

Abstract
Antiaromatic and open-shell molecules are attractive because of their distinct electronic and magnetic behaviour. However, their increased reactivity creates a challenge for probing their properties. Here, we describe the on-surface and in-solution generation and characterisation of a highly reactive antiaromatic molecule: indeno[1,2-b]fluorene (IF). In solution, we generated IF by KI-induced dehalogenation of a dibromo-substituted precursor molecule and found that IF survives for minutes at ambient conditions. Using atom manipulation at low temperatures we generated IF on Cu(111) and on bilayer NaCl. On these surfaces, we characterised IF by bond-order analysis using non-contact atomic force microscopy with CO-functionalised tips and by orbital imaging using scanning tunnelling microscopy. We found that the closed-shell configuration and antiaromatic character predicted for gas-phase IF are preserved on the NaCl film. On Cu(111), we observed significant bond-order reorganisation within the s-indacene moiety because of chemisorption, highlighting the importance of molecule surface interactions on the π-electron distribution.
Indeno[1,2-b]fluorene (IF) is an extremely reactive polycyclic conjugated hydrocarbon with antiaromatic character, thus it has not been detected to date. Here, the authors present the successful generation and characterisation of IF both on-surface and in-solution.
Introduction
Aromaticity is one of the most relevant, and intriguing, concepts in chemistry1,2. In 1931, Hückel suggested his well-known rule to explain the extra stability of planar monocyclic molecules that contain [4n + 2] π-electrons in a conjugated system3,4. With some limitations, the concept was later extended to polycyclic conjugated hydrocarbons (PCHs), based on the number of Clar sextets5, or the presence of conjugated circuits with [4n + 2] π-electrons within a particular structure6. In 1967, Breslow introduced the term antiaromaticity as the inverse of aromaticity, in order to explain the destabilisation of molecules with [4n] π-electrons in a cyclic conjugated system7. Among the PCHs with antiaromatic character8, indeno[1,2-b]fluorene (IF) is a remarkable example (Fig. 1)9,10. Compared with pentacene (1), the most prominent p-type organic semiconductor with five linearly fused six-membered rings and 22 π-electrons in its aromatic conjugate circuit ([4n + 2], n = 5), IF presents a 6-5-6-5-6 fused-ring motif and a formally antiaromatic 20 π-electron conjugate system ([4n] n = 5). The IF closed-shell configuration with a central eight π-electron para-quinodimethane core (in red, Fig. 1) and two Clar sextets (in blue) might be in resonance with the open-shell diradical configuration with three sextets11,12. As a result, unsubstituted IF is presumed to be an extremely reactive PCH and in fact has never been synthesised or even detected to date.
Fig. 1.
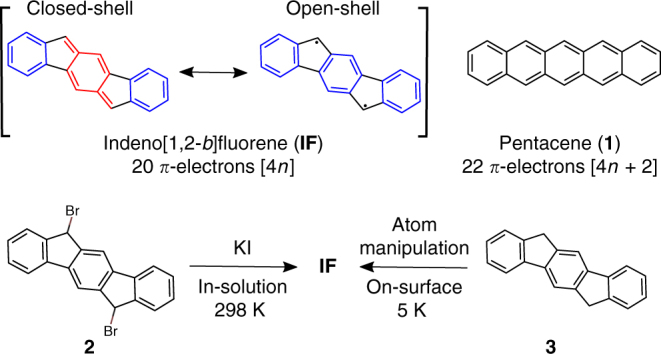
Molecular structures of indeno[1,2-b]fluorene (IF) and related compounds. The central eight π-electron para-quinodimethane core of IF is shown in red, the six π-electron Clar sextets are shown in blue
In contrast, in recent years large effort have been devoted to the preparation of substituted-IF derivatives that are stable enough to be isolated and studied in detail13–17. These antiaromatic PCHs have received special attention because of their distinct electron accepting, n-type semiconducting behaviour, based on their easy reduction to the corresponding aromatic dianions13,18. This feature, together with the narrow highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gap, make IF derivatives promising organic photoelectronic materials19. The high reactivity expected for unsubstituted IF implies a challenge for probing its structural and electrical properties. An interesting fact if that the parent structure of one of the most promising PCH cores for electronic devices has not yet been investigated20.
Advances in atomic force microscopy (AFM), particularly in resolving21 and modifying the structure of molecules at the atomic scale, have opened new routes in the chemistry of highly reactive compounds22–30. AFM with functionalised tips has been used to identify and characterise individual molecules31–34. One important aspect is the bond order, which can be resolved directly with AFM by comparing the apparent length and contrast of individual bonds within the molecule31, which has already been applied for the on-surface characterisation of biphenylenes35 and highly reactive molecules such as arynes29 or diradicals30.
