

ABSTRACT
Despite high vaccine coverage, pertussis cases in the United States have increased over the last decade. Growing evidence suggests that disease resurgence results, in part, from genetic divergence of circulating strain populations away from vaccine references. The United States employs acellular vaccines exclusively, and current Bordetella pertussis isolates are predominantly deficient in at least one immunogen, pertactin (Prn). First detected in the United States retrospectively in a 1994 isolate, the rapid spread of Prn deficiency is likely vaccine driven, raising concerns about whether other acellular vaccine immunogens experience similar pressures, as further antigenic changes could potentially threaten vaccine efficacy. We developed an electrochemiluminescent antibody capture assay to monitor the production of the acellular vaccine immunogen filamentous hemagglutinin (Fha). Screening 722 U.S. surveillance isolates collected from 2010 to 2016 identified two that were both Prn and Fha deficient. Three additional Fha-deficient laboratory strains were also identified from a historic collection of 65 isolates dating back to 1935. Whole-genome sequencing of deficient isolates revealed putative, underlying genetic changes. Only four isolates harbored mutations to known genes involved in Fha production, highlighting the complexity of its regulation. The chromosomes of two Fha-deficient isolates included unexpected structural variation that did not appear to influence Fha production. Furthermore, insertion sequence disruption of fhaB was also detected in a previously identified pertussis toxin-deficient isolate that still produced normal levels of Fha. These results demonstrate the genetic potential for additional vaccine immunogen deficiency and underscore the importance of continued surveillance of circulating B. pertussis evolution in response to vaccine pressure.
KEYWORDS: Bordetella pertussis, Fha, filamentous hemagglutinin, pertussis, whooping cough
INTRODUCTION
Bordetella pertussis is the causative agent of whooping cough (pertussis), a highly contagious respiratory disease most severe in unvaccinated infants. The introduction of vaccines against pertussis during the 1940s dramatically reduced disease incidence in the United States. Acellular pertussis (aP) vaccines replaced whole-cell preparations in the United States during the 1990s, and despite high vaccine coverage, pertussis cases have since risen, with notable recent epidemics (1–3). Multiple factors likely contribute to increased disease reporting, including heightened awareness, expanded surveillance, and improved laboratory diagnostic testing (3, 4). There is also growing evidence to suggest that waning protection conferred by aP vaccine formulations has led to increased disease rates among vaccinated individuals (3, 5–7).
The United States exclusively uses aP vaccines for childhood series and adolescent/adult boosters, composed of inactivated pertussis toxin (Pt), pertactin (Prn), and filamentous hemagglutinin (Fha), either with or without fimbria (Fim) types 2 and 3. However, genetic divergence of the circulating population has resulted in allelic mismatches with vaccine reference strains, which may contribute to disease resurgence within aP-vaccinated populations (8, 9). Mutations to vaccine immunogens, such as Pt (subunit ptxA and the promoter ptxP) and Fim (fimH), have spread quickly throughout the circulating population with little evidence of geographic restriction (10, 11), prompting the description of B. pertussis as a monomorphic pathogen. Yet pertussis epidemics are not clonal (1, 2, 12). Circulating isolates recovered in the United States have become predominantly Prn deficient by one of at least 16 independent mutations, including missense substitutions, insertions, deletions, and promoter disruptions, but most frequently through IS481 insertion at one of three positions (13). The global emergence of Prn deficiency is well documented (13, 14) but, although likely vaccine driven (15), does not appear to impact the effectiveness of aP vaccines (16). Together, such genetic shifts illustrate how circulating B. pertussis has significantly diverged from vaccine reference strains since widespread vaccine introduction, which many researchers have attributed to vaccine-driven immune selection (4, 10, 15, 17–20).
The regulation of virulence genes in B. pertussis, including aP vaccine immunogens, is controlled by the BvgAS two-component system (21, 22). Although immunogens are included in aP vaccine formulations for their antigenic properties, knowledge of their activity during infection varies. Fha is understood to function as both a surface-associated and secreted protein (23). Numerous studies have demonstrated that Fha contributes to both colonization and persistent infection, acting not only as an adhesin to mediate respiratory epithelium attachment (23, 24) and promote biofilm formation (25, 26) but also as a modulator of immune responses (24, 27–31). Mutants lacking Fha exhibit reduced colonization (25, 26) and elicit a more robust inflammatory response, leading to faster clearance compared to the wild type (27). It should be noted that much of this knowledge is derived from experiments in animal models, and often with the closely related Bordetella bronchiseptica, which do not exhibit the full spectrum of symptoms observed in human pertussis (32, 33). While Prn appears to be unessential for disease (13, 15), Fha on the other hand may critically mediate persistent infection such that the emergence of deficient isolates would represent a remarkable shift in B. pertussis host-pathogen interaction.
