

Abstract
Objective:
Association between chronic alcohol intake and cardiac abnormality is well known; however, the precise underlying molecular mediators involved in ethanol-induced heart abnormalities remain elusive. This study investigated the effect of chronic ethanol exposure on calcium/calmodulin-dependent protein kinase IIδ (CaMKIIδ) gene expression and monoamine oxidase (MAO) levels and histological changes in rat heart. It was also planned to find out whether Zingiber officinale (ginger) extract mitigated the abnormalities induced by ethanol in rat heart.
Methods:
Male wistar rats were divided into three groups of eight animals each: control, ethanol, and ginger extract treated-ethanol (GETE) groups.
Results:
After 6 weeks of treatment, the results revealed a significant increase in CaMKIIδtotal and isoforms δ2 and δ3 of CaMKIIδ gene expression as well as a significant decrease in the MAO levels in the ethanol group compared to that in the control group. Moreover, compared to the control group, the ethanol group showed histological changes, such as fibrosis, heart muscle cells proliferation, myocyte hypertrophy, vacuolization, and focal lymphocytic infiltration. Consumption of ginger extract along with ethanol ameliorated CaMKIIδtotal. In addition, compared to the ethanol group, isoforms gene expression changed and increased the reduced MAO levels and mitigated heart structural changes.
Conclusion:
These findings indicate that ethanol-induced heart abnormalities may, in part, be associated with Ca2+ homeostasis changes mediated by overexpression of CaMKIIδ gene and the decrease of MAO levels and that these effects can be alleviated by using ginger extract as an antioxidant and anti-inflammatory agent.
Keywords: ethanol, heart, oxidative stress, CaMKIIδ, rat, fibrosis, ginger
Introduction
Recent studies have demonstrated that chronic ethanol exposure leads to a wide range of functional and structural abnormities in the cardiovascular system (1-3). From the structural aspect, heart tissue fibrosis decreases the myocyte number, disrupts myofibrillar structure, and causes left ventricular hypertrophy and myocardial infarction. Moreover, even an increase in sudden death has been reported (1, 2). Large spectrums of functional alterations have also been reported as the result of chronic ethanol consumption. We have previously shown that ethanol consumption leads to an elevated systolic, diastolic, pulse, main arterial, and dicrotic pressure (4). In addition, reduced cardiac contractility, cardiac output, left ventricular ejection fraction, and abnormalities of the great vessels result from chronic ethanol exposure in animal and human models (5-7). Furthermore, alteration in Ca2+ transport, mitochondrial function, sarcoplasmic reticulum Ca2+ uptake/binding, and Ca2+ homeostasis have been demonstrated by several previous studies (3, 8, 9). Although different aspects of functional and structural cardiac alterations have been identified by early and recent studies, the precise mediating steps between exposure of heart muscle to ethanol and initiation of the cascade of responses leading to cardiac abnormality have not yet been completely clarified. Numerous mechanisms, such as oxidative stress, inflammatory reactions, toxicity of ethanol itself and its primary metabolite acetaldehyde, accumulation of fatty acid esters, and modification of lipoproteins, have been suggested to explain pathogenesis of chronic ethanol-induced abnormalities in the heart tissue (2, 4, 10, 11). However, studies have often suggested that mechanisms such as oxidative stress and inflammatory reactions explain alterations in heart structure and function following ethanol exposure and thus have not provided precise information concerning the specific molecules that could influence cardiac structure and functions molecular mediators after ethanol exposure. Among dozens of molecular mediators related to heart function, calcium/calmodulin-dependent protein kinase II, particularly CaMKIIδ isoform, is documented as an important mediator connecting pathological changes in sub-cellular environments to alterations in cardiomyocyte Ca2+ handling. CaMKII gene produces four isoforms (α, β, η, and δ) with different tissue distribution. The predominant form in the heart appears to be CaMKIIδ (12). Although numerous cellular functions, such as cell cycle, growth, and gene expression, are regulated by CaMKII in the heart, CaMKIIδ has a significant involvement in the regulation of Ca2+ homeostasis and cardiac contractility (13-16). Besides physiological functions, numerous studies have shown that overexpression of CaMKIIδ is a core mechanism for promoting heart diseases, such as myocardial hypertrophy, arrhythmias, myocyte apoptosis, defective ECG, Ca2+ homeostasis imbalances, and transition from hypertrophy to heart failure (17, 18). Monoamine oxidase (MAO) is another important mediator playing a prominent role in cardiac function. MAO belongs to a class of flavoenzymes located in the outer mitochondria membrane and is responsible for the deamination of neurotransmitters and re-uptake of catec-holamines (19). Because catecholamines released from the heart sympathetic nervous system influence myocardium continuously, their turnover and catabolism rate contribute to cardiac function and structural alterations. Regarding the role of MAO in catecholamines turnover, previous studies have demonstrated that MAO deletion or overproduction leads to cardiac abnormalities, such as cardiomyocyte hypertrophy, left ventricular dilation, and heart failure (20, 21). The fundamental role of CaMKIIδ and MAO in the initiation and development of cardiac abnormalities and heart failure, as mentioned above, prompted us to examine the following hypothesis: chronic ethanol consumption resulting in heart abnormalities is mediated, in part, by overexpression of CaMKIIδ related genes and alteration of MAO levels in the heart tissue. In addition, because of the well-documented oxidative and inflammatory nature of ethanol, a second aim of this work was to determine the possible protective effects of ginger extract against ethanol-induced histopathological alteration, CaMKIIδ gene expression changes. Among plants containing natural anti-oxidants, ginger exhibits unique antioxidant and anti-inflammatory properties with less unfavorable side effects (22). We also intended to investigate MAO levels alteration in the heart of male rats.
