal decomposition to provide isocyanates and nitrogen.1,2 Since then, this reaction known as Curtius rearrangement or Curtius reaction has been widely used in organic synthesis due to the usefulness of isocyanate intermediates.3–5 Isocyanates can be readily converted to a range of functionalities including amines, urethanes and ureas by reaction with appropriate nucleophiles such as water, alcohols or amines, respectively (Figure 1).6,7 These structural motifs frequently occur in natural products, pharmaceuticals and agrochemicals.8–10
Fig. 1.

The Curtius rearrangement route to isocyanates, amines and amine derivatives.
Since amines are key precursors for the synthesis of a variety of amine derivatives, many synthetic methods have been developed for their preparation.11,12 Amine synthesis using the Curtius rearrangement is particularly appealing since it is applicable to a wide range of carboxylic acids including aliphatic, aromatic, heterocyclic, saturated and unsaturated acids containing multiple functional groups.6,13,14 Furthermore, many mild methods have been developed for the conversion of carboxylic acids to their acyl azides, isocyanates and amine derivatives in a one-pot fashion.15–17 Another important feature of the Curtius rearrangement is that the reaction provides access to primary amines without contamination with secondary or tertiary amines.18,19 Furthermore, the reaction proceeds with complete retention of stereochemistry of the migrating carbon. This stereochemical integrity has been exploited for the generation of chiral nitrogen-containing centers. For these reasons, the Curtius rearrangement is often the method of choice for the synthesis of structurally complex amines.20–22
Curtius rearrangement has been extensively utilized in the synthesis of bioactive natural products and in drug development. However, the full potential of this reaction has not yet been recognized.23,24 In the present review, we will highlight the gradual development of the Curtius rearrangement, and chemical methodologies adopted for the synthesis of isocyanates, amines and amine derivatives. Furthermore, we plan to provide an overview of the applications of the Curtius rearrangement in total syntheses of natural products and their derivatives.
2. Mechanism of the Curtius Rearrangement
Over the years, the mechanism of the Curtius rearrangement has been a subject of great interest. Stieglitz in 1896, postulated that the mechanism of the Curtius rearrangement involved the loss of nitrogen from the acyl azide (2) and formation of an unstable acyl nitrene intermediate (7) where the R group migrates to the electron deficient nitrogen to give an isocyanate (3) (Figure 2).25 The migration of the R group happens with complete retention of stereochemistry.26 In 1891, Tiemann proposed a similar acyl nitrene intermediate for a related reaction, the Hofmann rearrangement.27 However, early efforts to trap the acyl nitrene intermediate failed and it was attributed to a short lifetime for the nitrene. This argued in favor of a concerted mechanism without involving this acyl nitrene intermediate.28–30
Fig. 2.

Stieglitz’s mechanism via carbonylnitrene intermediate.
Subsequent detailed mechanistic studies with a wide variety of acyl azides established that the intermediate acylnitrene can be trapped under photochemical conditions. However, the acyl nitrene could not be trapped under thermal conditions even though both thermal and photochemical decomposition of acyl azides were carried out at nearly identical reaction temperatures.28,29 In a series of studies, Lwowski and co-workers carried out thermolysis of pivaloyl azide in hydrocarbon solvents.29 In particular, thermolysis of pivaloyl azide in 2-methylbutane or in cyclohexene above room temperature provided quantitative yield of tert-butylisocyanate (Scheme 1).
Scheme 1.

Thermal and photochemical decomposition of pivaloyl azide.
There were virtually no insertion or addition products observed as the analytical method of detection would have identified 1% yield of any of these products.29 These experiments, along with a large number of other experimental,31 theoretical,32,33 and computational studies,34,35 concluded that the thermal Curtius rearrangement is a concerted reaction as shown in Scheme 2.31,32,36
Scheme 2.

Concerted thermal Curtius rearrangement mechanism.
The photochemical Curtius rearrangement, however, may take place in a concerted manner or it may involve a stepwise mechanism encompassing an acylnitrene intermediate. Lwowski and co-workers then carried out photolysis of pivaloyl azide in cyclohexane, 2-methylbutane, and cyclohexene as a solvent separately under a medium pressure mercury lamp at −15 °C to +5 °C temperature. Interestingly, these photochemical reactions furnished tert-butyl isocyanate 9 along with a range of products (10–13) arising from insertion of the nitrene into C-H bonds or its addition to the double bond.28,29 The yield of isocyanates was around 40% for all reactions even though nitrene traps were present in these reactions. Therefore, it has been postulated that the isocyanate and the trapping products were not derived from the same intermediate. Interestingly, photolysis of benzoyl azide and their derivatives resulted in 40–50% yields of isocyanates in the presence of solvents that trap acylnitrenes or inert solvents. Based upon these results, it was suggested that carboxylnitrenes do not undergo rearrangement to isocyanates at a rate competitive to their interception by the trapping agents.29,34,37,38
It is generally believed that the photochemical Curtius rearrangement proceeds via two separate pathways. These include, (i) a concerted pathway leading to an isocyanate and (ii) nitrene formation which may or may not provide an isocyanate. Pritchina and co-workers showed that Ar matrix photolysis of benzoyl azide provides phenyl isocyanate by a concerted mechanism as well as through a benzoylnitrene intermediate which was revealed by IR spectroscopy.39 Both experimental and computational work established that acylnitrenes have singlet ground states. Furthermore, calculations of the lowest singlet states of benzolynitrene and 2-naphthoylnitrene revealed that the structure of these carboxylnitrenes is an intermediate between nitrenes and oxazirenes with long N-O bonds with 1.73–1.82 Å distances. The calculated bond angle for NCO is nearly 90°. The singlet carbonylnitrene can be represented as a resonance hybrid of nitrene and oxazirene as shown in Scheme 3.31,40
Scheme 3.

Photochemical formation of acylnitrenes.
Wentrup and Bornemann31 showed that photolysis of benzoylazide isolated in an Ar matrix at 12 K at 308 nm provided phenyl isocyanate as the main product along with a small amount of phenyl cyanate with a characteristic C-O-C stretch of 1193 cm−1. Presumably, both of these products were derived from the direct excited state of the azide or from the rapid reaction via the nitrene.
Skell and Woodworth41 with Autrey and Schuster38 showed that acylnitrenes react with cis-alkenes to form cis-aziridine products. This is consistent with intercepting singlet state of acylnitrene. Interestingly, Liu and co-workers34 demonstrated that alkoxycarbonylazides upon photolysis in the presence of cis-alkenes resulted in both triplet and singlet acylnitrenes which provided a mixture of cis- and trans-aziridine derivatives (Scheme 4). Matrix spectroscopic experiments revealed triplet ESR spectra of alkoxycarbonylnitrenes during low temperature photolysis. Photolysis of benzoylazides however, failed to show ESR spectra characteristics of a benzoylnitrene.34 These spectroscopic studies suggest that acylnitrenes have singlet states. Recent theoretical studies of photochemical Curtius rearrangement on chlorodifluoroacetyl azide using the MS-CASPT2//CASSCF method along with density functional theory indicated that photo-induced Curtius rearrangement of F2ClC(O)N3 proceeds through a nitrene intermediate in a stepwise manner.42 A series of kinetic studies on the Curtius rearrangement have been proposed over the years.43–45
Scheme 4.

Photolysis of alkoxycarbonyl azide.
3. General procedures for the Curtius rearrangement
Since the Curtius rearrangement is the thermal decomposition of acyl azides, extensive research has been conducted for efficient methods for the synthesis of acyl azides, and several reviews regarding their preparation have been published.23,46 The methods for preparation mostly rely on functional group compatibilities. The two traditional routes for the preparation of acyl azides consist in i) the treatment of esters (21) with hydrazine to afford the corresponding hydrazides (22), which upon reacting with nitrous acid furnished the acyl azides (2) (Scheme 5A), and ii) the treatment of acid chlorides (23) with sodium azide (Scheme 5B). The required acid chlorides can easily be accessed from the corresponding carboxylic acids using thionyl or oxalyl chlorides.47
Scheme 5.

Preparation of acyl azides.
In 1942, Davis and Gardner prepared 8,9,15-trihydroxypentadecylamine from aleuritic acid using the Naegeli modification of the Curtius rearrangement (Scheme 6).48 N,N′-bis-8,9,15-Trihydroxypentadecyl urea 26 was formed upon heating of azide 24 in water. However, on refluxing 24 in anhydrous ethyl alcohol, 8,9,15-trihydroxypentadecylurethane 25 was observed. When the reaction was carried out using the Naegeli modification by heating 24 in anhydrous benzene, the corresponding isocyanate 28 was formed. The isocyanate was hydrolyzed with hot aqueous alkali to obtain 8,9,15-trihydroxypentadecylamine 27.
Scheme 6.