The emergence and spread of immunogen-deficient isolates raises concern about the stable production of antigenic proteins, and we have previously developed electrochemiluminescent antibody capture (ECL) assays for detecting Prn and Pt (34). To further improve surveillance of circulating B. pertussis, here we modified the same assay platform to screen 787 isolates for Fha production and identify rare Fha-deficient mutants. Subsequent genomic characterization of mutant isolates revealed putative, underlying genetic changes analogous to those contributing to the rise of Prn deficiency. These results demonstrate the potential for additional, multiple-immunogen deficiency, presenting an emerging public health challenge that emphasizes the importance of continued monitoring of circulating B. pertussis for production of all vaccine immunogens.
RESULTS
Isolates deficient in Fha production.
Seven hundred twenty-two isolates collected in the United States through disease surveillance from 2010 to 2016, as well as 65 historic clinical isolates and laboratory strains from the CDC collection, were screened for Fha production using an ECL assay. Among recent circulating strains, two isolates were identified with severely reduced Fha production (J014, J199) compared to the wild type (Fig. 1A). Three historic laboratory strains (B199, J042, and J043) were also deficient for Fha (Fig. 1A). Additionally, J014 and J199 were Prn deficient (Table 1) and J043 was deficient for both Prn and Pt (Fig. 1B; see also Table S1 in the supplemental material) when screened using the same platform (34). Altered Fha production was confirmed in all five isolates by Western blotting following growth in liquid culture (Fig. 2) and on agar media (data not shown). Under both growth conditions, no Fha or cross-reacting material was detected in B199, J014, J042, or J043 while a very low concentration of protein was observed in J199, consistent with initial screening results. Deficient clinical isolates were collected from cases of infected unvaccinated children, none of which were fatal. Clinical and epidemiologically derived data for each are listed in Table 1.
FIG 1.
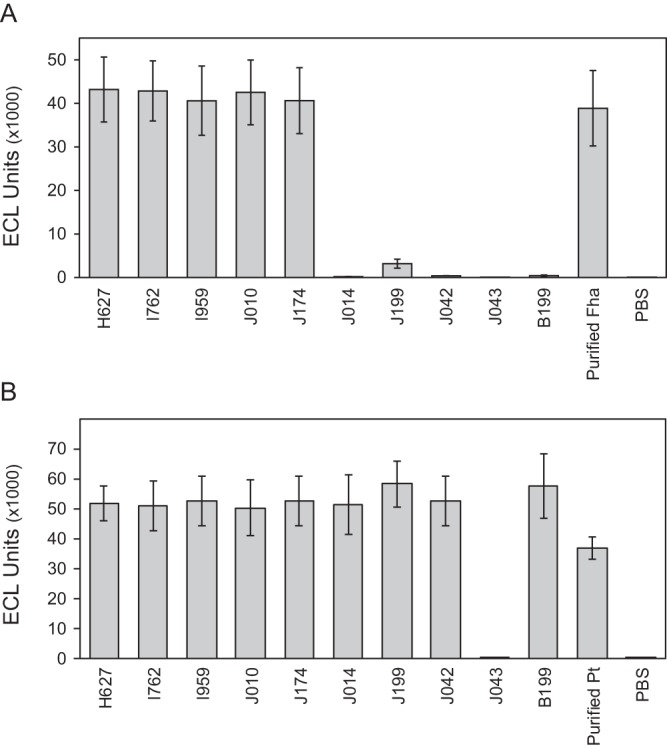
Vaccine immunogen detection by electrochemiluminescent assay. Fha (A) and Pt (B) protein production of Fha-deficient mutants J014, J199, J042, J043, and B199 compared to select Fha-producing isolates H627, I762, I959, J010, and J174 measured by secondary antibody capture. Purified protein and phosphate-buffered saline (PBS) were included for positive and negative control, respectively. Electrochemiluminescent (ECL) values are averages, and error bars represent the standard deviations for 50 replicates for Fha (A) and 18 replicates for Pt (B).
TABLE 1.
Epidemiologic data of vaccine immunogen-deficient clinical isolates
| Isolate | Phenotypec | Yr | State | Age of child | Vaccination status | Symptoms | Hospitalized |
|---|---|---|---|---|---|---|---|
| J014a | Prn-def, Fha-def | 2013 | Minnesota | <2 mo | Too young for vaccination | Paroxysmal cough, whooping cough, apnea, cyanosis | Yes |
| J199a | Prn-def, Fha-def | 2014 | Colorado | 4 yrs | Parental refusal | Paroxysmal cough, posttussive vomiting, apnea | No |
| J365 | Prn-def, Pt-defb | 2014 | California | <2 mo | Allegedly vaccinated | Paroxysmal cough | No |
FIG 2.