Methods
Animals and treatments
All experimental procedures described herein were performed in accordance with the Principles of Laboratory Animal Care (NIH publication, no.85–23, revised 1985) and were approved by the Urmia University of Medical Sciences Animal Care Committee. Overall, 24 male Wister rats with an initial body weight of 220±10 g were divided into three groups (n=8 in each group): control, ethanol, and ginger extract treated-ethanol (GETE) groups. For the rats in the ethanol group, ethanol was saluted in tap water (20% w/v) and gavaged intragastrically (4.5 g/kg) to rats in the ethanol group, 6 days a week for 6 weeks. For the rats in the GETE group, hydro-alcoholic extract of ginger was gavaged intragastrically (50 mg/kg) for 6 weeks. For the rats in the control group, tap water was gavaged.
Extract preparation
Dried ginger rhizome (originally Chinese) was purchased from a local market and coarsely powdered. Next, 3 kg of the powder was mixed with 6 L of 70% ethanol in a suitable container at room temperature for 3 days. After 3 days, it was filtrated through a filter paper and concentrated using a rotary evaporator. The yield of the extract was stored in a refrigerator at 4°C until use.
Sample preparation
After 6 weeks of treatment, the rats were anesthetized using 10% chloral hydrate (0.5 mL/100g body weight, IP). The anesthesia depth was assessed by pinching a hind paw. At termination, after weighing the animals, the thoracic cavity was opened and the heart was removed. The excised heart was freed from adventitial tissues, fat, and blood clots and was subsequently washed in ice-cold physiological saline and weighed. Next, the whole left ventricular wall (with septum) was excised from the heart and weighed. For total RNA isolation, 100 mg of ventricular tissue was immersed in 1 mL RiboxEX (total RNA isolation solution) (GeneALL, Seoul, Korea) and restored at −80°C until the time of RNA isolation. For biochemical analysis, other parts of the vent-ricles were washed with ice-cold physiological saline and dried on filter papers. Subsequently, an ice-cold extraction buffer (10% wt/vol) containing a 50 mM phosphate buffer (pH 7.4) was added and homogenized using Ultra Turrax (T10B, IKA, Germany). Next, the homogenates were centrifuged at 10,000×g at 4°C for 20 min. Finally, the supernatant sample was obtained and stored at −80°C until the time of analysis. For analyzing histopathological changes, a part of the ventricular was fixed in buffered formalin and embedded in paraffin after standard dehydration steps were taken.
Isolation of total RNA, amplification primers, and real-time polymerase chain reaction (RT-PCR)
The total RNA was obtained from 100 mg of the left ventricular frozen tissue using a kit (Gene all, South Korea, Cat no 305-101), in accordance with the manufacturer’s instructions. RNA concentration was verified by spectrophotometric measurement of the absorbance at 260–280 nm and determined by mixture of Tris base, acetic acid, and EDTA(TAE)-agarose gel electrophoresis.
Reverse transcription (RT) was performed using hyperscriptTM Reverse Transcriptase (Gene All, South Korea). RT-PCR was performed using an amplification reagent kit (Ampliqon, Denmark) by the XP-Cycler instrument (TCXPD, Bioer, USA) with CaMKIIδtotal, CaMKIIδ1, CaMKIIδ2, and the rat glyceraldehydes-3-phosphate dehydrogenase (GAPDH) primers. To amplify the cDNA, the 5’ and 3’ primer sequences (forward and reverse) of the CaMKIIδtotal, CaMKIIδ1, and CaMKIIδ2 designed via the Gene Bank (http://blast.ncbi.nlm.gov/Blast.cgi) revealed that the primers were gene specific. Furthermore, all the primers were verified using a Gene Runner software (Syngene, Cambridge, UK). Subsequently, the primers (forward and reverse) were synthesized to amplify the cDNA encoding GAPDH as a house keeping gene; the sequences of related primers are presented in Table 1.
Table 1.
Sequences of primers used to evaluate expression of GAPDH, CaMKIIδtotal, and CaMKIIδ1, CaMKIIδ2
| Product size | Primer sequence | Target Gene |
|---|---|---|
| 199 | ‘TGG CAA ACT AAA GAG GGA GC-3-’5 | CaMKIIδtotal (forward) |
| 5’-CCA AAA TCC CAA TGA GAA GCC C-3’ | (CaMKIIδtotal (reverse) | |
| 230 | 5’-AAC CGG ATG GGG TAA AGG AG-3’ | (CaMKIIδ2 (forward) |
| ‘CAA TGC TTC GGG TTC AAA GG-3-’5 | (CaMKIIδ2 (reverse) | |
| 164 | ‘CGG ATG GGG TAA AGA AAA GG-3-’5 | (CaMKIIδ3 (forward) |
| ‘CTC GAA GTC CCC ATTT GTT GA-3-’5 | (CaMKIIδ3 (reverse) | |
| 207 | ‘AGA CAG CCG CAT CTT CTT GT-3-’5 | GAPDH (forward) |
| 5’-CTT GCC GTG GGT AGA GTC AT-3’ | (GAPDH (reverse) |
Real-time quantification
Real-time quantification of the target genes was performed taking advantage of a Real-Time PCR Master Mix Green kit (Ampliqon, Denmark) in a total volume of 25 µL and in accordance with the manufacturer’s instructions. Furthermore, the mentioned genes expressions were analyzed using an iQ5 RT-PCR detection system (Bio-Rad, CA, USA). Next, the reactions were prepared for 10 min at 95°C in a 96-well optimal plate followed by 40 cycles of 20 sec at 59°C. To confirm the specificity of the amplification reactions, a melting curve was recorded. Each sample was replicated three times. The value of the threshold cycle (Ct) was the same as that of the corresponding mean. The relative expression of each mRNA was calculated by employing the 2-ΔΔCt method, with Ct being the threshold cycle. Next, the calculated levels were normalized to GAPDH. They were then analyzed for statistical significance applying a one-way analysis of variance.