Here we present the generation and characterisation of an antiaromatic PCH, i.e. IF comprising a π-system with 20 electrons. In-solution IF was generated by iodide-induced decomposition of dibromo-substituted precursor 2, whereas on-surface IF was obtained by tip-induced dehydrogenation of the polycyclic hydrocarbon 3. We found that IF survives for a few minutes in solution at ambient conditions. Using AFM and STM at low temperature and in ultra-high vacuum, we found that on bilayer NaCl on Cu(111) (denoted as 2 monolayers or 2 ML NaCl) IF preserves the closed-shell character predicted for the free molecule, whereas on Cu(111) the electronic configuration is significantly altered because of strong chemisorption to the surface.
Results
In-solution synthesis and lifetime of IF
We explored the in-solution generation of IF by KI-induced decomposition of 6,12-dibromo-6,12-dihydro indeno[1,2-b]fluorene (2) (Fig. 1). It is well known that highly reactive ortho-quinodimethane can be generated by iodide-induced debromination from the corresponding precursors36,37. Based on these precedents, we synthesised compound 2 (see Supplementary Methods for details) to study the generation of IF at room temperature by treatment with KI in a mixture of THF:CH3CN (1:1). The reaction was monitored by means of UV spectroscopy (Fig. 2). Before the KI treatment (t = 0), we did not observe any absorbance between 440 and 560 nm. However, immediately after adding KI, we observed a group of signals appearing between 460 and 520 nm (λmax ca. 507 nm), which are characteristic of the IF chromophore. Similar UV spectra were reported for 6,12-dimesityl-substituted IF, where there is a little electronic communication between the π-systems of the orthogonal mesityl groups and IF is very small18. The detected optical gap Egap and the position of the absorption peak λmax in our experiment are nearly identical with those of 6,12-dimesityl-substituted IF reported before (Table 1)18. The peak at 507 nm reaches its maximum intensity after 180 s of KI treatment; then it starts to vanish owing to the short lifetime of highly reactive IF (Supplementary Discussion).
Fig. 2.
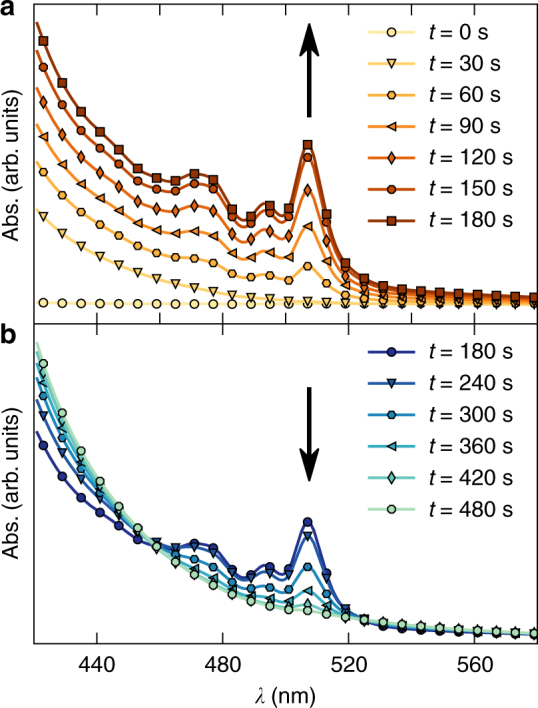
KI-induced dehalogenation of 2. The reaction was monitored by UV spectroscopy measured in a 1:1 mixture of THF:CH3CN at 20 °C. The first measurement (t = 0) was taken before the addition of KI. a Shows the time before the peak at 507 nm has reached its maximum and b after
Table 1.
Computational and optical data
On-surface generation and characterisation of IF
With the idea to increase the lifetime of IF and explore its aromaticity, we deposited compound 3 in ultra-high vacuum at low temperature on Cu(111) partly covered by bilayer NaCl islands. To dehydrogenate 3, we followed a similar procedure as that for the dehydrogenation of triangulene precursors38. A Cu-terminated tip was positioned above the centre of a precursor molecule 3 at a tip height corresponding to an STM setpoint of V = 0.1 V and I = 1 pA. At the opened-feedback loop the tip was retracted by 4–8 Å to limit the tunnelling current to a few picoamperes at elevated biases, then the sample voltage V was increased for 2 s. Typically, a sudden change in the tunnelling current occurred within two seconds at biases above 3.5 V, indicating a manipulation event24,27,28,38,39. The threshold for dehydrogenation (3.5 V) is consistent with previous experiments to remove a single H from doubly benzylic CH2 groups38 and with the dissociation energy of a C–H bond within the CH2 group of fluorene (80 kJ/mol or 3.47 eV)40. On Cu(111), two hydrogens were removed sequentially, giving rise to two distinct steps in the current. On 2 ML NaCl, IF formed directly without the formation of a stable intermediate indicating concerted dehydrogenation (see Supplementary Fig. 6 and its discussion for more details about the on-surface generation of IF).