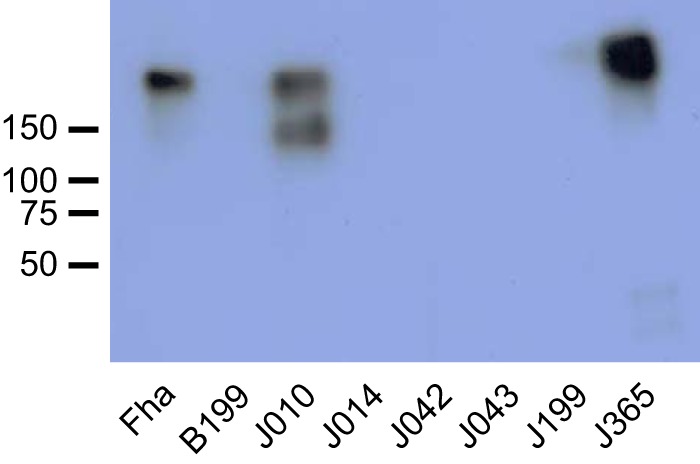
Fha detection by Western blotting. Fha production determined by ECL assay was found to be deficient in J014, J199, J042, J043, and B199 compared to Fha-producing isolates J010 and J365. Each well was loaded with 10 μg of total protein. The anti-Fha antibody recognized multiple polypeptides in J010, consistent with the frequent observation of various FhaB degradation products in B. pertussis cell extracts (28).
Mutation identification by whole-genome sequencing.
Shotgun sequencing reads from Fha-deficient isolates were mapped to the genome sequences of Fha-producing vaccine references Tohama I (E476) and 10536 to identify putative, underlying mutations. Genes encoding known proteins within the Fha biosynthesis pathway were further checked manually for nonsynonymous sequence variation or IS481 disruption. Identified mutations were confirmed to be absent in 148 Fha-producing isolates (Table S1) and are detailed in Table 2.
TABLE 2.
Characteristics, identified mutations, and genome sequence accession numbers of isolates analyzed in this studyg
| Isolate | Source (alias) | Yr isolated | PFGE profile | Detected mutations | GenBank accession no. | Reference |
|---|---|---|---|---|---|---|
| B199 | FDA (105) | 1935 | CDC059 | fhaB: IS481, pos 3124; 28 nonsynonymousb; 127-kb duplicationc; 34.5-kb duplicationc | CP022361 | This study |
| J014a | Minnesota, USA | 2014 | CDC237 | RD16_14675: del AGGCCC, pos 479 | CP012135 | This study |
| J042 | NIH (325) | 1947 | CDC232 | bipA: IS481, −88d; RD16_14485: C10T; RD16_09925: C352T; 57.5-kb duplicationc | CP019869 | This study |
| J043 | NIH (326) | 1947 | CDC372 | bvgS: ins C, pos 3312; fhaB: IS481, pos 9865; 58 nonsynonymousb; 6 IS481b | CP016887 | This study |
| J199a | Colorado, USA | 2014 | CDC237 | fhaB: del G, pos 1087; bipA: 5′-UTR C/Te | CP011245 | This study |
| J365 | California, USA | 2014 | CDC002 | 28-kb ptx-ptl delf; fhaB: IS481, pos 9865 | CP013867 | This study |
| 10536 | Sanofi vac | 1939 | CDC054 | NA | CP012128 | 37 |
| B1917 | Netherlands | 2000 | NT | NA | CP009751 | 78 |
| B1920 | Netherlands | 2000 | NT | NA | CP009752 | 78 |
| B3582 | Sweden | 2009 | NT | fhaB: ins G, pos 1087 | CP011443 | 35 |
| B3585 | Sweden | 2009 | NT | Unknown | CP011444 | 35 |
| C393 | Chinese vac | 1951 | CDC052 | NA | CP010963 | 12 |
| E476 | Tohama I | 1954 | CDC232 | NA | CP010964 | 12 |
| FR3749 | France | 2007 | CDC046 | NA | CP010966 | 45 |
| H378 | California, USA | 2010 | CDC253 | NA | CP010839 | 12 |
| H559 | California, USA | 2010 | CDC253 | NA | CP010844 | 12 |
| H622 | California, USA | 2010 | CDC217 | NA | CP010847 | 12 |
| H627 | California, USA | 2010 | CDC217 | NA | CP010962 | 12 |
| I468 | Vermont, USA | 2012 | CDC002 | NA | CP010251 | 12 |
| I472 | Vermont, USA | 2012 | CDC046 | NA | CP010253 | 12 |
| I483 | Vermont, USA | 2012 | CDC237 | NA | CP010256 | 12 |
| I518 | Vermont, USA | 2012 | CDC002 | NA | CP010259 | 12 |
| I521 | Vermont, USA | 2012 | CDC237 | NA | CP010260 | 12 |
| I762a | New York, USA | 2013 | CDC002 | NA | CP011745 | 36 |
| I959a | Oregon, USA | 2012 | CDC253 | NA | CP011746 | 36 |
| I979 | New York, USA | 2013 | CDC306 | NA | CP010966 | 45 |
| J010a | Connecticut, USA | 2013 | CDC237 | NA | CP012085 | 36 |
| J174a | Colorado, USA | 2014 | CDC237 | NA | CP013900 | 36 |
Isolates collected through Enhanced Pertussis Surveillance (61).
Details listed in Data Set S1 in the supplemental material.
Details listed in Data Set S3 in the supplemental material.
Details listed in Data Set S2 in the supplemental material.
164 bp upstream of the translational start site at position 1147474 (position 1169765 in E476).
Described by Williams et al. (45).
Abbreviations: del, deletion; ins, insertion; pos, position; vac, vaccine; UTR, untranslated region; NT, not tested; NA, not applicable.