MAO assay
MAO levels in the heart tissue was measured by the quantitative sandwich enzyme immunoassay method using a commercial rat MAO Elisa kit (ZellBio, Germany), in accordance with the manufacturer’s instructions.
Histopathological examinations
For histopathological staining, 5-µm thick histological sections from paraffin-embedded heart tissue were used. Proliferating cells were implemented, in accordance with our published protocol, by performing immunohistochemistry using an antibody against the proliferation cell nuclear antigen (PCNA) (10). Briefly, after taking tissue processing steps, such as deparaffinization, rehydration, and gradual ethanol passage, sections of the heart tissue (thickness, 5-µm) were stained using the Monoclonal Mouse anti-PCNA antibody (Dako, Catalog no: M0879, Copenhagen, Denmark). Optimal results were achieved using the EnVision™ visualization system. Furthermore, Hematoxylin was used as a counterstain. The assessment included proper negative controls. Moreover, all the slides were inspected by two expert pathologists independently. PCNA-positive indices were considered as indicators of heart cell proliferation. To assess PCNA-positive indices percentage, four non-overlapping fields of view per section from 2–3 sections per animal were analyzed. The number of positively stained cells and the total number of cells were counted for each field of view. In addition, for each animal, the number of positively stained cells was then presented as a percentage of the total number of counted cells. The criteria applied in scoring the quality of PCNA-positive indices were as follows: normal (i.e., PCNA-positive indices pre-sent in less than 5% of the heart cells), mild (i.e., PCNA-positive indices present in less than 25% of the heart cells), mild to mode-rate (i.e., PCNA-positive indices present in 25%–50% of the heart cells), moderate to severe (i.e., PCNA-positive indices present in 50%–75% of the heart cells), and severe (i.e., PCNA-positive indices present in 75%–100% of the heart cells) (10). To evaluate the heart tissue fibrosis, 5-µm heart tissue sections were stained using Masson Trichrome, in accordance with the manufacturer’s instructions (Asiapajohesh, Amol, Iran). The severity of tissue fib-rosis was estimated maintaining a semi-quantitative method explained by Ashcroft et al. (23) and our published protocol. A score ranging from zero (normal heart) to eight (total fibrosis) was set. The criteria appointed in scoring heart fibrosis were as follows: grade 0=normal heart; grade 1=minimal fibrosis thickening of heart tissue, grade 2 and 3=moderate thickening of heart tissue without obvious damage to the structure of heart tissue; grade 4 and 5=increased fibrosis with definite damage to architecture of the heart and formation of fibrosis bands or small fibrosis masses; grade 6 and 7=severe distortion of structure and large fibrosis areas; and grade 8=total fibrotic obliteration (23).
In addition, to assess general histological changes of heart tissue, paraffin-embedded sections of the heart tissue were stained with hematoxylin and All histological measurements in inter-intra observer variability were conducted by at least two independent expert examiners in a blinded manner and expressed in comparison to controls.
Statistical analyses
Normal distribution of data within each group was verified using Kolmogorov–Smirnov test. Statistical analyses were performed using the computer software SPSS 16.0 for Windows (SPSS, IBM, Chicago, USA). The statistical differences between the groups were tested using one-way ANOVA and then Tukey’s post hoc test. The data obtained from each test are presented as the mean±SD, and p<0.05 is considered as statistically significant.
Results
Biochemical and gene expression
The effects of ethanol consumption and treatment with ginger extract on the heart tissue MAO levels, gene expression of molecular markers of pathological cardiac hypertrophy, and left ventricular weight/body weight (LVW/BW) are shown in Table 2. Chronic ethanol administration, as an indicator of left ventricular hypertrophy, significantly increased the ratio of LVW/BW compared to that of the control group (p=0.05). Ginger extract administration along with ethanol reduced the ratio of LVW/BW significantly compared to that in the ethanol group (p>0.002), and no significant differences were found between the GETE and control groups. MAO levels in heart tissue was lower in the ethanol rats (p=0.05) than in the control group. Even though ginger extract administration along with ethanol increased the MAO levels in heart tissue, it was not significantly increased compared to that in the ethanol group. MAO levels was still significantly lower in the GETE group than in the control group (p=0.05). Chronic ethanol consumption significantly increased the expression of CaMKIIδtotal and isoforms δ2 and δ3 of CaMKIIδ related genes (mRNA) in the left ventricular of the ethanol group when compared with the control group (p>0.004). Although ginger extract administration along with ethanol reduced the CaMKIIδ isoform related genes expressions significantly compared to the ethanol group (p>0.004), they were still significantly higher than those in the control group (p=0.05).
Table 2.
Effect of ethanol and ethanol-ginger extract treatment on changes of heart tissue MAO, Gene expression of CaMKIIδtotal, CaMKIIδ1, CaMKIIδ2, PCNA-positive indices, and Left ventricular weight/body weight ratio
| Control | Ethanol | Ethanol-Ginger | |
|---|---|---|---|
| LVW/BW,mg/g | |||
| 2.04±0.02 | 2.34±0.1* | 2.03±0.03 | |
| CaMKIIδtotal, Fold | |||
| 1.23±0.15 | 8.22±0.23* | 2.09±0.98† | |
| CaMKIIδ2,Fold | 1.04±0.04 | ||
| 4.32±0.5* | 2.10±0.36† | ||
| CaMKII3, Fold | 1.15±0.09 | 3.70±0.9* | 2.20±0.34† |
| MAO, ng/ml | 4.91±0.0.64 | 2.85±0.42* | 3.2±0.33* |
| PCNA-positive indice,%, | 1 | 40.50±5* | 2† |
Values are mean±SE for eight rats per group.