Figure 3 shows constant-height AFM images before and after dehydrogenation on both surfaces and also of the intermediate 3′ after dissociation of only one H on Cu(111). AFM images were taken with a CO-functionalised tip at zero bias (V = 0). Height offsets Δz are denoted with respect to an STM setpoint of I = 1 pA at V = 0.1 V above the respective substrate surface. A positive Δz sign corresponds to an increase in the tip–sample separation. The structure of the precursor molecule adsorbed on Cu(111) was resolved using Δz = −1.3 Å. After complete dehydrogenation, molecular resolution was acquired at typically Δz = −2.5 Å, indicating a significantly reduced adsorption height. In contrast, on 2 ML NaCl we observed atomic resolution of 3 at Δz = 1.7 and of IF at 1.5 Å, suggesting only a minor change in the adsorption height due to dehydrogenation on NaCl. We quantified the change in the adsorption height with Δf(z) spectroscopy using z*, the tip height of which Δf(z) is minimal, as a measure of the relative adsorption height (Fig. 4)28. The z* map acquired on Cu (2 ML NaCl) shows that IF adsorbs 0.94 Å (0.18 Å) closer to the surface than 3. In addition, the z* maps indicate that IF adsorbs planarly on NaCl. On Cu(111) we observed a slight increase in the adsorption height at the outer benzene rings than at the molecular centre, similar to pentacene (1) adsorbed on Cu(111)21,28.
Fig. 3.

On-surface generation of indeno[1,2-b]fluorene (IF). 6,12-Dihydroindeno[1,2-b]fluorene (3) was dehydrogenated with bias pulses typically between 3.5 and 4.3 V. a–e Constant-height AFM images of the precursor molecule 3 (a, d), of radical 3′ (b) and IF (c, e) measured with a CO tip at V = 0 on Cu(111) (a–c) and on 2 ML NaCl (d, e), respectively. Images were taken at tip height offsets a Δz = −1.3 Å, b Δz = −2.0 Å, c Δz = −2.5 Å, d Δz = 1.7 Å and e Δz = 1.5 Å with respect to the STM setpoint of I = 1.0 pA and V = 0.1 V above the respective substrate. Scale bars, 500 pm
Fig. 4.

Δf(z) spectroscopy. a, c Δf(z) curves taken over the central benzene ring of 3 (yellow) and IF (purple) adsorbed on a Cu(111) and c 2 ML NaCl. The z scale is offset by z*(3) for the respective surface to show 3 at z = 0 as reference. b, d z* maps of IF adsorbed on b Cu(111) and d 2 ML NaCl. At z = −1.6 Å on Cu(111) and z = −0.7 Å on 2 ML NaCl (black regions), the approach was aborted to avoid tip instabilities, and z* was not reached above the bare surfaces. Scale bars, 500 pm
In solution, the IF core can be easily transformed into [4n + 2] aromatic dication (18 π-electrons) or dianion (22 π-electrons) by oxidation or reduction reactions, respectively13,18. On 2 ML NaCl, the charge state of the adsorbed molecule is governed by the work function of the surface and by the electron affinity level and the ionisation potential of the molecule41. We observed no interface state electron scattering at IF on 2 ML NaCl, demonstrating that the molecule remains neutral and therefore antiaromatic on 2 ML NaCl42.
To learn about the contributions of the open- and closed-shell resonant structures presented in Fig. 1, we probed the orbital configuration of IF on NaCl and carried out spin-polarised DFT calculations with a first-order perturbative correction (G0W0). For the calculations, we considered the molecule in its open-shell and closed-shell configurations (Methods). Their quasiparticle energies are shown in Fig. 5a. The zero of the energy scale has been adjusted to match the experimentally determined work function of 2 ML NaCl on Cu(111) (Φ = 4.0 eV)43. Based on the level alignments, IF is predicted to remain uncharged in both cases considered. α and β are the frontier molecular orbitals of IF. In the closed-shell configuration, α is fully occupied (closed) and is the highest occupied molecular orbital, whereas β is the lowest unoccupied molecular orbital. Note that non-polarised energy minimisation also leads to the closed-shell configuration. In the open-shell configuration, α and β are non-degenerate singly occupied molecular orbitals (SOMOs). We imaged the negative ion resonance (NIR) at positive sample bias, with its onset at V = 1.0 V (Fig. 5b). We were unable to image the positive ion resonance (PIR) because the molecule started to move at increased negative biases before the bias voltage reached the onset of the PIR. Because IF is neutral, the NIR is attributed to electron tunnelling into the lowest-lying empty state(s) of the free molecule42. In the closed-shell case, the NIR is expected to closely resemble the shape of the LUMO (orbital β), whereas in the open-shell case, the NIR is related to the empty spin-down channel of the α orbital (Fig. 5a). In Fig. 5c, d, simulated STM images are shown of orbitals α and β. We considered an extended s-like wave function for the tip to simulate the STM appearance of orbitals38,44. The experimentally observed image concurs with the simulated LUMO (β orbital) STM image of the molecule in the closed-shell resonance structure, indicating that IF is in the closed-shell configuration.