Isolate J199 produced very little Fha, and genome sequencing revealed a single deleted G at position 1087 in fhaB, which encodes the precursor to mature Fha (Fig. 3). This deletion occurred within a homopolymeric tract, typically 10 bp long in Fha-producing isolates, causing a frameshift mutation and presumably a truncated protein product. The Swedish Fha-deficient isolate B3582 sequenced by Bart et al. (35) contained an inserted G within the same homopolymeric tract, also causing a frameshift mutation (Fig. 3). Because the PacBio RSII platform is prone to single-base insertion-deletion errors in homopolymeric tracts, Illumina reads were manually checked to confirm the deletion in 36 (64.3%) of 56 reads mapped to this locus. The genome sequence of J199 did not contain any additional disruptive IS481 insertions but did include a strain-specific single nucleotide polymorphism (SNP) in the 5′ untranslated region (5′ UTR) of the BvgAS-regulated bipA (RD16_05540 in E476 [Table 2]). The presence of opposing variation in the same homopolymeric tract within fhaB in genomes from two Fha-deficient isolates suggests that this locus is subject to strand slippage during replication.
FIG 3.

Observed mutations in fhaB. Mutations within domains of the FhaB preprotein: signal peptide (SP), two-partner secretion (TPS), mature C-terminal (MCD), N-terminal prodomain (PNT), proline-rich region (PRR), and extreme C-terminal (ECT) domains. Genomes of isolates J199, B199, and B3585 exhibited mutations that altered mature Fha, while J043 and J365 included IS481 insertion within the C-terminal prodomain. Predicted IS481 insertion target sites at positions 2785, 3124, and 9865 (RD16_09375 in E476) are indicated with red arrows.
The fhaB coding region includes three predicted IS481 target sites (36), and an insertion was observed at position 3124 in B199, disrupting the central β-helical shaft domain of mature Fha (Fig. 3). B199 is most closely related to vaccine strain 10536 (37) and differed in genome sequence by an additional 83 variants, including 28 nonsynonymous substitutions, none of which were located in genes known to be associated with Fha biosynthesis (see Data Set S1 in the supplemental material). In J043, IS481 insertion was observed in fhaB at position 9865, near the C-terminal end of the FhaB prodomain (Fig. 3). However, this insertion was also observed in J365, which produced Fha (Fig. 2), indicating that IS481 at this site does not lead to Fha deficiency.
No mutations were detected in any genes of the known Fha biosynthesis pathway in the deficient clinical isolate J014. Instead, a 6-bp in-frame deletion was identified within a gene encoding a possible alternative sigma factor (RD16_14675 in E476). This mutation was not observed in any of the 148 Fha-producing isolates and removed two amino acids adjacent to a predicted Sigma70 region 4 (SR4) DNA-binding motif (see Fig. S1 in the supplemental material). Additional annotation suggested that this sigma factor belonged to the extracytoplasmic function (ECF) class (38).
Historic laboratory strains J042 and J043 are phylogenetically related to Tohama I, and their genomes differed from that of E476 by 2 and 106 sequence variants, respectively. In J042, one nonsynonymous variant was detected in a predicted amidotransferase (RD16_14485 in E476) and the other within a hypothetical membrane protein pseudogene (RD16_09925 in E476 [Table 2]). Three additional IS481 insertions were present in J042: two adjacent to existing insertions in E476 and one within the 5′ UTR of bipA (Table 2). The insertion upstream of bipA not only disrupted known BvgA binding sites (39, 40) but also facilitated a unique ∼22-kb inversion, separating the bipA promoter and transcriptional start site from its coding region (see Fig. S1 and Data Set S2 in the supplemental material).
Historic laboratory strain J043 was deficient for Prn, Fha, and Pt. Of the observed sequence variants, 58 were predicted to cause amino acid changes or frameshift mutations in encoded proteins (Data Set S1). Among these was a single C insertion at position 3307 within a homopolymeric tract in bvgS, the central histidine kinase of the B. pertussis virulence regulon (Table 2; Fig. S1). Illumina sequencing reads mapped to this locus confirmed the insertion in 89 (87.3%) of 102 reads. The mutation to bvgS occurred between the receiver (REC) and histidine phosphotransfer (Hpt) domains, which are necessary for phosphorylation of BvgA to activate expression of Prn, Pt, and Fha. The genome of J043 also contained seven additional IS481 insertions compared to E476, including within fhaB (Data Set S1; Fig. 3). IS481 insertion was also detected in the methyl-directed mismatch repair gene mutS, the disruption of which increases mutation rates in various bacterial species (41). Insertion into mutS was not observed in any other Fha-deficient or Fha-producing isolates.
Large genome duplications and deletions.