Denotes significant difference compared to the control
Denotes significant difference compared to the ethanol group
Histopathological alterations
General histological changes
Results from the heart tissue histopathological examination are given in Figures 1–3. Compared to the control group, several histopathological changes, such as myocyte hypertrophy with enlarged nuclei and some areas of irregularity arrangement, were observed in the ethanol-treated group. Scattered cytoplasmic vacuoles, infiltrated polymorphonuclear leukocytes (PMN), and focal lymphocytic infiltration were also observed in the ethanol-treated group. There were no significant differences in terms of heart tissue structure between the GETE and control groups (Fig. 1).
Figure 1.
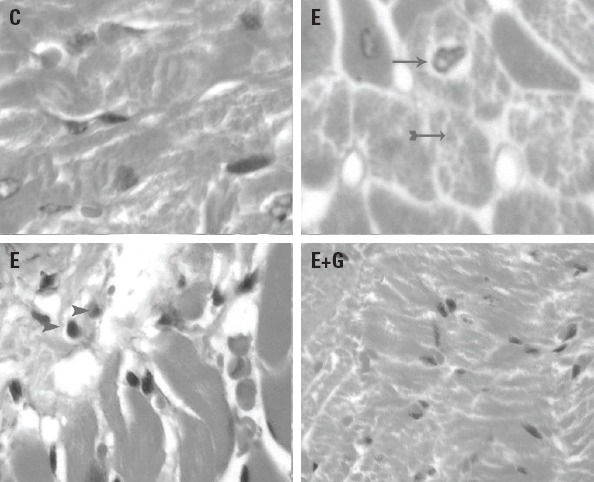
Hematoxylin-eosin (H&E) staining shows infiltration of focal PMN, increase of, vacuolization of cells, and heart muscle cell atrophy were observed in heart tissue in ethanol-treated group (E) compared to control group animals (C). There were no significant differences in terms of heart tissue structure between the GETE (E+G) and control groups. (Original magnification, 400×). Vacuolization ( ), PMN(
), PMN( ), Hyperhrophy (
), Hyperhrophy ( )
)
Figure 2.

Immunohistochemical staining of heart tissue by proliferating cell nuclear antigen (PCNA) antibody showed mild to moderate heart muscle cell proliferation (E) compared to the control group (C). Ginger extract treatment along with ethanol normalized cell proliferation in heart tissue (E+G). (Original magnification, 400×). PCNA-positive indices ( )
)
Figure 3.

Photomicrograph of heart tissue of rats (Masson trichrome staining). in (C), sample obtained from control group; in (E), sample obtained from Ethanol group; in(E+G), sample obtained from GETE group. (Original magnification, 400×). Fibrosis ( )
)
Heart cells proliferation
Heart cells proliferation, as detected by the percentage of cells stained positive for the nuclear antigen (PCNA) are given in Figure 2 and Table 2. The PCNA-positive indices were dramatically increased (mild to moderate) in the ethanol-treated group compared to that in the control group (p=0.001). Ginger extract administration along ethanol reduced PCNA-positive indices significantly compared to the ethanol group, and there were no significant differences between the GETE and the control group.
Heart tissue fibrosis
Figure 3 shows microscopic fibrosis scores in heart tissue obtained from different study groups. There were no lesion scores in heart tissue of the control group (grade 0). The mic-roscopic lesion score in the heart tissue from ethanol-treated group was 4–5, which indicates increased fibrosis with definite damage to the heart architecture and formation of fibrosis bands or small fibrosis masses. Moreover, there were no significant differences found between the GETE and the control group.