Fig. 5.

Spin-polarised DFT calculations. a Energy-level diagram obtained from DFT calculations carried out with the G0W0 approximation. Marked energy levels correspond to the lowest-laying unoccupied states. b Constant-current STM image of IF taken with a metal tip at the energy corresponding to the negative ion resonance (NIR) (V = 1.0 V and I = 1.0 pA). c, d Simulated STM images of orbitals α (c) and β (d). Scale bar, 500 pm
In addition, we carried out a bond-order analysis by AFM, which can be used to investigate the contributions of open- and closed-shell resonant structures, as these resonant structures lead to qualitatively different bond-order relations in the s-indacene moiety. The bond-order analysis of IF on 2 ML NaCl also shows excellent agreement with the closed-shell configuration. Figure 6e and f shows bond orders derived from the relaxed geometries using Pauling’s empirical model29,45–47. In experiment, we compare the brightness (Δf) and apparent lengths of bonds to deduce bond-order relations28,29,47. Bonds of greater bond order are imaged more brightly owing to greater repulsive forces and they appear shorter47. In the high-resolution AFM image on 2 ML NaCl shown in Fig. 6c, we compared bonds that show a very similar chemical environment, i.e. bond a with b and bond c with d, respectively. For IF on NaCl (Fig. 6c, d) the AFM measurements indicate greater bond order of b than of a and greater bond order of d than of c, both in line with the closed-shell configuration of IF. In our calculations, the energy of the closed-shell configuration is by 0.92 eV lower than that of the open-shell for the gas-phase molecule. Our findings are in good agreement with the previously calculated low diradical character for IF12.
Fig. 6.
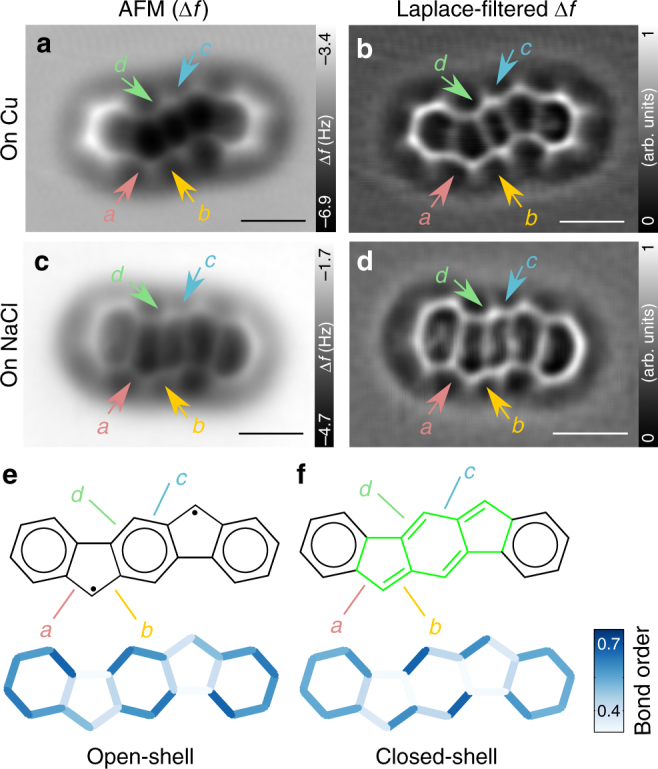
Bond-order analyses. a, c Constant-height AFM images of IF taken with CO tip at V = 0 V on Cu(111) (Δz = −2.1 Å) (a) and on 2 ML NaCl (Δz = 1.5 Å) (c). b, d To emphasise the structure of the molecule, Laplace-filtered AFM images are also shown. e, f Bond orders of IF in its open-shell (e) and closed-shell (f) resonance structures. The s-indacene moiety is highlighted in green (f). On the bond-order scale, 0 refers to a single bond and 1 to a double-bond45. Bond orders were determined by using the bond lengths of relaxed geometries. Scale bars, 500 pm
On Cu(111), we were not able to probe the frontier molecular orbitals because of the strong electronic coupling between the molecule and the metal substrate42. However, bond-order analysis by AFM can be used to gain information about the resonance character of IF. AFM images appear less distorted on Cu(111) then on NaCl48. On Cu(111), bonds a and b appear with similar contrast and also their apparent bond lengths are comparable as well (Fig. 6a, b). On Cu(111), bond c, located in the central benzene ring of the s-indacene moiety, has a brighter appearance and a shorter apparent length than bond d. Thus AFM indicates that bonds a and b have even bond orders and c has a greater bond order than d. The bond-order relations found on Cu(111) match neither with the closed-shell nor with the open-shell configurations shown in Fig. 6e, f. As discussed above, the adsorption height of IF on Cu(111) decreases significantly with respect to its precursor 3, indicating a strong molecule–surface interaction after dehydrogenation. Reduction of the adsorption height was shown for olympicene when the physisorbed state was changed into a chemisorbed configuration by transforming the molecule into a π-radical by atomic manipulation28. Similarly, we attribute the origin of the reduced adsorption height and bond-order reorganisation to chemisorption.