The chromosomal structure of the B. pertussis genome is subject to IS481-mediated rearrangement (36), and the genomes of historic Fha-deficient laboratory strains B199 and J042 included large chromosomal duplications (Table 2). These duplications were not resolvable by high-throughput sequencing alone, and assembly required enzyme mapping with high-molecular-weight genomic DNA. The genome of J042 included the direct duplication of a 57.5-kb repeat, while B199 harbored a more complex repeat structure; three copies of a 34.5-kb repeat nested within one copy of a larger 127-kb duplication. Accurate assembly of the complex repeat in B199 could be determined only with the application of Nabsys HD-Mapping. The gene content within the duplications differed between the two strains, including a locus of flagellar biosynthesis in B199 and adenylate-cyclase hemolysin (ACT) in J042 (see Data Set S3 in the supplemental material). Duplications of genes in both regions have been reported previously (42–44) and were observed in a small number of Fha-producing isolates here based on high Illumina sequencing read coverage (Table S1), suggesting that neither confers Fha deficiency. Conversely, the genome of Pt-deficient isolate J365 contained a 28-kb deletion surrounding both the Pt biosynthetic (ptx) and transport (ptl) operons. The same deletion was reported in Pt-deficient isolates from New York (I979) and France (FR3749) (45). In the genomes of B199, J042, and J365, copies of IS481 were observed flanking both ends of the duplication or deletion, suggesting that these large structural mutations were the result of recombination events.
Phylogenetic and structural ancestry.
The phylogenetic placement of identified Fha-deficient B. pertussis isolates among a representative subset of circulating and reference isolates was reconstructed from 584 variable core positions using maximum parsimony (Fig. 4). Both of the recent Fha-deficient clinical isolates J014 and J199 belonged to a monophyletic clade sharing pulsed-field gel electrophoresis (PFGE) profile CDC237. All sequenced individuals in this clade share a conserved IS481 disruption of prn at position 1613 and a common chromosome structure, without rearrangement (36). Historic laboratory strains B199, J042, and J043 did not share SNP profiles or genome structures with recent circulating isolates; rather, they more closely matched the vaccine reference strains. Specifically, J042 and J043 were related to Tohama I (E476) and B199 was closest to vaccine strain 10536 (Fig. 4). Previously reported Fha-deficient Swedish isolates B3582 and B3585 were not related, by SNP phylogeny or genome structure, to deficient isolates recovered in the United States or each other. Pt-deficient isolate J365 did share a SNP phylogeny, genome structure, and prn mutation with I979, which harbors the same ptx-ptl deletion (45).
FIG 4.

SNP phylogeny and molecular typing of Fha-deficient isolates. Phylogenetic reconstruction of Fha-deficient isolates and select references from 584 variable nucleotides using maximum parsimony. Vaccine immunogen production, gene alleles, and PFGE profile are listed next to the tree as indicated in the key. Deficient isolates sequenced as part of the current study are highlighted in red. The scale bar indicates the number of substitutions per site.
DISCUSSION
In this study, we utilized an ECL assay to measure the production of the aP vaccine immunogen Fha and screened 787 isolates, many collected recently through disease surveillance. The vast majority of recent B. pertussis isolates produced Fha, consistent with existing knowledge of its central role in persistent infection, and only two deficient clinical isolates were observed. A genome sequence comparison of the Fha-deficient isolates identified here to a large collection of Fha-producing controls revealed putative underlying mutations. The results here demonstrate the potential for Fha deficiency and illustrate parallels to the recent emergence of Prn deficiency.
Mutations identified here underlying Fha deficiency included types also known to confer Prn deficiency, such as homopolymeric tract variation and IS481 insertion, indicating that both phenotypes can derive from a common mutation spectrum, as expected. Homopolymeric tracts are prone to strand slippage during replication, and changes in repeat length modulate phenotypic phase variation in diverse bacterial species (46, 47). In B. pertussis, homopolymeric tracts appear in coding regions or promoters of numerous genes, including virulence determinants, and some polymorphisms have been linked to variable expression (48, 49). J199 and Swedish isolate B3582 (35) exhibited mutations to the same homopolymeric tract within fhaB. A similar mutation to bvgS was observed in J043 and has been reported to control phenotypic switching (50). Fha deficiency resulting from such mutations may be reversible, potentially yielding a mixed population of Fha-producing and -deficient bacteria within the infected host. Although the recovery of single isolates by culture makes this impossible to determine, polymorphisms were observed among sequencing reads from J199 and J043, suggesting that reversions to the wild type were present during laboratory growth.
Genomes of B. pertussis harbor >240 copies of IS481, and approximately 250 additional target sites for insertion have been predicted, including three each within fhaB and prn (36). Insertion at any of the positions in prn constitutes a significant source of Prn deficiency, occurring repeatedly within the circulating population (13, 36). In fhaB, insertion at either of two sites present in the β-helical shaft domain would in all likelihood abolish Fha function, consistent with the observed Fha deficiency in B199. However, the third insertion site is located within the C-terminal prodomain that is degraded during secretion (23). Although deletion of this prodomain has previously been shown to not impede production of mature Fha or prevent Fha-mediated adherence of B. bronchiseptica to the mouse respiratory tract, full-length FhaB may play other roles in persistent infection (51). Therefore, while the presence of multiple target sites suggests that fhaB is vulnerable to frequent IS481 disruption, like that to prn seen in Prn deficiency, insertion may confer varied phenotypes depending on its position. The ECL assay here detected Fha production in J365, despite IS481 disruption of the C-terminal prodomain, highlighting its limitation to screening for mature Fha production only, not all FhaB-related activity.