Discussion
Although several studies have reported that chronic ethanol ingestion is an identifiable cause of heart tissue disease and heart function abnormalities, none have focused on the precise molecular mediating steps between exposure of heart muscle to ethanol and initiation of the cascade of responses leading to cardiac abnormalities (6-9). A large amount of data shows that the CaMKIIδ pathway is one of the hallmarks of molecular alteration that promotes myocardial hypertrophy and heart failure. Alterations in the CaMKIIδ expression, particularly two major splices of CaMKIIδ in heart tissue including CaMKIIδ2 and CaMKIIδ3, and its associations with shifts in cardiac function have been reported in some pathologic conditions, such as dilated cardiomyopathy, myocardial infarction, early after depolarization(EAD), arrhythmia, and heart failure upon injuries, such as pressure overload and ischemia-reperfusion (24-26). Similarly, the protective effect of CaMKIIδ gene knockout mice against cardiac dysfunction and interstitial fibrosis after pressure overload and β-adrenergic stimulation provide strong evidence for CaMKIIδ maladaptive functions in cardiac pathogenesis (27). The overexpression of CaMKIIδ induces heart abnormalities through multiple processes. Overexpression and activation of CaMKIIδ due to reactive oxygen species shift Ina availability and enhance accumulation of Na channels in the intermediated state; leading to intracellular Na and Ca overloading (27). Intracellular overloaded Ca is then transmitted to the nucleus and activates the nuclear localized isoform of CaMKII, which plays a predominant role in Ca-mediated transcriptional genes associated with cardiac hypertrophy (28). In addition, recently it has been reported that activation of CaMKII by redox signaling induces AngII stimulation that causes cardiomyocyte mitogen activated protein kinase (MAPK) activation and apoptosis during heart failure (29). Furthermore, numerous studies have verified that overexpression of CaMKII induces activation of hypertrophic genes program and development of heart hypertrophy (30-32). Interestingly, results of the present study showed significant increase in the left vent-ricular weight/body weight ratio (as an indicator of heart hypertrophy), heart tissue proliferation, and heart tissue fibrosis along with the overexpression of two major splices of CaMKIIδ in the heart tissue, including CaMKIIδ2 and CaMKIIδ3 in the heart obtained from the ethanol group, compared to those in the control group. Heart cells proliferation and fibrosis induced by ethanol exposure has been previously reported (2). The precise mechanism of heart cells proliferation and fibrosis induced by ethanol is not fully understood; however, it may be due to oxidative stress and inflammatory reactions. The results of the present study demonstrated infiltrated leukocytes along scattered cytoplasmic vacuoles in the heart tissue obtained from ethanol-treated group compared to the control group. Infiltrated leukocytes and their PMN induce inflammation process through several ways. Previous studies have revealed that infiltrated leukocytes and the resulting PMNs promote the formation of fibrosis and proliferation in the skin tissue of the cultured cells (33). In addition, PMNs stimulate the generation of ROS and arouse inflammatory responses (34). For this, PMNs generate lipid mediator of leukotrine B4, a substance that stimulates generation of ROS and constitutes a chemotactic factor for neutrophils and other leukocytes (34). Moreover, PMNs induce production of several cytokines that have a pro-inflammatory role in promoting systemic inflammatory responses and recruiting inflammatory cells locally (34). Therefore, leukocytes infiltration in the heart tissue may predispose development of tissue fibrosis and proliferation via contribution of inflammatory responses in the heart.
To our knowledge, this is the first in vivo study to show that ethanol exposure increases the overexpression of CaMKIIδ splices along with cardiac hypertrophy in rats.
Another important finding of this study was the significant decrease in MAO levels in the ethanol group as compared to those in the control group. Although previous reports have indicated that alcoholism leads to a decrease in the MAO activity in platelets and the brain (35, 36), to the best of our knowledge, this is the first report to show that ethanol consumption lowers the MAO levels in heart tissue. As a promising biological marker, MAO plays an important role in neurotransmitters metabolism and turnover. In cardiac tissue, MAO substrates catecholamines play a prominent role in the regulation of cardiac function (37). Catecholamines activity and availability depend on local degradation as well as a release from the sympathetic nervous end. Because of the continuous influence of norepinephrine and sympathomimetic amines on myocardium function, their turnover and catabolism rate affect cardiac function and structure, particularly in presence of excessive norepinephrine availability (37). Previous studies have verified the association between norepinephrine neuronal re-uptake impairment and chronic heart failure in human and animal models (38, 39). In addition, deletion of MAO activity in mice caused elevation of norepinephrine in the heart, leading to cardiac abnormalities, such as cardiomyocyte hypertrophy, left ventricular dilation, and a lower left ventricular contractility (20, 40). Moreover, elevated norepinephrine reportedly results in cardiomyocyte apoptosis, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation, intracellular Ca-overload, myocardial cell damage, and ventricular arrhythmia (37). Furthermore, previous studies demonstrated that ethanol increases urinary excretion of catecholamines and raises plasma concentration of catecholamines (41-43). Studies have also shown that ethanol inhibits noradrenaline release from peripheral nerves (44, 45). Another study indicated that ethanol consumption resulted in decreased circulatory clearance of noradrenaline, suggesting that increased noradrenaline level in plasma is due to the inhibitory action on uptake and/or metabolism of noradrenaline rather than from an increase of sympathetic nervous outflow (42).
As mentioned above, the deleterious effects of low MAO activity and a high norepinephrine level in heart function and structure and ethanol-induced heart structural abnormalities along with reduced heart tissue MAO levels prompted us to form a hypothesis that ethanol may, at least in part, induce its hazardous effects on the heart by reducing MAO activity in the heart tissue.
The second issue addressed in the present study was the mitigating effect of ginger extract against CaMKIIδ isoforms, gene expression transition, MAO levels reduction, and heart structural alteration induced by ethanol exposure in the heart tissue. Antioxidant and anti-inflammatory properties of ginger supplementation are well established in previous studies (46).
Our recent works have demonstrated that ginger supplementation mitigates oxidative DNA damage and NADPH oxidase. In addition, it increases the total antioxidant capacity and reduces lipid and protein oxidation as two main ROS generator sources in diabetics and other oxidative stress conditions, such as ethanol exposure (46-48). In addition, it has been shown that ginger supplementation inhibits inflammation process by suppressing pro-inflammatory cytokine expressions, such as tumor necrosis factor alpha (TNF-α), arachidonic acid cascade, interleukin-1beta (IL-1β), and macrophage chemoattractant protein-1(MCP-1), and inhibits prostaglandin and leukotriene biosynthesis via suppression of 5-lipooxygenase synthetize activities (49). Chronic ethanol consumption reportedly induces some functional and structural abnormalities in different organs, such as the heart, brain, and kidney, through oxidative stress and inflammation (2, 47). Moreover, recent studies have revealed that conditions such as oxidative stress predispose heart failure by damaging membrane, proteins, and DNA and by redox signaling or even activating physiological signaling pathways (50). Reactive oxygen species activate CaMKII by oxidation dependent pathways, and kinase activated by oxidative stress affects cardiac function through increasing AngII and MAPK activation and apoptosis during transition to HF (29). Accordingly, if ethanol induces functional and structural abnormalities through oxidative stress, mediated CaMKII overexpression, and also reduced MAO levels, the effect of ginger supplementation on rescue abnormalities will be due to its antioxidant and anti-inflammatory properties.