Preparation of 8,9,15-trihydroxypentadecylamine using the Naegeli modification.
As mentioned earlier, one of the usual methods to prepare the acyl azides for the Curtius rearrangement is the treatment of the corresponding acid chloride with sodium azide. However, carboxylic acids may undergo decomposition or isomerization in the presence of mineral acids. In such cases, the conversion of the esters to the corresponding hydrazides, represented the only alternative.49
In 1936, Darapsky applied the Curtius rearrangement for preparing glycine from cyanoacetic ester.50 Since then, this method has been widely applied for the synthesis of amino acids from α-cyanoesters.51 As shown in Scheme 7, ester 29 was converted to the acylhydrazine derivative 30 by reaction with hydrazine. The acylhydrazine, upon reaction with nitrous acid, afforded the acyl azide 31. Curtius rearrangement of the acyl azide in presence of ethanol furnished carbamate 32, which upon acidic hydrolysis provided derivative 33 with the free amine and carboxylic functionalities.
Scheme 7.

Synthesis of amino acid derivatives from α-cyanoacetic esters.
Mixed carboxylic-carbonic anhydrides were popular in peptide synthesis for the preparation of amides and esters of sensitive carboxylic acids, as this avoided the ‘acid chloride route’. In 1960, Weinstock and collaborators observed that acid azides could be formed under very mild conditions upon reaction of mixed anhydrides (for example, anhydride derived from the carboxylic acid and ethyl chloroformate) with sodium azide. The azide could be converted to the isocyanate by Curtius rearrangement and hydrolyzed to the amine, without requiring the isolation of any of the intermediates. Employing this methodology, they converted cis-2-phenylcyclopropanecarboxylic acid to the corresponding amine, without isomerization to the trans isomer, which would not be possible employing the ‘acid chloride route’.49
In 1972, Yamada and collaborators disclosed the use of diphenylphosphoryl azide (DPPA, 35) and proposed its application for Curtius rearrangement.14,52 The reagent is a stable, nonexplosive liquid, bp 157 °C (0.17 mmHg), and was easily prepared by reacting diphenylphosphorochloridate (34) with a slight excess of sodium azide in acetone at room temperature (Scheme 8A). More recently, a procedure for large-scale synthesis of analytically pure DPPA has also been reported by Wolff and Waldvogel.53
Scheme 8.

Synthesis of DPPA and modified Curtius rearrangement.
Yamada and co-workers proposed that DPPA could be used for direct conversion of carboxylic acids to urethanes, through the intermediate carboxylic acid azide (Scheme 8B). The reactivity of DPPA is due to the oxophilic nature of the phosphorus center. Since this discovery, literature has often referred this procedure as the “modified Curtius rearrangement”.
Yamada also used DPPA as the azido group source for racemization-free peptide synthesis.52 When the acylamino carboxylic acid was reacted with DPPA in the presence of triethylamine and the free amino acid or peptide ester hydrochloride at 0 °C, amide bond formation was accomplished. However, when the same transformation was carried out at high temperatures, the Curtius rearrangement would take place, forming the isocyanate, and eventually resulting in formation of urea with the amino acid. In 1993, Thompson and co-workers reported a direct conversion of activated alcohols to azides using DPPA.54
Overman and co-workers developed a general procedure for the preparation of dienyl carbamates using the Curtius rearrangement. A representative example is shown in Scheme 9 starting from carboxylic acid 36, through acyl azide 37, finally providing benzyl carbamate 38.55,56
Scheme 9.

Overman procedure for the Curtius rearrangement.
Weinstock also reported a procedure for the preparation of amines from mixed carboxylic-carbonic anhydrides, similar to Overman procedure; however, they used triethylamine as the base.57,58
More recently, Lebel and Leogane developed a single-pot Curtius rearrangement which proceeded under very mild conditions to afford Boc-protected amines (39, Scheme 10).59
Scheme 10.

General procedure developed by Lebel.
Aliphatic carboxylic acids were converted to the alkyl azides in the presence of di-tert-butyl dicarbonate and sodium azide. These alkyl azides undergo the Curtius rearrangement in the presence of TBAB and zinc (II) triflate spontaneously at 40 °C to furnish the corresponding isocyanate. The isocyanate is then trapped by the tert-butanol present in the reaction mixture, which is the slowest step of the reaction. TBAB and zinc triflate also accelerate this process, probably due to the formation of a zinc carbamoyl bromide species.
An iron(II)-catalyzed Curtius-like rearrangement of hydroxamates to isocyanates was recently reported by Li and co-workers.60 Iron-catalyzed cleavage of the N–O bonds of functionalized heteroauxin hydroxamates (40) resulted in an iron-nitrenoid complex (41), which then decomposed to form the isocyanates (43) (Scheme 11).
Scheme 11.

Protocol for the Curtius rearrangement by Li and co-workers.
Among a few other procedures developed for the Curtius rearrangement, trapping of the isocyanate with 2-trimethylsilylethanol, leading to trimethylsilylethyl carbamate intermediates19 and radical azidonation of aldehydes61,62 were also reported. A new protocol for the Curtius rearrangement was reported by Augustine and co-workers describing the conversion of carboxylic acids to carbamates in the presence of propylphosphonic acid anhydride (T3P®) and azidotrimethylsilane in a single step.63 A one-pot domino reaction for the conversion of acrylic acid derivatives to novel photochromic oxazines was developed by Zhao and Carreira.64 Recently, Ley and co-workers developed a modular mesofluidic flow reactor for performing Curtius rearrangement as a continuous flow process.65
4. Application of Curtius rearrangement in development of synthetic methodologies
Knoechel and co-workers developed a copper-catalyzed anti SN2′ allylic substitution reaction on pentafluorobenzoates of trisubstituted allylic alcohols (44) to generate quaternary carbon centers with high stereoselectivity (45).66 These products could be converted to isocyanates (46) and amines (47) possessing a tertiary chiral center with complete retention of configuration (Scheme 12).67
Scheme 12.

Knoechel protocol for quaternary carbon centers.
A convenient synthesis of orthogonally protected N(α)-Boc2-N(β)-Cbz-2,3-diaminopropionic acid (DAP, 49) was developed by Appella. DAP is a frequently used probe for studying protein structure and function. The Curtius rearrangement was used as a key step to introduce the 3-amino group on acid derivative 48. This strategy was much less expensive and time-consuming compared to previous reported methods (Scheme 13).68
Scheme 13.

Synthesis of orthogonally protected diaminopropionic acid.
Curtius rearrangement has also been applied to the synthesis of optically active cyclopropylamine derivatives. Alkaline hydrolysis of cyclopropane carboxylic acid esters or amides (eg. 50) furnished the free acid, which subsequently underwent Curtius rearrangement to yield cyclopropylamines (eg. 51) (Scheme 14A).69,70 Another approach was the Sharpless oxidative degradation of the phenyl group of cyclopropane carboxylic acid ester (eg. 52), followed by Curtius rearrangement, thus proving cyclopropylamine derivatives (eg. 53) (Scheme 14B).71
Scheme 14.

Synthesis of optically active cyclopropylamines.
Curtius rearrangement has also found application in the synthesis of species containing a metal-nitrogen double bond, through the thermolysis/photolysis of the corresponding azides,72 in the thermolysis of silicon azides to give sila-imines, in the photolysis of germanium isologues,73,74 and phosphinic azides,75 and in the photolytic rearrangement of penta-coordinate phosphorus species.76
A highly effective chiral auxiliary was developed by Ghosh and co-workers for asymmetric alkylation and asymmetric syn-aldol reactions. The β-ketoester 54 was subjected to Baker’s yeast reduction to provide alcohol 55, followed by ester hydrolysis and Curtius rearrangement to furnish the cyclopentano-oxazolidinone chiral auxiliary 56 (Scheme 15).77 Similarly, the chiral auxiliary p-menthane-3-carboxaldehyde was developed by Spino and collaborators using Curtius rearrangement as the key step.78
Scheme 15.

Synthesis of a cyclopentano-oxazolidinone.
Taubinger and collaborators developed an interesting procedure for ring-opening of α-amino acid derived β-lactams with various O-, N- or S-nucleophiles. These compounds are of considerable interest on account of their possible utilization as peptidomimetics.79 Reaction of lactam 57 with amino ester 58 in the presence of stoichiometric sodium azide afforded urea derivative 59 in good yield (Scheme 16).
Scheme 16.