In conclusion, we have shown a successful on-surface and in-solution generation and characterisation of highly reactive antiaromatic indeno[1,2-b]fluorene (IF). We generated IF by iodide-induced debromination in solution, whereas on surface we used tip-induced dehydrogenation. In solution, we found that IF survives for a few minutes even at ambient conditions. On surface, the molecule shows its antiaromatic, closed-shell configuration on 2 ML NaCl. This is in contrast to IF adsorbed on Cu(111), where bond-order analysis indicates significant deviations from the closed-shell configuration, demonstrating the importance of molecule–surface interactions on the π-electron distribution.
Methods
AFM/STM experiments
Experiments were carried out using a home-built combined STM/AFM under ultrahigh vacuum conditions (below 10−10 mbar) at a temperature of 5 K. The bias voltage V was applied to the sample. A qPlus sensor49 (stiffness k = 1800 N/m, eigenfrequency f0 = 25 kHz, quality factor Q = 2 × 105) operated in frequency-modulation mode50 was used to perform AFM measurements. A focused ion beam setup was used to cut and sharpen the PtIr tip. The oscillation amplitude was 0.5 Å. A Cu(111) single crystal was cleaned by several sputtering and annealing cycles. Ultrathin NaCl films were grown on Cu(111) by thermal evaporation of NaCl at a temperature of about 270 K. Low coverages of 3 and CO molecules were deposited while the sample temperature was kept below 10 K. CO tips were prepared by picking up a single CO molecule from NaCl21.
DFT calculations
DFT calculations were performed using the FHI-AIMS code51. The geometry of the isolated molecule was optimised with the tight basis defaults. For structural relaxation, the Perdew–Burke–Ernzerhof exchange-correlation functional was applied52 with vdW correction53. The convergence criterion for the total forces was 10−3 eV/Å, and for the total energy it was set to 10−5 eV. The closed-shell configuration was calculated by performing unrestricted spin-polarised energy minimisation or spin-unpolarised calculations. The open-shell configuration was considered by keeping a spin multiplicity of 3 for the total spin of the molecule during the spin-polarised total energy minimisation.
Hybrid functional Heyd, Scuseria and Ernzerho (HSE)54,55 with a mixing coefficient of 0.3 was applied for the computational calculations of the HOMO–LUMO gaps presented in Table 1. The mixing coefficient was adjusted to have the best match between the optical gap and the calculated gap of 6,12-mesityl-IF and 2. The convergence criterion for the total forces was 10−3 eV/Å, and for the total energy it was set to 10−5 eV.
In-solution lifetime measurements
UV/Vis spectra were recorded in a Jasco V-630 spectrophotometer. See Supplementary Methods for more details about in-solution experiments and the synthesis of the IF precursors 2 and 3.
Data availability
All experimental and theoretical data presented here are available from the corresponding authors on reasonable request.
Electronic supplementary material
Acknowledgements
We thank B. Schuler, S. Fatayer and R. Allenspach for discussions. We acknowledge financial support from the European Research Council Advanced Grant CEMAS (291194), the European Union project PAMS (610446) and the ERC Consolidator Grant AMSEL (682144) programs, the Agencia Estatal de Investigación (MAT2016-78293-C6-3-R and CTQ2016-78157-R), the Xunta de Galicia (Centro singular de investigación de Galicia accreditation 2016-2019, ED431G/09) and the European Regional Development Fund (ERDF).
Author contributions
Z.M., N.P., G.M. and L.G. performed and analysed the AFM/STM measurements. Z.M., N.P. and N.M. carried out the DFT calculations. M.V.-V., D.Pér., E.G. and D.Peñ. synthesised precursor molecules and performed the in-solution experiments. All authors contributed to writing the paper.
Competing interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary Information accompanies this paper at 10.1038/s41467-018-03368-9.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Zsolt Majzik, Email: maj@zurich.ibm.com.