Some Fha-deficient isolates lacked mutations in genes in the known Fha biosynthesis pathway, such as fhaB and bvgS, but did contain specific mutations elsewhere. In contrast, all identified mutations responsible for Prn deficiency occur within the prn coding region or promoter (13, 14, 36). Fha biosynthesis is complex, regulated transcriptionally by BvgAS (21, 22) and posttranslationally through a series of degradation and translocation steps (23, 24), and so additional opportunities for indirect disruption likely exist. In J014, mutation was observed to an undescribed ECF sigma factor, a small regulator from a family of proteins that often coordinate transcriptional responses to extracellular signals (38). Such regulators have previously been shown to control virulence gene expression, such as type III secretion, in Bordetella spp. (52, 53) but not Fha production. Likewise, a unique IS481 insertion in J042 was observed within the 5′ UTR of bipA, disrupting the intricate promoter architecture (39, 40). The function of BipA remains undetermined, but the predicted protein sequence includes secretion signals and surface-associated domains (54). Although transcription of both fhaB and bipA increases rapidly upon Bvg+ phase activation (55), no interaction between Fha and BipA has been reported. Mutations in this novel ECF sigma factor and bipA, whose specific functions are unknown, suggest that Fha regulation is more complex than currently understood and perhaps so too are Fha deficiency phenotypes, at least compared to Prn deficiency.
A phylogenetic reconstruction of Fha-deficient isolates corroborated that each arose through independent mutation rather than transmission of common genotypes, much like the emergence of Prn deficiency. Only recent clinical isolates J014 and J199 were closely related and belonged to a monophyletic clade that shares the predominant PFGE profile CDC237 (36). Recent whole-genome sequence comparison indicated that isolates with profile CDC237 share SNP patterns, a conserved IS481 insertion into prn, and a common chromosome structure (36). Given the rarity of Fha deficiency, it is unclear whether this genetic background facilitated the loss of Fha production or, alternatively, the shared ancestry of J014 and J199 simply reflected the higher abundance of CDC237 in the circulating U.S. population of isolates (56). Analogously, the emergence and spread of Prn deficiency has primarily occurred in the predominant genetic background defined by prn2-ptxP3-ptxA1-ptxB2 (13). Previous SNP analyses of large isolate collections have indicated that the recent genetic history of B. pertussis has been punctuated by successive selective sweeps separated by periods of allelic radiation (10, 11). Perhaps the observed independent mutations in J014 and J199 represent another period of diverging variation following the sweeping expansion of CDC237. Determining whether such mutations toward Fha deficiency result from vaccine-driven immune selection will depend on future observation of additional deficient clinical isolates.
Genome reduction has been credited with B. pertussis speciation (57) and IS481 continues to reshape the genome through deletion (58, 59), a process exemplified by J365. This isolate exhibited defects in aP vaccine immunogens Prn and Pt and potentially full-length FhaB activity, which were all acquired through successive IS481-mediated mutations. However, the application of complete genome assemblies for analyses here serves as a reminder that IS481-mediated structural variation also includes large duplications, albeit more rarely. The chromosomes of historic Fha-deficient laboratory strains B199 and J042, as well as some Fha-producing isolates, contained previously reported (42, 44, 60), yet phenotypically uncharacterized, duplication of the flagellar and ACT biosynthesis loci, respectively. Importantly, such insight into the genome evolution of circulating B. pertussis through gene deletion and duplication, potentially impacting vaccine efficacy, currently depends on bacterial culture, a diagnostic practice often omitted in favor of PCR.
Conclusions.
The rapid emergence and spread of Prn deficiency raise concerns about the stable production of other immunogenic proteins used in current aP vaccines. The ECL assay described here for Fha builds on previous applications of the same platform for measuring Prn and Pt, which together enable rapid, sensitive screening to identify deficient isolates. Clinical Fha-deficient mutants are currently rare; whether this is due to the significance of Fha for persistent infection, to the absence of selection pressure elicited by aP vaccines, or to the initially limited circulation remains unanswered, given the available data. However, their observed emergence by parallel mutation within a predominant genetic background mirrors trends underlying the recent spread of Prn deficiency, which was recognized only through retrospective analyses. These results emphasize the importance of screening for production of all aP vaccine immunogens during disease surveillance.
MATERIALS AND METHODS
Bacterial strains and characterization.
Recent U.S. Bordetella pertussis isolates screened for Fha production were collected by state public health laboratories during 2010 to 2016 either through routine surveillance and outbreaks or through the Enhanced Pertussis Surveillance/Emerging Infections Program, which combines systematic case ascertainment and augmented data collection with clinical isolate submission (61), and were forwarded to the Centers for Disease Control and Prevention (CDC) in Atlanta, GA. Historic B. pertussis clinical isolates and laboratory strains were taken from the CDC collection, dating back to 1935. Laboratory strain B199 was provided by Bruce Meade of the U.S. Food and Drug Administration (FDA). Laboratory strains J042 and J043, originally from the U.S. National Institutes of Health (NIH), were given by the University of Michigan. Basic characteristics of identified Fha-deficient isolates and select references are listed in Tables 1 and 2. Additional Fha-producing isolates sequenced for mutation identification are listed in Table S1.