Study limitations
Our study had a few limitations. First, as a molecular underlying for heart failure, along with CaMKIIδ gene expression, the protein levels of this key enzyme was not analyzed in the present study. We did not study alterations of calcium ion homeostasis or norepinephrine, which are important hallmarks of molecular alteration in heart failure. Second, we did not assess acute phase inflammatory protein changes, such as alpha and beta globulins, in plasma of the rats after the treatment.
Conclusions
In conclusion, according to results of the present study, we conclude that ethanol exerts its deleterious effect on the heart, at least in part, by CaMKIIδtotal and splicing genes overexpression and lowering the heart tissue MAO levels mediated by oxidative stress. However, further research is still required to elucidate the comprehensive details of the mechanisms through which ethanol consumption exerts its deleterious effects on heart causing abnormalities. Furthermore, whether ethanol exposure induces heart failure via overexpression of CaMKIIδ genes needs to be discovered by studies using knockout of CaMKIIδ gene expression analysis to elucidate the underlying molecular mechanism of the subjects.
Acknowledgment:
This work derived from a Master of Science thesis in Nutrition by Urmia University of Medical Sciences, Urmia, Iran.
Footnotes
Funding: None declared.
Conflict of interest: None declared.
Peer-review: Externally peer-reviewed.
Authorship contributions: Concept – A.S.; Design – E.H.; Supervision – A.S.; Materials – M.A.; Data collection &/or processing – F.K.; Analysis &/or interpretation – F.H.G.; Literature search – A.S.; Writing – A.S.; Critical review – A.S.
References
- 1.Thomas AP, Rozanski DJ, Renard DC, Rubin E. Effects of ethanol on the contractile function of the heart:a review. Alcohol Clin Exp Res. 1994;18:121–31. doi: 10.1111/j.1530-0277.1994.tb00891.x. [DOI] [PubMed] [Google Scholar]
- 2.Shirpoor A, Nemati S, Ansari MH, Ilkhanizadeh B. The protective effect of vitamin E against prenatal and early postnatal ethanol treatment-induced heart abnormality in rats:a 3-month follow-up study. Int Immunopharmacol. 2015;26:72–9. doi: 10.1016/j.intimp.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Ni Y, Feng-Chen KC, Hsu L. A tissue culture model for studying ethanol toxicity on embryonic heart cells. Cell Biol Toxicol. 1992;8:1–11. doi: 10.1007/BF00119291. [DOI] [PubMed] [Google Scholar]
- 4.Shirpoor A, Salami S, Khadem-Ansari MH, Heshmatian B, Ilkhanizadeh B. Long-term ethanol consumption initiates atherosclerosis in rat aorta through inflammatory stress and endothelial dysfunction. Vascul Pharmacol. 2012;57:72–7. doi: 10.1016/j.vph.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Norouzi L, Shirpoor A, Khadem Ansari MH, Ilkhanizadeh B. Vitamin E attenuates alcohol-induced aortic wall damage in rats. Artery Research. 2015;10:20–6. [Google Scholar]
- 6.Davidson DM. Cardiovascular effects of alcohol. West J Med. 1989;151:430–9. [PMC free article] [PubMed] [Google Scholar]
- 7.Kojima S, Kawano Y, Abe H, Sanai T, Yoshida K, Imanishi M, et al. Acute effects of alcohol ingestion on blood pressure and erythrocyte sodium concentration. J Hypertens. 1993;11:185–90. doi: 10.1097/00004872-199302000-00011. [DOI] [PubMed] [Google Scholar]
- 8.Staley NA, Tobin JD., Jr Reversible effects of ethanol in utero on cardiac sarcoplasmic reticulum of guinea pig offspring. Cardiovasc Res. 1991;25:27–30. doi: 10.1093/cvr/25.1.27. [DOI] [PubMed] [Google Scholar]
- 9.Altura MB, Zhang A, Cheng TP, Altura BT. Exposure of piglet coronary arterial muscle cells to low alcohol results in elevation of intracellular free Ca2+:relevance to fetal alcohol syndrome. Eur J Pharmacol. 1996;314:R9–11. doi: 10.1016/s0014-2999(96)00739-x. [DOI] [PubMed] [Google Scholar]
- 10.Shirpoor A, Salami S, Khadem Ansari MH, Ilkhanizadeh B, Abdollahzadeh N. Ethanol promotes rat aortic vascular smooth muscle cell proliferation via increase of homocysteine and oxidized-low-density lipoprotein. J Cardiol. 2013;62:374–8. doi: 10.1016/j.jjcc.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 11.Hannuksela ML, Liisanantti MK, Savolainen MJ. Effect of alcohol on lipids and lipoproteins in relation to atherosclerosis. Crit Rev Clin Lab Sci. 2002;39:225–83. doi: 10.1080/10408360290795529. [DOI] [PubMed] [Google Scholar]
- 12.Zhang T, Kohlhaas M, Backs J, Mishra S, Phillips W, Dybkova N, et al. CaMKIIdelta isoforms differentially affect calcium handling but similarly regulate HDAC/MEF2 transcriptional responses. J Biol Chem. 2007;282:35078–87. doi: 10.1074/jbc.M707083200. [DOI] [PubMed] [Google Scholar]