Diego Peña, Email: diego.pena@usc.es.
References
- 1.Krygowski T, Cyrañski M, Czarnocki Z, Häfelinger G, Katritzky AR. Aromaticity: a theoretical concept of immense practical importance. Tetrahedron. 2000;56:1783–1796. doi: 10.1016/S0040-4020(99)00979-5. [DOI] [Google Scholar]
- 2.Peeks MD, Claridge TDW, Anderson HL. Aromatic and antiaromatic ring currents in a molecular nanoring. Nature. 2017;541:200–203. doi: 10.1038/nature20798. [DOI] [PubMed] [Google Scholar]
- 3.Hückel E. Quantentheoretische Beiträge zum Benzolproblem. Z. Phys. 1931;70:204–286. doi: 10.1007/BF01339530. [DOI] [Google Scholar]
- 4.Zhao L, Grande-Aztatzi R, Foroutan-Nejad C, Ugalde JM, Frenking G. Aromaticity, the Hückel 4n + 2 rule and magnetic current. Chem. Sel. 2017;2:863–870. [Google Scholar]
- 5.Clar, E. The Aromatic Sextet (John Wiley & Sons, London, 1972).
- 6.Randić M. Aromaticity and conjugation. J. Am. Chem. Soc. 1977;99:444–450. doi: 10.1021/ja00444a022. [DOI] [Google Scholar]
- 7.Breslow R, Brown J, Gajewski JJ. Antiaromaticity of cyclopropenyl anions. J. Am. Chem. Soc. 1967;89:4383–4390. doi: 10.1021/ja00993a023. [DOI] [Google Scholar]
- 8.Zeng Z, et al. Pro-aromatic and anti-aromatic π-conjugated molecules: an irresistible wish to be diradicals. Chem. Soc. Rev. 2015;44:6578–6596. doi: 10.1039/C5CS00051C. [DOI] [PubMed] [Google Scholar]
- 9.Frederickson CK, Rose BD, Haley MM. Explorations of the indenofluorenes and expanded quinoidal analogues. Acc. Chem. Res. 2017;50:977–987. doi: 10.1021/acs.accounts.7b00004. [DOI] [PubMed] [Google Scholar]
- 10.Tobe Y. Non-alternant non-benzenoid aromatic compounds: past, present, and future. Chem. Rec. 2015;15:86–96. doi: 10.1002/tcr.201402077. [DOI] [PubMed] [Google Scholar]
- 11.Kubo T. Recent progress in quinoidal singlet biradical molecules. Chem. Lett. 2015;44:111–112. doi: 10.1246/cl.140997. [DOI] [Google Scholar]
- 12.Fukuda K, Nagami T, Fujiyoshi J, Nakano M. Interplay between open-shell character, aromaticity, and second hyperpolarizabilities in indenofluorenes. J. Phys. Chem. A. 2015;119:10620–10627. doi: 10.1021/acs.jpca.5b08520. [DOI] [PubMed] [Google Scholar]
- 13.Rose BD, et al. Experimental and computational studies of the neutral and reduced states of indeno[1,2-b]fluorene. J. Am. Chem. Soc. 2014;136:9181–9189. doi: 10.1021/ja503870z. [DOI] [PubMed] [Google Scholar]
- 14.Chase DT, Fix AG, Rose BD, Weber CD. Electron accepting 6,12-diethynylindeno [1,2-b] fluorenes: synthesis, crystal structures, and photophysical properties. Angew. Chem. Int. Ed. 2011;50:11103–11106. doi: 10.1002/anie.201104797. [DOI] [PubMed] [Google Scholar]
- 15.Rudebusch GE, et al. Diindeno-fusion of an anthracene as a design strategy for stable organic biradicals. Nat. Chem. 2016;8:753. doi: 10.1038/nchem.2518. [DOI] [PubMed] [Google Scholar]
- 16.Nishida Ji, Tsukaguchi S, Yamashita Y. Synthesis, crystal structures, and properties of 6,12-diaryl-substituted indeno[1,2-b]fluorenes. Chem. Eur. J. 2012;18:8964–8970. doi: 10.1002/chem.201200591. [DOI] [PubMed] [Google Scholar]
- 17.Frederickson CK, Zakharov LN, Haley MM. Modulating paratropicity strength in diareno-fused antiaromatics. J. Am. Chem. Soc. 2016;138:16827–16838. doi: 10.1021/jacs.6b11397. [DOI] [PubMed] [Google Scholar]
- 18.Chase DT, et al. 6,12-Diarylindeno[1,2-b]fluorenes: syntheses, photophysics, and ambipolar OFETs. J. Am. Chem. Soc. 2012;134:10349–10352. doi: 10.1021/ja303402p. [DOI] [PubMed] [Google Scholar]
- 19.Anthony JE. The larger acenes: versatile organic semiconductors. Angew. Chem. Int. Ed. 2008;47:452–483. doi: 10.1002/anie.200604045. [DOI] [PubMed] [Google Scholar]
- 20.Liu J, et al. π-Extended and curved anti-aromatic polycyclic hydrocarbons. J. Am. Chem. Soc. 2017;139:7513–7521. doi: 10.1021/jacs.7b01619. [DOI] [PubMed] [Google Scholar]