Synthesis of peptidomimetic derivatives.
A unique macrocyclization of unprotected peptide isocyanates was developed by Vinogradov and co-workers to prepare macrocyclic peptides of varying ring size, rigidity, topology and having a range of biological activities.80 Peptides containing two glutamic acid γ-hydrazides (60) were converted to the acyl azides (61), and Curtius rearrangement of the acyl azides led to the corresponding isocyanates (62). The isocyanates reacted with bifunctional nucleophiles to furnish the unprotected peptide macrocycles (63), which were more stable than their linear analogs (Scheme 17).
Scheme 17.

Synthesis of macrocylic derivatives.
The Curtius rearrangement was used to prepare spirocyclic and fused cyclic lactams (66) in an efficient manner. The isocyanates (65) formed upon the Curtius rearrangement of acids 64 underwent a cascade intramolecular nucleophilic addition through the enol carbon in the same pot (Scheme 18).81
Scheme 18.

Synthesis of spirocyclic lactam derivatives.
5. Application of the Curtius Rearrangement in Total synthesis
5.1 Triquinacene
In 1964, Woodward and co-workers synthesized triquinacene (70) to gain information about the postulated phenomenon of homoaromaticity and the nature of homoallylic participation in olefin reactivity.82 For their synthesis, they applied the Curtius rearrangement for the conversion of diacid 67 to bis-urethane 69, which could be reduced to the corresponding bis-amine. The bis-amine oxide underwent Hofmann elimination to afford triquinacene (Scheme 19).
Scheme 19.

Synthesis of triquinacene.
5.2 Haemanthidine
Hendrickson and co-workers utilized the Curtius rearrangement for the stereospecific insertion of the nitrogen in the total synthesis of haemanthidine (74).83 For inserting the nitrogen into the molecule, it was envisioned that azide attack on the less hindered carbonyl of the anhydride 71, followed by Curtius rearrangement would give the required product. However, azide ring opening was not successful, and the methoxide ion attacked the more hindered carbonyl, leading to the thermodynamic product. The free acid could then be converted to the acid chloride, followed by acyl azide, which was then subjected to the Curtius rearrangement to furnish the isocyanate 72. The isocyanate was cyclized to the lactam 73 by polyphosphoric acid, and the lactam was converted to haemanthidine in a few steps (Scheme 20).
Scheme 20.

Synthesis of haemanthidine.
5.3 Saxitoxin
Kishi et al. used the Curtius rearrangement for the first total synthesis of d,l-saxitoxin (77).84 The thiourea ester 75 was converted to the thiourea urea 76, which could be converted to saxitoxin in a few more steps (Scheme 21).
Scheme 21.

Synthesis of saxitoxin.
5.4 Colchicine
Evans et al reported the total synthesis of (±)-colchicine (81), where the amine functionality was introduced via a Curtius rearrangement.85 Treatment of carboxylic acid 78 with DPPA and triethylamine in t-butyl alcohol gave carbamate 79. The Boc group was removed with simultaneous hydrolysis of the ether to provide (±)-desacetylcolchicine 80, which was converted to (±)-colchicine (Scheme 22).
Scheme 22.

Synthesis of colchicine.
5.5 Streptonigrin
The antitumor antibiotic streptonigrin (84) was synthesized in an efficient manner by Kende and co-workers, where one of the amine groups was introduced by the Yamada modification of the Curtius rearrangement.86 Similarly, in the synthesis of streptonigrone (87) by Boger and co-workers, the pyridine C5 amine was introduced by the Shioiri-Yamada modification of the Curtius rearrangement.87 In this case, the intermediate isocyanate was found to be very stable. Lithium hydroxide mediated hydrolysis provided the free amine 86. Interestingly, when the Curtius reaction was attempted without the MOM protection, the free hydroxyl attacked the acyl azide intramolecularly to form the corresponding lactone, before the rearrangement could take place. A similar strategy was also used by Ciufolini and co-workers in their total synthesis of streptonigrone (Scheme 23).88
Scheme 23.

Synthesis of streptonigrin and streptonigrone.
5.6 Camptothecin
Vollhardt developed a method for the synthesis of 5-indolizinones by the cycloaddition of isocyanatoalkynes with alkynes through a cobalt-catalyzed reaction (Scheme 24).89 They also applied this to the total synthesis of antitumor alkaloid camptothecin (91). The isocyanatoalkyne (88) was prepared by the Curtius rearrangement from the corresponding carboxylic acid.89
Scheme 24.

Synthesis of camptothecin.
5.7 (−)-Huperzine A
(−)-Huperzine A (93) diffuses across the blood-brain barrier and is a selective, reversible inhibitor of acetylcholinesterase (AChE), used to increase the levels of cerebral acetylcholine for the treatment of symptomatic Alzheimer’s disease.90,91 (−)-Huperzine A was found to protect neuronal and glial cells against the cytotoxicity of β-amyloid plaques.92
Kozikowski and collaborators developed an enantioselective synthesis of (−)-huperzine A. In the last step of the synthesis, Curtius rearrangement of carboxylic acid 92 furnished (−)-huperzine A (Scheme 25).93 (+)-Huperzine A was also synthesized following the same protocol. In vitro studies revealed (−)-huperzine A to be 33 times more potent than its enantiomer. Fukuyama also introduced the amino group in (−)-huperzine A through a Curtius rearrangement.94 However, he used DPPA for converting the carboxylic acid to the acyl azide, and trapped the isocyanate with methanol to obtain the corresponding methyl carbamate. Recently, Leman and co-workers proposed the synthesis of isotopically labelled (−)-[d3]huperzine A through a Curtius rearrangement.95
Scheme 25.

Synthesis of huperzine A.
5.8 (+)-Calyculin A
For the synthesis of the C26–C32 γ-amino oxazole fragment of (+)-calyculin A (97), the Curtius rearrangement was utilized to introduce the nitrogen that formed the γ-amine, and ultimately led to the amide bond in (+)-calyculin A.96 As shown, Curtius rearrangement of acid 94 efficiently installed the C32 amino group in protected form as in intermediate 95. This latter was then converted into the required C26–C32 γ-amino oxazole fragment 96 for the synthesis of (+)-calyculin A (Scheme 26).
Scheme 26.

Synthesis of calyculin A.
5.9 (+)-Zampanolide
For the total synthesis of the nonnaturally occurring (+)-zampanolide (100), Curtius rearrangement was employed to install the N-acyl hemiaminal moiety.97,98 The one-pot Curtius rearrangement protocol of Yamada using DPPA led to decomposition of the alkoxy acid. Therefore, the α-alkoxy acid 98 was converted to the acyl azide following Weinstock’s procedure.49 Curtius rearrangement of the azide formed the isocyanate, which was captured with 2-(trimethylsilyl)-ethanol to afford the α-alkoxy Teoc-carbamate 99 with complete retention of configuration (Scheme 27).
Scheme 27.

Synthesis of zampanolide.
5.10 Salicylihalamide A
Brabander and collaborators synthesized salicylihalamide A (104) and its congeners, which are potent vacuolar (H+)-ATPase inhibitors for possible treatment of osteoporosis and cancer.99 The salicylihalamide class of compounds contain a highly unsaturated N-acyl enamine side chain. This was introduced by addition of metallo-hexadiene (103) to an E-alkenyl isocyanate. The isocyanate 102 was derived from the corresponding α,β-unsaturated carboxylic acid 101 via a Curtius rearrangement (Scheme 28).
Scheme 28.

Synthesis of salicylihalamide.
5.11 (±)-Gelsemine
Overman and co-workers devised an aza-Cope rearrangement-Mannich cyclization sequence for construction of complex tertiary amines.100 For synthesis of the precursors for the aza-Cope rearrangement, the Curtius rearrangement was used. Curtius rearrangement of carboxylic acid 105, followed by trapping the isocyanate with PMB-alcohol yielded the p-methoxybenzyl carbamate (Moz) 106, which was converted to aza-Cope rearrangement precursor 107 by standard synthetic manipulations. Base-promoted aza-Cope rearrangement, followed by quenching of the rearrangement product with methyl chloroformate, and cleavage of the resulting carbonate afforded carbamate 108. Incorporation of bromine followed by reflux in TFA afforded azatricyclo[4.4.0.0]decanone 109, an advanced intermediate for the total synthesis of (±)-gelsemine (110) (Scheme 29).
Scheme 29.