Pulsed-field gel electrophoresis was performed by using restriction enzyme XbaI (62), based on the method developed by Gautom et al. (63) and similar to that currently used by U.S. state health departments participating in CDC's PulseNet for the typing of foodborne pathogens (64). PFGE patterns were compared to a database of B. pertussis isolate profiles maintained at CDC, and profiles were assigned on the basis of bands in the 125- to 450-kb range using BioNumerics v5.01 (Applied Maths, Austin, TX, USA).
Electrochemiluminescent assay.
Isolates were cultured on Regan-Lowe agar without cephalexin for 72 h at 37°C. Cells were collected and suspended in 0.01 M phosphate-buffered saline (PBS) to a turbidity of 1.0, measured using a Dade Behring MicroScan Turbidity Meter (Siemens Global, Munich, Germany). Cells were pelleted by centrifugation at 10,000 × g and resuspended in Xtractor lysis buffer (Clontech, California, USA) containing 25 U of Benzonase (Sigma-Aldrich, Missouri, USA) at a 1:1 ratio (vol/vol). The mixture was vortexed gently for 10 min at room temperature and centrifuged, and lysis buffer supernatant was discarded. Cell lysate pellets were resuspended in PBS for analysis.
The electrochemiluminescent antibody capture (ECL) assay was performed as previously described (34), using the Sector S6000 platform (Meso-Scale Discovery, Maryland, USA). Briefly, 96-well plates (Meso-Scale Discovery) were coated with 30 μl of capture sheep Fha antiserum (NIBSC, UK), at a 1/33,000 working dilution and incubated overnight at 4°C. Plates were washed with PBS containing 0.05% Tween 20 and blocked with PBS containing 5% dry skim milk powder (Fisher Scientific, Pennsylvania, USA), 2% goat serum (Sigma-Aldrich, Missouri, USA), and 2% Blocker A (Meso-Scale Discovery). The blocking solution was discarded and replaced with 25 μl cell lysate. Plates were washed again, and detection was done with 25 μl of mouse anti-Fha detection antibody (NIBSC, United Kingdom), at a 1/5,000 dilution. Goat anti-mouse Sulfotag secondary antibody (Meso-Scale Discovery) was then added at 1 μg/ml. Luminescence was measured with a Sector Reader (Meso-Scale Discovery). Purified Fha protein (GlaxoSmithKline, UK) at 1 μg/ml was used as a positive control. A 2-fold dilution series with 2 μg/ml purified Fha was performed and fit with a four-parameter logistic (4-PL) regression using the instrument software to confirm the absence of nonspecific cross-reactivity. Isolates were qualitatively defined as either Fha producing or Fha deficient based on their signal intensity compared to positive and negative controls measured on the same plate to account for variability between plates (34). For any isolate producing <50% signal intensity compared to the control, the assay was repeated for confirmation.
Western blotting.
Isolates were cultured on agar medium and prepared in a manner similar to that described above. However, final cell lysate pellets were instead resuspended in SDS Sample Buffer (Bio-Rad, California, USA) and both the lysis buffer supernatant and pellet suspension were used for SDS-PAGE. Cells from Regan-Lowe agar plates were also inoculated into Stainer-Scholte broth (65) and incubated at 37°C to an optical density at 650 nm (OD650) of approximately 2.3. Liquid cultures were then diluted 1:100 into fresh medium, incubated at 37°C to OD650 of 0.5 to 1.0, and centrifuged at 10,000 × g. Liquid culture cell pellets were lysed in the same manner as described above, while potential proteins remaining in the broth supernatant were precipitated by addition of 100% (wt/vol) trichloroacetic acid (TCA) according to Green and Sambrook (66). TCA was removed by centrifugation at 10,000 × g, and the pellet was washed three times with acetone before resuspension in SDS Sample Buffer. Sample preparations derived from the lysis buffer supernatant, cell pellet, and liquid culture broth precipitate were all used for SDS-PAGE.
The protein concentrations of sample preparations were quantified using a Nanodrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., Wilmington, DE). Ten micrograms of protein was denatured by boiling for 5 min in SDS Sample Buffer, loaded to a 4 to 20% SDS polyacrylamide gradient gel (Bio-Rad, California, USA), and run for 1 h at 200 V. Separated proteins were transferred to a polyvinylidene difluoride (PVDF) membrane using the Mini Trans-Blot Cell (Bio-Rad). The ∼220-kDa Fha protein was detected using a mouse anti-Fha antibody (NIBSC, UK) at a 1:1,000 dilution in nonfat dry milk (5%) followed by 1:5,000 horseradish peroxidase (HRP)-conjugated anti-mouse antibody. The blots were applied to Clarity Wester chemiluminescent (Bio-Rad) substrate, and images were captured using an X-OMAT 2000A (Kodak, New York, USA).