- 13.Masse T, Kelly PT. Overexpression of Ca2+/calmodulin-dependent protein kinase II in PC12 cells alters cell growth, morphology, and nerve growth factor-induced differentiation. J Neurosci. 1997;17:924–31. doi: 10.1523/JNEUROSCI.17-03-00924.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Planas-Silva MD, Means AR. Expression of a constitutive form of calcium/calmodulin dependent protein kinase II leads to arrest of the cell cycle in G2. EMBO J. 1992;11:507–17. doi: 10.1002/j.1460-2075.1992.tb05081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nghiem P, Ollick T, Gardner P, Schulman H. Interleukin-2 transcriptional block by multifunctional Ca2+/calmodulin kinase. Nature. 1994;371:347–50. doi: 10.1038/371347a0. [DOI] [PubMed] [Google Scholar]
- 16.Braun AP, Schulman H. The multifunctional calcium/calmodulin-dependent protein kinase:from form to function. Annu Rev Physiol. 1995;57:417–45. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- 17.Zhang T, Johnson EN, Gu Y, Morissette MR, Sah VP, Gigena MS, et al. The cardiac-specific nuclear delta (B) isoform of Ca2+/calmodu-lin-dependent protein kinase II induces hypertrophy and dilated cardiomyopathy associated with increased protein phosphatase 2A activity. J Biol Chem. 2002;277:1261–7. doi: 10.1074/jbc.M108525200. [DOI] [PubMed] [Google Scholar]
- 18.Hund TJ, Mohler PJ. Role of CaMKII in cardiac arrhythmias. Trends Cardiovasc Med. 2015;25:392–7. doi: 10.1016/j.tcm.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edmondson DE, Mattevi A, Binda C, Li M, Hubalek F. Structure and mechanism of monoamine oxidase. Curr Med Chem. 2004;11:1983–93. doi: 10.2174/0929867043364784. [DOI] [PubMed] [Google Scholar]
- 20.Lairez O, Calise D, Bianchi P, Ordener C, Spreux-Varoquaux O, Guilbeau-Frugier C, et al. Genetic deletion of MAO-A promotes serotonin-dependent ventricular hypertrophy by pressure overload. J Mol Cell Cardiol. 2009;46:587–95. doi: 10.1016/j.yjmcc.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 21.Ojaimi C, Qanud K, Hintze TH, Recchia FA. Altered expression of a limited number of genes contributes to cardiac decompensation during chronic ventricular tachypacing in dogs. Physiol Genomics. 2007;29:76–83. doi: 10.1152/physiolgenomics.00159.2006. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Hong Y, Han Y, Wang Y, Xia L. Chemical characterization and antioxidant activities comparison in fresh, dried, stir-frying and carbonized ginger. J Chromatogr B Analyt Tecnol Biomed Life Sci. 2016;1011:223–32. doi: 10.1016/j.jchromb.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 23.Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988;41:467–70. doi: 10.1136/jcp.41.4.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoch B, Meyer R, Hetzer R, Krause EG, Karczewski P. Identification and expression of delta-isoforms of the multifunctional Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human myocardium. Circ Res. 1999;84:713–21. doi: 10.1161/01.res.84.6.713. [DOI] [PubMed] [Google Scholar]
- 25.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–74. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maier LS. CaMKIIdelta overexpression in hypertrophy and heart failure:cellular consequences for excitation-contraction coupling. Braz J Med Biol Res. 2005;38:1293–302. doi: 10.1590/s0100-879x2005000900002. [DOI] [PubMed] [Google Scholar]
- 27.Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:468–73. doi: 10.1016/j.yjmcc.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang T, Miyamoto S, Brown JH. Cardiomyocyte calcium and calcium/calmodulin-dependent protein kinase II:friends or foes? Recent Prog Horm Res. 2004;59:141–68. doi: 10.1210/rp.59.1.141. [DOI] [PubMed] [Google Scholar]
- 29.Palomeque J, Rueda OV, Sapia L, Valverde CA, Salas M, Petroff MV, et al. Angiotensin II-induced oxidative stress resets the Ca2+dependence of Ca2+-calmodulin protein kinase II and promotes a death pathway conserved across different species. Circ Res. 2009;105:1204–12. doi: 10.1161/CIRCRESAHA.109.204172. [DOI] [PubMed] [Google Scholar]
- 30.Hagemann D, Bohlender J, Hoch B, Krause EG, Karczewski P. Expression of Ca2+/calmodulin-dependent protein kinase II delta-subunit isoforms in rats with hypertensive cardiac hypertrophy. Mol Cell Biochem. 2001;220:69–76. doi: 10.1023/a:1010899724222. [DOI] [PubMed] [Google Scholar]
- 31.Boknik P, Heinroth-Hoffmann I, Kirchhefer U, Knapp J, Linck B, Luss H, et al. Enhanced protein phosphorylation in hypertensive hypertrophy. Cardiovasc Res. 2001;51:717–28. doi: 10.1016/s0008-6363(01)00346-7. [DOI] [PubMed] [Google Scholar]
- 32.Colomer JM, Mao L, Rockman HA, Means AR. Pressure overload selectively up-regulates Ca2+/calmodulin-dependent protein kinase II in vivo. Mol Endocrinol. 2003;17:183–92. doi: 10.1210/me.2002-0350. [DOI] [PubMed] [Google Scholar]