Synthesis of gelsemine.
5.12 Methoxatin
Weinreb and co-workers described the first total synthesis of the bacterial coenzyme, methoxatin (113), where they used the Curtius rearrangement to introduce the nitrogen towards the beginning of the synthesis.101 Curtius rearrangement of benzoic acid 111, derived from 2,3-dimethoxytoluene, led to aniline 112, which was converted to the required pyrroloquinoline quinone structure 113 through a series of steps (Scheme 30).
Scheme 30.

Synthesis of methoxatin.
5.13 (+)-Sinefungin
Nucleoside antibiotic sinefungin (118) was synthesized by Ghosh and co-workers starting from D-ribose.102 Carboxylic acid 115 was prepared using a highly diastereoselective allylation as the key step. The C-6 amine was then introduced by a Curtius rearrangement of carboxylic acid 115, which proceeded with retention of configuration. The C-9 stereogenic center was set by a rhodium chiral bisphosphine-catalyzed asymmetric hydrogenation of an α-(acylamino)acrylate derivative. Anomeric adenosylation using Vorbruggen’s protocol completed the total synthesis of (+)-sinefungin (Scheme 31).
Scheme 31.

Synthesis of sinefungin.
5.14 AI-77-B
Ghosh and co-workers reported the synthesis of the gastroprotective agent AI-77-B (122) in a highly stereoselective manner, using an ester-derived titanium-enolate mediated syn-aldol reaction.103 The β-hydroxy acid derived from 119 was converted to the β-amino acid moiety using the Curtius rearrangement at the key step. Oxazolidinone 120 was converted to acid 121 by applying Dondoni’s aldehyde homologation as a key step. Acid 121 was then extended to complete the synthesis of AI-77-B (Scheme 32).
Scheme 32.

Synthesis of AI-77-B.
5.15 Brostallicin
Beria and collaborators proposed the synthesis of distamycin-like derivatives as novel cytotoxic DNA minor groove binders.104 Brostallicin behaves as a DNA minor grove binder active against a broad spectrum of tumor cell lines with an excellent cytotoxicity and myelotoxicity ratio. Synthesis of brostallicin (126) has been achieved by these authors starting from the antibiotic distamycin A (123). Brostallicin behaves as a DNA minor groove binder active against a broad spectrum of tumor cell lines with an excellent cytotoxicity/myelotoxicity ratio. A key Curtius rearrangement was carried out on the carboxylic acid 124 obtained from distamycin. The intermediate isocyanate was efficiently trapped by the neighboring carboxamide functional group, to furnish the acyl imidazolidinone 125 in 62% yield (Scheme 33). This represents a practical application of intramolecular isocyanate trapping by a carboxamide in the degradation of an oligopeptide natural product.104
Scheme 33.

Synthesis of brostallicin.
5.16 Belactosin A
Belactosin A (131) and its analogs are known to be potential antitumor agents and have also shown to effect proteasome inhibition. An efficient route was developed by Armstrong and co-workers for the stereocontrolled synthesis of the unique 3-(trans-2-aminocyclopropyl)alanine amino acid 130 present in the enantiomer of the molecule.105 The Wadsworth-Emmons cyclopropanation, which is the reaction of a protected glycidol with triethyl phosphonoacetate, was used to access cyclopropanecarboxylate 128. Curtius rearrangement was applied to convert the cyclopropane derivative 128 to aminocyclopropane 129, which was then converted to the required amino acid in a few steps (Scheme 34).
Scheme 34.

Synthesis of belactosin A.
5.17 Himandrine skeleton
The hexacyclic himandrine skeleton 135 was prepared starting from benzobicyclooctene 132. Diels-Alder cycloaddition of 132 afforded carboxylic acid 133, which was transformed via the Curtius rearrangement to carbamate 134. Carbamate 134 was then converted to the himandrine skeleton, using a Birch reduction, an intramolecular nucleophilic amination and a Pd-catalyzed alkene amination as the key steps (Scheme 35).106
Scheme 35.

Synthesis of the himandrine skeleton.
5.18 Welwitindolinones
Rawal and co-workers described a synthesis of the core bicylco[4.3.1]decane ring system of welwitindolinones (139).107 N-methylwelwitindolinone C isothiocyanate is known to reverse multidrug resistance (MDR) in chemotherapeutic cancer treatment.108 The authors used an intramolecular Pd-mediated enolate arylation to construct the bicyclic skeleton 137. The bridgehead methyl ester served as the masked form of the isocyanate unit, which was introduced in the last step by the Curtius rearrangement from the corresponding carboxylic acid 138 (Scheme 36).
Scheme 36.

Synthesis of welwitindolinones.
5.19 Diisocyanodociane
A formal total synthesis of diisocyanoadociane (143), a potent antimalarial diterpenoid, was reported by Mander and co-workers.109 As shown, phenanthrenoid 140 was converted to pyrene 141 by an intramolecular Michael reaction. To maintain stereochemical control of the insertion of the isonitrile groups, they applied double Curtius rearrangement of the corresponding acyl azides to furnish the diamine 142. The diamine was converted to diisocyanoadociane using a previously reported route (Scheme 37).
Scheme 37.

Synthesis of diisocyanoadociane.
5.20 Mycalamide A
A novel Yb(OTf)3-TMSCl catalytic system was developed for a cross-aldol reaction without epimerization for the synthesis of 144. Cyclization of 144 followed by stereoselective allylation furnished trioxadecalin ring system 145. To prepare the aminal moiety, alcohol 145 was oxidized with Jones’ reagent to provide the corresponding carboxylic acid. Curtius rearrangement of the carboxylic acid stereoselectively introduced the required aminal to afford the Teoc-carbamate 146, thus installing the nitrogen functionality for the amide in mycalamide A (147) (Scheme 38).110
Scheme 38.

Synthesis of mycalamide A.
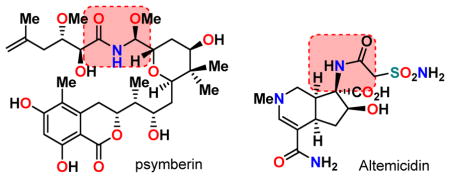
5.21 Altemicidin skeleton
Altemicidin (152) is a monoterpene alkaloid possessing acaricidal activity. It also inhibits tumor cell growth. Kan et al reported a stereocontrolled construction of the core framework of altemicidin.111 The bicyclo[3.3.0] scaffold 148, obtained by an intramolecular C-H insertion reaction, was converted to the cyclic enamide-containing carboxylic acid 149 through a series of steps. To incorporate the nitrogen atom onto the quaternary carbon, the Curtius rearrangement was employed. The in situ generated isocyanate was trapped by the primary alcohol, resulting in the formation of an oxazolidinone derivative 150, thus generating the β-hydroxy α-disubstituted-α-amino acid framework with the required stereochemistry. Functional group interconversions and introduction of the C1 unit into the enamide led to compound 151, which represents the altemicidin skeleton (Scheme 39).111
Scheme 39.

Synthesis of altemicidin core.
5.22 Pancratistatin
Pancratistatin (158), a potent and selective anticancer agent, was synthesized by Cho et al. using the cycloaddition reaction of 3,5-dibromo-2-pyrone (153) with the β-silyl styrene derivative 154 as the key step. The cycloadduct 155 was converted to the corresponding carboxylic acid 156, which upon Curtius rearrangement followed by treatment with sodium methoxide afforded carbamate 157. Further functional group manipulations, including a modified Bischler-Napieralski reaction were then performed in order to complete the total synthesis of (±)-pancratistatin (Scheme 40). The same strategy was also used for the installation of the nitrogen atoms of (±)-α-lycorane (159), (±)-lycorine (160), and (±)-1-deoxylycorine (161).112,113,114
Scheme 40.

Synthesis of pancratistatin.
5.23 NP25302
NP25302 (166), an alkaloid displaying an acylaminotetrahydropyrrolizine core, was shown to inhibit the growth of chronic myeloid leukemia cells. It was synthesized by Robertson and co-workers using a 5-endo-dig N-cyclization and a Curtius rearrangement as the key steps.115 The novel 5-endo-dig cyclization was applied to pyrrolidine derivative 162 and for the first time in the synthesis of a pyrrolizidine-type system. Alkaline hydrolysis of 163 with potassium hydroxide afforded the potassium carboxylate which was nucleophilic enough in DMF to react with DPPA producing the acyl azide 164. Interestingly, when lithium hydroxide was used for the hydrolysis, the corresponding carboxylate was unreactive, even with ethyl chloroformate. The Curtius rearrangement was applied in order to achieve the isocyanate intermediate, however attempts to trap the isocyanate with an isobutenyl organometallic led to its rapid polymerization. The isocyanate was then converted to the corresponding amine, finally acylated with 165 to provide NP25302 (Scheme 41).115
Scheme 41.