- 21.Gross L, Mohn F, Moll N, Liljeroth P, Meyer G. The chemical structure of a molecule resolved by atomic force microscopy. Science. 2009;325:1110–1114. doi: 10.1126/science.1176210. [DOI] [PubMed] [Google Scholar]
- 22.Lauhon LJ, Ho W. Single-molecule chemistry and vibrational spectroscopy: pyridine and benzene on Cu(001) J. Phys. Chem. A. 2000;104:2463–2467. doi: 10.1021/jp991768c. [DOI] [Google Scholar]
- 23.Hla SW, Bartels L, Meyer G, Rieder KH. Inducing all steps of a chemical reaction with the scanning tunneling microscope tip: towards single molecule engineering. Phys. Rev. Lett. 2000;85:2777–2780. doi: 10.1103/PhysRevLett.85.2777. [DOI] [PubMed] [Google Scholar]
- 24.Zhao A, et al. Controlling the Kondo effect of an adsorbed magnetic ion through its chemical bonding. Science. 2005;309:1542–1544. doi: 10.1126/science.1113449. [DOI] [PubMed] [Google Scholar]
- 25.Baadji N, et al. Controlled sequential dehydrogenation of single molecules by scanning tunneling microscopy. Phys. Rev. B. 2010;82:115447. doi: 10.1103/PhysRevB.82.115447. [DOI] [Google Scholar]
- 26.Mohn F, et al. Reversible bond formation in a gold-atom–organic-molecule complex as a molecular switch. Phys. Rev. Lett. 2010;105:266102. doi: 10.1103/PhysRevLett.105.266102. [DOI] [PubMed] [Google Scholar]
- 27.Lit J, et al. Suppression of electron–vibron coupling in graphene nanoribbons contacted via a single atom. Nat. Commun. 2013;4:2023. doi: 10.1038/ncomms3023. [DOI] [PubMed] [Google Scholar]
- 28.Schuler B, et al. Adsorption geometry determination of single molecules by atomic force microscopy. Phys. Rev. Lett. 2013;111:086101. doi: 10.1103/PhysRevLett.111.086101. [DOI] [PubMed] [Google Scholar]
- 29.Pavliček N, et al. On-surface generation and imaging of arynes by atomic force microscopy. Nat. Chem. 2015;7:623–628. doi: 10.1038/nchem.2300. [DOI] [PubMed] [Google Scholar]
- 30.Schuler B, et al. Reversible Bergman cyclization by atomic manipulation. Nat. Chem. 2016;8:220–224. doi: 10.1038/nchem.2438. [DOI] [PubMed] [Google Scholar]
- 31.Gross L, et al. Organic structure determination using atomic-resolution scanning probe microscopy. Nat. Chem. 2010;2:821–825. doi: 10.1038/nchem.765. [DOI] [PubMed] [Google Scholar]
- 32.Kawai S, et al. Thermal control of sequential on-surface transformation of a hydrocarbon molecule on a copper surface. Nat. Commun. 2016;7:12711. doi: 10.1038/ncomms12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rogers C, et al. Closing the nanographene gap: surface-assisted synthesis of peripentacene from 6,6-bipentacene precursors. Angew. Chem. Int. Ed. 2015;54:15143. doi: 10.1002/anie.201507104. [DOI] [PubMed] [Google Scholar]
- 34.de Oteyza DG, et al. Direct imaging of covalent bond structure in single-molecule chemical reactions. Science. 2013;340:1434–1437. doi: 10.1126/science.1238187. [DOI] [PubMed] [Google Scholar]
- 35.Kawai S, et al. Competing annulene and radialene structures in a single anti-aromatic molecule studied by high-resolution atomic force microscopy. ACS Nano. 2017;11:8122–8130. doi: 10.1021/acsnano.7b02973. [DOI] [PubMed] [Google Scholar]
- 36.Cava MP, Napier DR. Condensed cyclobutane aromatic systems. II. Dihalo derivatives of benzocyclobutene and benzocyclobutadiene dimer. J. Am. Chem. Soc. 1957;79:1701–1705. doi: 10.1021/ja01564a048. [DOI] [Google Scholar]
- 37.Segura JL, Martín N. o-Quinodimethanes: efficient intermediates in organic synthesis. Chem. Rev. 1999;99:3199–3246. doi: 10.1021/cr990011e. [DOI] [PubMed] [Google Scholar]
- 38.Pavliček N, et al. Generation and characterization of triangulene. Nat. Nanotechnol. 2017;12:308–311. doi: 10.1038/nnano.2016.305. [DOI] [PubMed] [Google Scholar]