- 33.Sasaki T, Hori H, Arai K, Hattori S, Nagai Y. Effects of a factor derived from polymorphonuclear leukocytes on the growth and collagen metabolism in normal and scleroderma skin fibroblast cultures. J Dermatol Sci. 1996;11:10–8. doi: 10.1016/0923-1811(95)00410-6. [DOI] [PubMed] [Google Scholar]
- 34.Di Gennaro A, Kenne E, Wan M, Soehnlein O, Lindbom L, Haeggstrom JZ. Leukotriene B4-induced changes in vascular permeability are mediated by neutrophil release of heparin-binding protein (HBP/CAP37/azurocidin) FASEB J. 2009;23:1750–7. doi: 10.1096/fj.08-121277. [DOI] [PubMed] [Google Scholar]
- 35.Whitfield JB, Pang D, Bucholz KK, Madden PA, Heath AC, Statham DJ, et al. Monoamine oxidase:associations with alcohol dependence, smoking and other measures of psychopathology. Psychol Med. 2000;30:443–54. doi: 10.1017/s0033291799001798. [DOI] [PubMed] [Google Scholar]
- 36.Anthenelli RM, Tipp J, Li TK, Magnes L, Schuckit MA, Rice J, et al. Platelet monoamine oxidase activity in subgroups of alcoholics and controls:results from the Collaborative Study on the Genetics of Alcoholism. Alcohol Clin Exp Res. 1998;22:598–604. doi: 10.1111/j.1530-0277.1998.tb04298.x. [DOI] [PubMed] [Google Scholar]
- 37.Kaludercic N, Mialet-Perez J, Paolocci N, Parini A, Di Lisa F. Monoamine oxidases as sources of oxidants in the heart. J Mol Cell Cardiol. 2014;73:34–42. doi: 10.1016/j.yjmcc.2013.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Himura Y, Felten SY, Kashiki M, Lewandowski TJ, Delehanty JM, Liang CS. Cardiac noradrenergic nerve terminal abnormalities in dogs with experimental congestive heart failure. Circulation. 1993;88:1299–309. doi: 10.1161/01.cir.88.3.1299. [DOI] [PubMed] [Google Scholar]
- 39.Nozawa T, Igawa A, Yoshida N, Maeda M, Inoue M, Yamamura Y, et al. Dual-tracer assessment of coupling between cardiac sympathetic neuronal function and downregulation of beta-receptors during development of hypertensive heart failure of rats. Circulation. 1998;97:2359–67. doi: 10.1161/01.cir.97.23.2359. [DOI] [PubMed] [Google Scholar]
- 40.Kaludercic N, Takimoto E, Nagayama T, Feng N, Lai EW, Bedja D, et al. Monoamine oxidase A-mediated enhanced catabolism of norepinephrine contributes to adverse remodeling and pump failure in hearts with pressure overload. Circ Res. 2010;106:193–202. doi: 10.1161/CIRCRESAHA.109.198366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Adams MA, Hirst M. Adrenal and urinary catecholamines during and after severe ethanol intoxication in rats:a profile of changes. Pharmacol Biochem Behav. 1984;21:125–31. doi: 10.1016/0091-3057(84)90141-2. [DOI] [PubMed] [Google Scholar]
- 42.Eisenhofer G, Lambie DG, Johnson RH. Effects of ethanol on plasma catecholamines and norepinephrine clearance. Clin Pharmacol Ther. 1983;34:143–7. doi: 10.1038/clpt.1983.143. [DOI] [PubMed] [Google Scholar]
- 43.Howes LG, Reid JL. Changes in plasma free 3,4-dihydroxyphenylethylene glycol and noradrenaline levels after acute alcohol administration. Clin Sci (Lond) 1985;69:423–8. doi: 10.1042/cs0690423. [DOI] [PubMed] [Google Scholar]
- 44.Gothert M, Duhrsen U, Rieckesmann JM. Ethanol, anaesthetics and other lipophilic drugs preferentially inhibit 5-hydroxytryptamine- and acetylcholine-induced noradrenaline release from sympathetic nerves. Arch Int Pharmacodyn Ther. 1979;242:196–209. [PubMed] [Google Scholar]
- 45.Gothert M, Thielecke G. Inhibition by ethanol of noradrenaline output from peripheral sympathetic nerves:possible interaction of ethanol with neuronal receptors. Eur J Pharmacol. 1976;37:321–8. doi: 10.1016/0014-2999(76)90040-6. [DOI] [PubMed] [Google Scholar]
- 46.Ilkhanizadeh B, Shirpoor A, Khadem Ansari MH, Nemati S, Rasmi Y. Protective Effects of Ginger (Zingiber officinale) Extract against Diabetes-Induced Heart Abnormality in Rats. Diabetes Metab J. 2016;40:46–53. doi: 10.4093/dmj.2016.40.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shirpoor A, Rezaei F, Fard AA, Afshari AT, Gharalari FH, Rasmi Y. Ginger extract protects rat's kidneys against oxidative damage after chronic ethanol administration. Biomed Pharmacother. 2016;84:698–704. doi: 10.1016/j.biopha.2016.09.097. [DOI] [PubMed] [Google Scholar]
- 48.Afshari A T, Shirpoor A, Farshid A, Saadatian R, Rasmi Y, Saboory E, et al. The effect of ginger on diabetic nephropathy, plasma antioxidant capacity and lipid peroxidation in rats. Food Chemistry. 2007;101:148–53. [Google Scholar]
- 49.Isa Y, Miyakawa Y, Yanagisawa M, Goto T, Kang MS, Kawada T, et al. 6-Shogaol and 6-gingerol, the pungent of ginger, inhibit TNF-alpha mediated downregulation of adiponectin expression via different mechanisms in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2008;373:429–34. doi: 10.1016/j.bbrc.2008.06.046. [DOI] [PubMed] [Google Scholar]
- 50.Hafstad AD, Nabeebaccus AA, Shah AM. Novel aspects of ROS signalling in heart failure. Basic Res Cardiol. 2013;108:359. doi: 10.1007/s00395-013-0359-8. [DOI] [PubMed] [Google Scholar]