Synthesis of NP25032.
5.24 Dievodiamine
The first total synthesis of racemic dievodiamine (173) was performed in a protecting-group-free manner by Taylor and co-workers.116 Indole 167 was converted into lactam 168 by a Curtius rearrangement as the key step. The lactam was then heated with dimethyl anthranilate 169 in the presence of phosphorus oxychloride to afford dehydroevodiamine hydrochloride (170, DHED·HCl), which was converted to stannane intermediate 171 using an organometallic addition as the key step. Stille coupling with derivative 172 completed the total synthesis of (±)-dievodiamine (Scheme 42).
Scheme 42.

Synthesis of dievodiamine.
5.25 (−)-Lyconadin C
An enantioselective total synthesis of (−)-lyconadin C (178) was achieved by Waters and collaborators using a one-pot, tandem Curtius rearrangement/6−-electrocyclization to construct the 2-pyridone ring system.117 Luciduline congener 174, prepared through a Mannich-type cycloaddition, was converted to dienyl carboxylic acid 175 in a few steps. The carboxylic acid was converted to the acyl azide, and Curtius rearrangement of the acyl azide afforded isocyanate 176, which was strategically used for the 6−-electrocyclization in the same pot. The conjugated dienyl isocyanate served as the perfect substrate for the electrocyclic cyclization due to system planarity and optimal orbital alignment. When the intermediate acyl azide was heated to 80 °C, tandem Curtius rearrangement and 6−-electrocyclization resulted in the pyridone ring of lyconadin C (177). Deprotection of the carboxybenzyl group afforded enantiopure (−)-lyconadin C (Scheme 43).
Scheme 43.

Synthesis of lyconadin C.
5.26 (±)-Lundurine B
Nishida and co-workers reported the total synthesis of (±)-lundurine B (182).118,119 The cyclopropane-fused indoline was obtained by using the Curtius rearrangement as one of the key steps. Saponification of the lactone-ester 179 yielded the lactone-carboxylic acid, that furnished the Boc-protected amine upon Curtius rearrangement. Bromination of the aromatic ring provided bromo-derivative 180 that was submitted to a copper-mediated cyclization to afford the tetracyclic compound 181. This latter compound was finally converted into lundurine B after a series of further steps (Scheme 44).
Scheme 44.

Synthesis of lundurine B.
5.27 Axamide-1 and axisonitrile-1
For the total synthesis of axamide-1 (187) and axisonitrile-1 (188), Piers and co-workers employed the Curtius rearrangement for introducing the nitrogen atom.120 The carboxylic acid 183 was transformed into the acyl azide 184 by standard reactions. Curtius rearrangement of the acyl azide, followed by trapping the resulting isocyanate with 2-trimethylsilylethanol, furnished the carbamate 185 with retention of configuration. Treatment of 185 with TBAF afforded the primary amine 186, which was then formylated to provide (±)-axamide-1. Dehydration of (±)-axamide-1 furnished crystalline (±)-axisonitrile-1 (Scheme 45).
Scheme 45.

Synthesis of axisonitrile and axamide.
5.28 Aspeverin
Aspeverin (192), a prenylated indole alkaloid, was synthesized using the Curtius rearrangement as a key step.121 The bicyclic urethane linkage functionality is distinctive of this class of natural products. To this aim ester 189 was converted into the corresponding acyl azide 190. Following thermolysis in the presence of 2-(trimethylsilyl)ethanol yielded the desired carbamate 191. A unique iodine(III)-mediated cyclization and a novel approach to introduce the geminal dimethyl group were then used to finalize the desired bicyclic urethane linkage (Scheme 46).
Scheme 46.

Synthesis of aspeverin.
5.29 Cylindricine alkaloid
The synthesis of the cylindricine alkaloid core (198) reported by Dalton and collaborators envisaged a key Curtius rearrangement step.122,123 A Negishi coupling between the organozinc intermediate 193 and iododerivative 194, followed by hydrolysis provided the carboxylic acid, finally converted into the desired isocyanate 195 upon Curtius rearrangement. A Rh(I)·CKphos catalyzed [2+2+2] cycloaddition of the alkenyl isocyanate 195 and alkyl alkyne 196 selectively provided the vinylogous amide indolizidinone cycloadduct 197. The cycloaddition created a quaternary stereocenter with excellent enantioselectivity. The tricyclic core of the cylindricine alkaloids (198) was then finalized by reduction with DIBAL-H and subsequent cyclization in the presence of potassium t-butoxide with excellent regioselectivity and enantioselectivity (Scheme 47).
Scheme 47.

Synthesis of cyclindricine core.
5.30 (−)-Cephalotaxine core
For the enantioselective synthesis of (−)-cephalotaxine (199), one of the most efficient strategies involved the synthetic manipulation of derivatives of the (R)-1-azaspiro[4.4]nonane-2,6-dione system 200 (Scheme 48).124
Scheme 48.

Retrosynthetic approach to cephalotaxine.
For the synthesis of cephalotaxine, spiro lactam 204 was synthesized from optically active keto diester 201. To differentiate the two ester groups, diester 201 was converted to bicyclic lactone 202. The Curtius rearrangement was then carried out by converting the carboxylic acid to the corresponding acyl azide using DPPA, followed by rearrangement to the benzyl carbamate 203. This was converted to the azaspirononane derivative 204 (Scheme 49).
Scheme 49.

Synthesis of azaspiro[4.4]nonane-2,6-dione 204.
5.31 (−)-Esermethole
Badiola and co-workers proposed a total synthesis of (−)-esermethole (208) involving a Curtius rearrangement step.125 α-Hydroxyketone 205 underwent oxidative cleavage to furnish carboxylic acid 206. The Curtius rearrangement of carboxylic acid afforded the corresponding methyl carbamate 207, which underwent reductive cyclization on treatment with lithium aluminum hydride to furnish (−)-esermethole in high ee (Scheme 50).
Scheme 50.

Synthesis of (−)-esermethole.
The following examples show the construction of natural compounds containing N,O-aminals/hemiaminals through a stereoselective Curtius rearrangement, with retention of the stereochemistry at the α-carbon.
5.32 (+)-Psymberin (Irciniastatin A)
The marine sponge cytotoxin (+)-psymberin (irciniastatin A, 211) was synthesized Smith and co-workers using a late-stage Curtius rearrangement to install the sensitive N,O-aminal moiety.126 Curtius rearrangement of carboxylic acid 209 followed by trapping of the resulting amine isocyanate with 2-trimethylsilylethanol provided the Teoc-protected aminal 210, which was converted to the natural product (+)-psymberin (Scheme 51).
Scheme 51.

Synthesis of psymberin.
5.33 Pederin
The acylaminal group at C-10 is a common structural feature in pederins and mycalamide classes of natural products. However, the C-10 aminal unit is reported to be unstable under acidic, basic and neutral conditions, leading to mixtures of the corresponding amide. Roush and co-workers developed a stereoselective synthesis of the trioxodecalin nucleus of mycalamide (213), encompassing a stereocontrolled insertion of the C(10) amine as a carbamate through the Curtius rearrangement (Scheme 52).127
Scheme 52.

Synthesis of the mycalamide nucleus.
In 2007, Rawal installed the nitrogen atom in pederin (216, a potent insect toxin, with anticancer properties) by a Curtius rearrangement, which proceeded stereospecifically.128 Saponification of the ester 214, followed by a Curtius rearrangement protocol provided the Teoc-protected pederamine (215) with complete retention of stereochemistry of the aminal group. Pederamine was then converted into pederin after few additional steps (Scheme 53).
Scheme 53.

Synthesis of pederin.
5.34 Mycalamide B
A similar strategy was undertaken by Rawal et al. for the synthesis of mycalamide B (220).129 However, final coupling of the amine with pederic acid chloride was not successful. An alternative route is shown in Scheme 54. As shown, hydrogenolysis of benzyl ether 217 and saponification of the ester were followed by treatment with DPPA and triethylamine, providing the corresponding acyl azide, which underwent the Curtius rearrangement upon heating. The resulting isocyanate 218 was trapped intramolecularly by the primary alcohol to yield the required cyclic carbamate 219. The cyclic carbamate was converted to mycalamide B (Scheme 54). The use of “chemical handcuffs” to temporarily restrict portions of the molecule thus enabled the convergent synthesis of mycalamide B.
Scheme 54.