- 39.Majzik Z, et al. Synthesis of a naphthodiazaborinine and its verification by planarization with atomic force microscopy. ACS Nano. 2016;10:5340–5345. doi: 10.1021/acsnano.6b01484. [DOI] [PubMed] [Google Scholar]
- 40.McMahon TB, Kebarle P. Intrinsic acidities of carbon and nitrogen acids measured by gas phase proton transfer equilibria. Substituent effects on the stabilities of gas phase carbanions and nitrogen anions. J. Am. Chem. Soc. 1976;98:3399–3406. doi: 10.1021/ja00428a001. [DOI] [Google Scholar]
- 41.Swart I, Sonnleitner T, Repp J. Charge state control of molecules reveals modification of the tunneling barrier with intramolecular contrast. Nano Lett. 2011;11:1580–1584. doi: 10.1021/nl104452x. [DOI] [PubMed] [Google Scholar]
- 42.Repp J, Meyer G, Stojković SM, Gourdon A, Joachim C. Molecules on insulating films: scanning-tunneling microscopy imaging of individual molecular orbitals. Phys. Rev. Lett. 2005;94:086101. doi: 10.1103/PhysRevLett.94.026803. [DOI] [PubMed] [Google Scholar]
- 43.Bennewitz R, et al. Aspects of dynamic force microscopy on NaCl/Cu(111): resolution, tip–sample interactions and cantilever oscillation characteristics. Surf. Interface Anal. 1999;27:462–466. doi: 10.1002/(SICI)1096-9918(199905/06)27:5/6<462::AID-SIA543>3.0.CO;2-0. [DOI] [Google Scholar]
- 44.Pavliček N, Swart I, Niedenführ J, Meyer G, Repp J. Symmetry dependence of vibration-assisted tunneling. Phys. Rev. Lett. 2013;110:136101. doi: 10.1103/PhysRevLett.110.136101. [DOI] [PubMed] [Google Scholar]
- 45.Pauling L, Brockway LO, Beach JY. The dependence of interatomic distance on single bond-double bond resonance. J. Am. Chem. Soc. 1935;57:2705–2709. doi: 10.1021/ja01315a105. [DOI] [Google Scholar]
- 46.Sedlar J, Anelić I, Gutman I, Vukičević D, Graovac A. Vindicating the Pauling-bond-order concept. Chem. Phys. Lett. 2006;427:418–420. doi: 10.1016/j.cplett.2006.06.026. [DOI] [Google Scholar]
- 47.Gross L, et al. Bond-order discrimination by atomic force microscopy. Science. 2012;337:1326–1329. doi: 10.1126/science.1225621. [DOI] [PubMed] [Google Scholar]
- 48.Neu M, et al. Image correction for atomic force microscopy images with functionalized tips. Phys. Rev. B. 2014;89:205407. doi: 10.1103/PhysRevB.89.205407. [DOI] [Google Scholar]
- 49.Giessibl FJ. Atomic resolution on Si(111)–(7 × 7) by noncontact atomic force microscopy with a force sensor based on a quartz tuning fork. Appl. Phys. Lett. 2000;76:1470–1472. doi: 10.1063/1.126067. [DOI] [Google Scholar]
- 50.Albrecht TR, Grütter P, Horne D, Rugar D. Frequency modulation detection using high-Q cantilevers for enhanced force microscope sensitivity. J. Appl. Phys. 1991;69:668–673. doi: 10.1063/1.347347. [DOI] [Google Scholar]
- 51.Blum V, et al. Ab initio molecular simulations with numeric atom-centered orbitals. Comput. Phys. Commun. 2009;180:2175–2196. doi: 10.1016/j.cpc.2009.06.022. [DOI] [Google Scholar]
- 52.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 53.Tkatchenko A, Scheffler M. Accurate molecular van Der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009;102:073005. doi: 10.1103/PhysRevLett.102.073005. [DOI] [PubMed] [Google Scholar]
- 54.Krukau AV, Vydrov OA, Izmaylov AF, Scuseria GE. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006;125:224106. doi: 10.1063/1.2404663. [DOI] [PubMed] [Google Scholar]
- 55.Heyd J, Scuseria GE, Ernzerhof M. Hybrid functionals based on a screened Coulomb potential. J. Chem. Phys. 2003;118:8207–8215. doi: 10.1063/1.1564060. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All experimental and theoretical data presented here are available from the corresponding authors on reasonable request.