Synthesis of mycalamide B.
5.35 Echinocandin C
Messik and collaborators proposed a total synthesis of echinocandin C (223) featuring a stereoselective Curtius rearrangement step for the synthesis of the crucial N-acyl hemiaminal fragment 222. Accordingly, treatment of the crude carboxylic acid 221 with isobutyl chloroformate and subsequent reaction with aqueous sodium azide produced the corresponding acyl azide, which was heated in the presence of 2-(trimethylsilyl)ethanol to afford the Teoc-protected N-acyl hemiaminal 222 in very good yield (Scheme 55).130 A number of other syntheses of echinocandin C has also been reported.131,132
Scheme 55.

Synthesis of echinocandin C.
Conclusion
Although, the Curtius rearrangement was discovered more than a century ago, this reaction remains an important strategy for the synthesis of a wide variety of nitrogen-containing functionalities in organic synthesis. One of the intriguing aspects of the Curtius rearrangement is the transformation of acyl azide intermediate to isocyanate from carboxylic acid. Even though the reaction is extensively utilized, the mechanism of this reaction remains a subject of great interest. The reaction proceeds via a concerted mechanism and the rearrangement occurs with complete retention of stereochemistry of the migrating carbon. Thus, the stereochemical integrity of this rearrangement can be exploited for the generation of chiral nitrogen containing centers. The abundance of carboxylic acids and their relatively simple conversion to acyl azides also contribute to increasing the scope of this reaction. Over the years, numerous mild methods have been developed, mostly to be compatible with the functional groups present within the molecules. This review highlights the gradual development of Curtius rearrangement, and chemical methodologies adopted for the synthesis of isocyanates, amines and amine derivatives. Amine functionalities, such as urethanes, ureas and amides are widely prevalent in naturally occurring and therapeutically relevant biomolecules, therefore the Curtius rearrangement has been strategically applied to their synthesis. The review provides an overview of the applications of the Curtius rearrangement in total synthesis of natural products and their structural derivatives. The review also provides a broad picture of the Curtius rearrangement and we hope that it will stimulate further development, particularly in the areas of asymmetric synthesis and process development.
Acknowledgments
Financial support of this research by the National Institutes of Health (GM53386) and Purdue University is gratefully acknowledged. We would like to thank Ms. Anne Veitschegger for helpful discussions.
Abbreviations
- AChE
Acetylcholinesterase
- Boc
tert-butyloxycarbonyl
- CASSCF
Complete Active State SCF
- DAP
N(α)-Boc2-N(β)-Cbz-2,3-diaminopropionic acid
- DCM
Dichloromethane
- DHED·HCl
Dehydroevodiamine hydrochloride
- DIBAL-H
Diisobutylaluminium hydride
- DMAP
4-Dimethylaminopyridine
- DMF
Dimethylformamide
- DNA
Deoxyribonucleic acid
- DPPA
Diphenylphosphoryl azide
- ESR
Electron spin resonance
- LAH
Lithium aluminum hydride
- MDR
Multidrug resistance
- Moz
p-Methoxybenzyl carbamate
- MS-CASPT2
Multi-State Complete Active State Perturbation Theory 2nd Order
- PMB
p-Methoxy benzyl
- TBAF
Tetra-n-butylammonium fluoride
- TBS
tert-Butyldimethylsilyl
- Teoc
2-Trimethylsilylethoxycarbonyl
- THF
Tetrahydrofuran
- TMS
Trimethylsilyl chloride
- T3P®
Propylphosphonic acid anhydride
Footnotes
Conflicts of interest
Authors declare no competing interests
Notes and references
- 1.Curtius T. Ber Dtsch Chem Ges. 1890;23:3023–3033. [Google Scholar]
- 2.Curtius T. J Prakt Chem. 1894;50:275–294. [Google Scholar]
- 3.Stollé RMNJ. Prakt Chem. 1928;119:275–278. [Google Scholar]
- 4.Wilds AL, Woolsey NF, Berghe JVD, Winestock CH. Tetrahedron Lett. 1965:4841–4846. [Google Scholar]
- 5.Saikachi H, Kitagawa T. Chem Pharm Bull (Tokyo) 1978;26:1054–1060. [Google Scholar]
- 6.Smith PAS. Org React. 1946;3:337–449. [Google Scholar]
- 7.Saunders JH, Slocombe RJ. Chem Rev. 1948;43:203–218. doi: 10.1021/cr60135a001. [DOI] [PubMed] [Google Scholar]
- 8.Aubin Y, Fischmeister C, Thomas CM, Renaud JL. Chem Soc Rev. 2010;39:4130–4145. doi: 10.1039/c003692g. [DOI] [PubMed] [Google Scholar]
- 9.Amino Group Chemistry. From Synthesis to the Life Sciences. Wiley-VCH; Weinheim: 2008. [Google Scholar]
- 10.Amines: Synthesis, Properties and Applications. Cambridge University Press; Cambridge: 2004. [Google Scholar]
- 11.Salvatore RN, Yoon CH, Jung KW. Tetrahedron. 2001;57:7785–7811. [Google Scholar]
- 12.Lawrence SA. Amines: Synthesis, Properties and Applications. Cambridge University Press; 2004. [Google Scholar]
- 13.Banthrope DV. The Chemistry of the Azido Group’. Interscience; New York: 1971. [Google Scholar]
- 14.Ninomiya KS, Yamada TS. Tetrahedron. 1974;30:2151–2157. [Google Scholar]
- 15.Murato K, Shioiri T, Yamada S-I. Chem Pharm Bull (Tokyo) 1975;23:1738–1740. [Google Scholar]
- 16.Leogane O, Lebel H. Synthesis. 2009:1935–1940. [Google Scholar]
- 17.Lebel H, Leogane O, Huard K, Lectard S. Pure Appl Chem. 2006;78:363–375. [Google Scholar]
- 18.Bauer L, Exner O. Angew Chem. 1974;13:376–384. [Google Scholar]
- 19.Capson TL, Poulter CD. Tetrahedron Lett. 1984;25:3515–3518. [Google Scholar]
- 20.Imof R, Ladner DW, Muchowski JM. J Org Chem. 1977;41:3709–3713. [Google Scholar]
- 21.Sasmal S, Geyer A, Maier ME. J Org Chem. 2002;67:6260–6263. doi: 10.1021/jo025889b. [DOI] [PubMed] [Google Scholar]
- 22.Patil BS, Vasanthakumar GR, Babu VVS. J Org Chem. 2003;68:7274–7280. doi: 10.1021/jo020516w. [DOI] [PubMed] [Google Scholar]
- 23.Scriven EFV, Turnbull K. Chem Rev. 1988;88:297–368. [Google Scholar]
- 24.L’abbé G. Chem Rev. 1969;69:345–363. [Google Scholar]
- 25.Stieglitz J. Am Chem J. 1896;18:756. [Google Scholar]
- 26.Campell A, Kenyon J. J Chem Soc. 1946:25–27. [Google Scholar]
- 27.Tiemann F. Ber Chem. 1891;24:4162–4167. [Google Scholar]
- 28.Linke S, Tisue GT, Lwowski W. J Am Chem Soc. 1967;89:6308–6310. [Google Scholar]
- 29.Lwowski W, Tisue GT. J Am Chem Soc. 1965;87:4022–4023. [Google Scholar]
- 30.Abramovitch RA, Davis BA. Chem Rev. 1964;64:149–185. [Google Scholar]
- 31.Wentrup C, Bornemann H. Eur J Org Chem. 2005:4521–4524. doi: 10.1021/jo050429e. [DOI] [PubMed] [Google Scholar]
- 32.Vyas S, Kubicki J, Luk HL, Zhang YL, Gritsan NP, Hadad CM, Platz MS. J Phys Org Chem. 2012;25:693–703. [Google Scholar]
- 33.Tarwade V, Dmitrenko O, Bach RD, Fox JM. J Org Chem. 2008;73:8189–8197. doi: 10.1021/jo801104t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu J, Mandel S, Hadad CM, Platz MS. J Org Chem. 2004;69:8583–8593. doi: 10.1021/jo048433y. [DOI] [PubMed] [Google Scholar]
- 35.Kakkar R, Zaidi S, Grover R. Int J Quantum Chem. 2009;109:1058–1069. [Google Scholar]
- 36.Banert K, Berndt C, Hagedorn M, Liu H, Anacker T, Friedrich J, Rauhut G. Angew Chem Int Ed Engl. 2012;51:4718–4721. doi: 10.1002/anie.201200029. [DOI] [PubMed] [Google Scholar]
- 37.Lwowski W. Azides and Nitrenes - Reactivity and Utility. Academic Press; New York: 1984. [Google Scholar]
- 38.Autrey T, Schuster GB. J Am Chem Soc. 1987;109:5814–5820. [Google Scholar]
- 39.Klimenko LS, Pritchina EA, Gritsan NP. Chem Eur J. 2003;9:1639–1644. doi: 10.1002/chem.200390188. [DOI] [PubMed] [Google Scholar]
- 40.Gritsan NP, Pritchina EA. Mendeleev Commun. 2001:94–95. [Google Scholar]
- 41.Skell PS, Woodworth RC. J Am Chem Soc. 1956;78:4496–4497. [Google Scholar]
- 42.Peng XL, Ding WL, Li QS, Li ZS. Org Chem Front. 2017;4:1153–1161. [Google Scholar]
- 43.Yukawa Y, Tsuno Y. J Am Chem Soc. 1957;79:5530–5534. [Google Scholar]
- 44.Yukawa Y, Tsuno Y. J Am Chem Soc. 1959;81:2007–2012. [Google Scholar]
- 45.Fahr E, Neumann L. Angew Chem. 1965;77:591–591. [Google Scholar]
- 46.Brase S, Gil C, Knepper K, Zimmermann V. Angew Chem Int Ed Engl. 2005;44:5188–5240. doi: 10.1002/anie.200400657. [DOI] [PubMed] [Google Scholar]
- 47.Trost BM, Fleming I. Comprehensive Organic Synthesis [Google Scholar]
- 48.Davis AL, Gardner WH. J Am Chem Soc. 1942;64:1902–1905. [Google Scholar]
- 49.Weinstock J. J Org Chem. 1961;26:3511–3511. [Google Scholar]
- 50.Darapsky A. J Prakt Chem. 1936;146:250–267. [Google Scholar]
- 51.Gagnon PE, Boivin PA, Craig HM. Can J Chem. 1951;29:70–75. doi: 10.1139/v51-023. [DOI] [PubMed] [Google Scholar]
- 52.Shioiri T, Ninomiya K, Yamada S. J Am Chem Soc. 1972;94:6203–6205. doi: 10.1021/ja00772a052. [DOI] [PubMed] [Google Scholar]
- 53.Wolff O, Waldvogel SR. Synthesis. 2004:1303–1305. [Google Scholar]
- 54.Thompson AS, Humphrey GR, Demarco AM, Mathre DJ, Grabowski EJJ. J Org Chem. 1993;58:5886–5888. [Google Scholar]
- 55.Jessup PJ, Petty CB, Roos J, Overman LE. Org Synth. 1988;50–9:95–101. [Google Scholar]
- 56.Jessup PJ, Petty CB, Roos J, Overman LE. Org Synth. 1979;59:1. [Google Scholar]
- 57.Kaiser C, Weinstock J. Org Synth. 1971;51:48. [Google Scholar]
- 58.Kaiser C, Weinstock J. Org Synth. 1988;6:910. Coll. [Google Scholar]
- 59.Lebel H, Leogane O. Org Lett. 2005;7:4107–4110. doi: 10.1021/ol051428b. [DOI] [PubMed] [Google Scholar]
- 60.Li D, Wu T, Liang K, Xia C. Org Lett. 2016;18:2228–2231. doi: 10.1021/acs.orglett.6b00864. [DOI] [PubMed] [Google Scholar]
- 61.Marinescu L, Thinggaard J, Thomsen IB, Bols M. J Org Chem. 2003;68:9453–9455. doi: 10.1021/jo035163v. [DOI] [PubMed] [Google Scholar]
- 62.Shinomoto Y, Yoshimura A, Shimizu H, Yamazaki M, Zhdankin VV, Saito A. Org Lett. 2015;17:5212–5215. doi: 10.1021/acs.orglett.5b02543. [DOI] [PubMed] [Google Scholar]
- 63.Augustine JK, Bombrun A, Mandal AB, Alagarsamy P, Atta RN, Selvam P. Synthesis. 2011:1477–1483. [Google Scholar]
- 64.Zhao W, Carreira EM. Org Lett. 2011;13:5084–5087. doi: 10.1021/ol2019482. [DOI] [PubMed] [Google Scholar]
- 65.Baumann M, Baxendale IR, Ley SV, Nikbin N, Smith CD, Tierney JP. Org Biomol Chem. 2008;6:1577–1586. doi: 10.1039/b801631n. [DOI] [PubMed] [Google Scholar]
- 66.Harrington-Frost N, Leuser H, Calaza MI, Kneisel FF, Knochel P. Org Lett. 2003;5:2111–2114. doi: 10.1021/ol034525i. [DOI] [PubMed] [Google Scholar]
- 67.Leuser H, Perrone S, Liron F, Kneisel FF, Knochel P. Angew Chem Int Ed Engl. 2005;44:4627–4631. doi: 10.1002/anie.200500672. [DOI] [PubMed] [Google Scholar]
- 68.Englund EA, Gopi HN, Appella DH. Org Lett. 2004;6:213–215. doi: 10.1021/ol0361599. [DOI] [PubMed] [Google Scholar]
- 69.Ogasawara D, Suzuki T, Mino K, Ueda R, Khan MN, Matsubara T, Koseki K, Hasegawa M, Sasaki R, Nakagawa H, Mizukami T, Miyata N. Bioorg Med Chem. 2011;19:3702–3708. doi: 10.1016/j.bmc.2010.12.024. [DOI] [PubMed] [Google Scholar]
- 70.Chanthamath S, Nguyen DT, Shibatomi K, Iwasa S. Org Lett. 2013;15:772–775. doi: 10.1021/ol303404c. [DOI] [PubMed] [Google Scholar]
- 71.Miller JA, Hennessy EJ, Marshall WJ, Scialdone MA, Nguyen ST. J Org Chem. 2003;68:7884–7886. doi: 10.1021/jo0344079. [DOI] [PubMed] [Google Scholar]
- 72.Paetzold PI, Grundke H. Synthesis. 1973:635. [Google Scholar]
- 73.Parker DR, Sommer LH. J Organomet Chem. 1976;120:C1. [Google Scholar]
- 74.Baceiredo A, Bertrand G, Mazerolles P. Tetrahedron Lett. 1981;22:2553. [Google Scholar]
- 75.Harger MJP, Westlake S. Tetrahedron Lett. 1982;23:3621. [Google Scholar]
- 76.Baceiredo A, Bertrand G, Majoral JP, Wermuth U, Schmutzler R. J Am Chem Soc. 1984;106:7065–7068. [Google Scholar]
- 77.Ghosh AK, Cho H, Onishi M. Tetrahedron-Asymmetry. 1997;8:821–824. doi: 10.1016/S0957-4166(97)00065-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Spino C, Godbout C, Beaulieu C, Harter M, Mwene-Mbeja TM, Boisvert L. J Am Chem Soc. 2004;126:13312–13319. doi: 10.1021/ja046084j. [DOI] [PubMed] [Google Scholar]
- 79.Taubinger AA, Fenske D, Podlech J. Synlett. 2008:539–542. [Google Scholar]
- 80.Vinogradov AA, Choo ZN, Totaro KA, Pentelute BL. Org Lett. 2016;18:1226–1229. doi: 10.1021/acs.orglett.5b03626. [DOI] [PubMed] [Google Scholar]
- 81.Yang W, Sun X, Yu W, Rai R, Deschamps JR, Mitchell LA, Jiang C, MacKerell AD, Jr, Xue F. Org Lett. 2015;17:3070–3073. doi: 10.1021/acs.orglett.5b01350. [DOI] [PubMed] [Google Scholar]
- 82.Woodward RB, Fukunaga T, Kelly RC. J Am Chem Soc. 1964;86:3162–3164. [Google Scholar]
- 83.Hendrickson JB, Bogard TL, Fisch ME, Grossert S, Yoshimura N. J Am Chem Soc. 1974;96:7781–7789. [Google Scholar]
- 84.Tanino H, Nakata T, Kaneko T, Kishi Y. J Am Chem Soc. 1977;99:2818–2819. doi: 10.1021/ja00450a079. [DOI] [PubMed] [Google Scholar]
- 85.Evans DA, Tanis SP, Hart DJ. J Am Chem Soc. 1981;103:5813–5821. [Google Scholar]
- 86.Kende AS, Lorah DP, Boatman RJ. J Am Chem Soc. 1981;103:1271–1273. [Google Scholar]
- 87.Boger DL, Cassidy KC, Nakahara S. J Am Chem Soc. 1993;115:10733–10741. [Google Scholar]
- 88.Chan BK, Ciufolini MA. J Org Chem. 2007;72:8489–8495. doi: 10.1021/jo701435p. [DOI] [PubMed] [Google Scholar]
- 89.Earl RA, Vollhardt KPC. J Am Chem Soc. 1983;105:6991–6993. [Google Scholar]
- 90.Kozikowski AP, Miller CP, Yamada F, Pang YP, Miller JH, McKinney M, Ball RG. J Med Chem. 1991;34:3399–3402. doi: 10.1021/jm00116a010. [DOI] [PubMed] [Google Scholar]
- 91.McKinney M, Miller JH, Yamada F, Tuckmantel W, Kozikowski AP. Eur J Pharmacol. 1991;203:303–305. doi: 10.1016/0014-2999(91)90730-e. [DOI] [PubMed] [Google Scholar]
- 92.Wang R, Zhang HY, Tang XC. Eur J Pharmacol. 2001;421:149–156. doi: 10.1016/s0014-2999(01)01030-5. [DOI] [PubMed] [Google Scholar]
- 93.Yamada F, Kozikowski AP, Reddy ER, Pang YP, Miller JH, Mckinney M. J Am Chem Soc. 1991;113:4695–4696. [Google Scholar]
- 94.Koshiba T, Yokoshima S, Fukuyama T. Org Lett. 2009;11:5354–5356. doi: 10.1021/ol9022408. [DOI] [PubMed] [Google Scholar]
- 95.Leman L, Kitson SL, Brown RT, Cairns J, Watters W, McMordie A, Murrell VL, Marfurt J. J Labelled Compd Rad. 2011;54:720–730. [Google Scholar]
- 96.Evans DA, Gage JR, Leighton JL. J Am Chem Soc. 1992;114:9434–9453. [Google Scholar]
- 97.Smith AB, 3rd, Safonov IG, Corbett RM. J Am Chem Soc. 2002;124:11102–11113. doi: 10.1021/ja020635t. [DOI] [PubMed] [Google Scholar]
- 98.Smith AB, 3rd, Safonov IG, Corbett RM. J Am Chem Soc. 2001;123:12426–12427. doi: 10.1021/ja012220y. [DOI] [PubMed] [Google Scholar]
- 99.Wu Y, Liao X, Wang R, Xie XS, De Brabander JK. J Am Chem Soc. 2002;124:3245–3253. doi: 10.1021/ja0177713. [DOI] [PubMed] [Google Scholar]
- 100.Earley WG, Jacobsen JE, Madin A, Meier GP, O’Donnell CJ, Oh T, Old DW, Overman LE, Sharp MJ. J Am Chem Soc. 2005;127:18046–18053. doi: 10.1021/ja055710p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gainor JA, Weinreb SM. J Org Chem. 1981;46:4317–4319. [Google Scholar]
- 102.Ghosh AK, Liu W. J Org Chem. 1996;61:6175–6182. doi: 10.1021/jo960670g. [DOI] [PubMed] [Google Scholar]
- 103.Ghosh AK, Bischoff A, Cappiello J. Org Lett. 2001;3:2677–2680. doi: 10.1021/ol0101279. [DOI] [PubMed] [Google Scholar]
- 104.Beria I, Nesi M. Tetrahedron Lett. 2002;43:7323–7327. [Google Scholar]
- 105.Armstrong A, Scutt JN. Org Lett. 2003;5:2331–2334. doi: 10.1021/ol0346887. [DOI] [PubMed] [Google Scholar]
- 106.O’Connor PD, Mander LN, McLachlan MM. Org Lett. 2004;6:703–706. doi: 10.1021/ol036308n. [DOI] [PubMed] [Google Scholar]
- 107.MacKay JA, Bishop RL, Rawal VH. Org Lett. 2005;7:3421–3424. doi: 10.1021/ol051043t. [DOI] [PubMed] [Google Scholar]
- 108.Zhang X, Smith CD. Mol Pharmacol. 1996;49:288–294. [PubMed] [Google Scholar]
- 109.Fairweather KA, Mander LN. Org Lett. 2006;8:3395–3398. doi: 10.1021/ol061228f. [DOI] [PubMed] [Google Scholar]
- 110.Kagawa N, Ihara M, Toyota M. Org Lett. 2006;8:875–878. doi: 10.1021/ol052943c. [DOI] [PubMed] [Google Scholar]
- 111.Kan T, Kawamoto Y, Asakawa T, Furuta T, Fukuyama T. Org Lett. 2008;10:169–171. doi: 10.1021/ol701940f. [DOI] [PubMed] [Google Scholar]
- 112.Jung YG, Kang HU, Cho HK, Cho CG. Org Lett. 2011;13:5890–5892. doi: 10.1021/ol202525a. [DOI] [PubMed] [Google Scholar]
- 113.Jung YG, Lee SC, Cho HK, Darvatkar NB, Song JY, Cho CG. Org Lett. 2013;15:132–135. doi: 10.1021/ol303157b. [DOI] [PubMed] [Google Scholar]
- 114.Shin HS, Jung YG, Cho HK, Park YG, Cho CG. Org Lett. 2014;16:5718–5720. doi: 10.1021/ol502792p. [DOI] [PubMed] [Google Scholar]
- 115.Stevens K, Tyrrell AJ, Skerratt S, Robertson J. Org Lett. 2011;13:5964–5967. doi: 10.1021/ol202381m. [DOI] [PubMed] [Google Scholar]
- 116.Unsworth WP, Kitsiou C, Taylor RJK. Org Lett. 2013;15:3302–3305. doi: 10.1021/ol4013469. [DOI] [PubMed] [Google Scholar]
- 117.Cheng X, Waters SP. Org Lett. 2013;15:4226–4229. doi: 10.1021/ol401954f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hoshi M, Kaneko O, Nakajima M, Arai S, Nishida A. Org Lett. 2014;16:768–771. doi: 10.1021/ol4034786. [DOI] [PubMed] [Google Scholar]
- 119.Arai S, Nakajima M, Nishida A. The Alkaloids. Vol. 78. Academic Press; London: 2017. pp. 167–204. [DOI] [PubMed] [Google Scholar]
- 120.Piers E, Yeung BWA, Rettig SJ. Tetrahedron. 1987;43:5521–5535. [Google Scholar]
- 121.Levinson AM. Org Lett. 2014;16:4904–4907. doi: 10.1021/ol5024163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dalton DM, Rovis T. Org Lett. 2013;15:2346–2349. doi: 10.1021/ol400529k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dalton DM, Rovis T. Org Lett. 2013;15:4915–4915. doi: 10.1021/ol400529k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pizzonero M, Dumas F, d’Angelo J. Heterocycles. 2005;66:31–37. [Google Scholar]
- 125.Badiola E, Fiser B, Gomez-Bengoa E, Mielgo A, Olaizola I, Urruzuno I, Garcia JM, Odriozola JM, Razkin J, Oiarbide M, Palomo C. J Am Chem Soc. 2014;136:17869–17881. doi: 10.1021/ja510603w. [DOI] [PubMed] [Google Scholar]
- 126.Smith AB, 3rd, Jurica JA, Walsh SP. Org Lett. 2008;10:5625–5628. doi: 10.1021/ol802466t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Roush WR, Marron TG. Tetrahedron Lett. 1993;34:5421–5424. [Google Scholar]
- 128.Jewett JC, Rawal VH. Angew Chem Int Ed Engl. 2007;46:6502–6504. doi: 10.1002/anie.200701677. [DOI] [PubMed] [Google Scholar]
- 129.Jewett JC, Rawal VH. Angew Chem Int Ed Engl. 2010;49:8682–8685. doi: 10.1002/anie.201003361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Messik F, Oberthur M. Angew Chem Int Ed Engl. 2013;52:5871–5875. doi: 10.1002/anie.201301262. [DOI] [PubMed] [Google Scholar]
- 131.Roush WR, Pfeifer LA. Org Lett. 2000;2:859–862. doi: 10.1021/ol005629l. [DOI] [PubMed] [Google Scholar]
- 132.Crimmins MT, Stevens JM, Schaaf GM. Org Lett. 2009;11:3990–3993. doi: 10.1021/ol901655e. [DOI] [PMC free article] [PubMed] [Google Scholar]