Genomic DNA preparation.
Isolates were cultured on Regan-Lowe agar without cephalexin for 72 h at 37°C. Genomic DNA isolation and purification were conducted according to the Qiagen Gentra Puregene Yeast/Bacteria kit's standard protocol with slight modification (Qiagen, Valencia, CA). Briefly, two aliquots of approximately 1 × 109 bacterial cells were harvested and resuspended in 500 μl of 0.85% sterile saline and then pelleted by centrifugation for 1 min at 16,000 × g. Recovered genomic DNA was resuspended in 100 μl of DNA hydration solution. Aliquots were quantified and qualified using a Nanodrop 2000 instrument (Thermo Fisher Scientific Inc., Wilmington, DE).
Genome sequencing and assembly.
Whole-genome shotgun sequencing was performed using a combination of the PacBio RSII (Pacific Biosciences, Menlo Park, CA), Illumina HiSeq/MiSeq (Illumina, San Diego, CA), and Argus (OpGen, Gaithersburg, MD) platforms as described previously (12). Briefly, genomic DNA libraries were prepared for PacBio sequencing runs using the SMRTbell Template Prep kit 1.0 and Polymerase Binding kit P4 or P6, while Illumina libraries were prepared using the NEB Ultra Library Prep kit (New England BioLabs, Ipswich, MA). De novo genome assembly of filtered reads was performed using the Hierarchical Genome Assembly Process (HGAP, v3; Pacific Biosciences) (67) and, when necessary, the A-Hybrid-Assembly workflow (AHA; Pacific Biosciences). The resulting consensus sequences were determined with Quiver (v1), manually checked for circularity, and then reordered to start at the coding region for glucose-inhibited cell division protein A (gidA), consistent with the available genome sequences of B. pertussis. Assemblies were confirmed by comparison to restriction digest optical maps using the Argus system (OpGen) with MapSolver (v.2.1.1; OpGen). Final PacBio assemblies were further polished by mapping either Illumina HiSeq PE-100, MiSeq PE-150, or MiSeq PE-300 reads using CLC Genomics Workbench (v10.0.1; CLC bio, Boston, MA) and annotated using the NCBI automated Prokaryotic Genome Annotation Pipeline (PGAP). Whole-genome shotgun sequencing of Fha-producing isolates was performed using Illumina HiSeq/MiSeq only, and reads were quality trimmed and filtered with CLC Genomics Workbench (CLC bio) as described previously (12).
Duplication assembly in J042 was resolved manually with the aid of BridgeMapper (v1; Pacific Biosiences) and MapSolver (OpGen). The more complex duplication in B199 was manually assembled based on Nabsys HD-Mapping (Nabsys 2.0 LLC, Providence, RI). Following culture as described above, genomic DNA isolation was performed at CDC according to a Nabsys solution-based protocol modified from the bacterial DNA protocol for AXG 20 columns and the NucleoBond Buffer Set III (Macherey-Nagel, Bethlehem, PA). Briefly, modifications to reduce DNA shearing included increasing lysate incubation to 60°C for 60 min, reducing vortex mixing to 5 s, and using wide-bore pipette tips. Purified DNA was sent to Nabsys for nicking, tagging, coating, and processing on an HD-Mapping instrument using the enzymes Nb.BsmI, Nt.Bpu10I, and Nt.BspQI. The resulting de novo-assembled HD maps, raw data, and data remapped to PacBio de novo assemblies were provided by Nabsys for further analyses and sequence assembly confirmation at CDC with NPS Analysis (v1.2.1661; Nabsys). Duplication assembly in both J042 and B199 was further confirmed by successfully mapping filtered PacBio sequencing reads of >10 kb across each of the novel duplication boundaries using BLASR (v1) (68) and visualized with SMRT View (v2.3.0; Pacific Biosciences).
Genome sequence and structure variation.
Phylogenetic reconstruction of assembled genomes and related references was calculated using kSNP3 (69), with k of 23 and maximum parsimony of core SNPs after masking the pertactin gene (prn) and all IS element sequences with ‘N′s. Internal nodes with <50% bootstrap support were collapsed into multifurcations in Archaeopteryx v0.9901 (70), and tree annotation was performed with iTOL v3.0 (71). Variation in genome structure was detected by pairwise alignment of assembled genomes with closely related reference strains using progressiveMauve (72) with optimized parameters (−seed-weight = 16; −hmm-identity = 0.85).
Sequence variation was detected by mapping filtered Illumina reads from Fha-deficient and Fha-producing isolates to the reference strain E476 (CP010964) using the Basic Variant Detection tool in CLC Genomics Workbench (CLC bio) as described previously (12). Mutations putatively responsible for disrupting Fha production were identified by comparing the variant profile of each Fha-deficient mutant to those found in 148 Fha-producing isolates. Predicted proteins containing nonsynonymous mutations were further annotated with a betaproteobacterium-specific subset of EggNOG v4.1 (73) using HMMER v3.1bs (http://hmmer.org) as well as manual query of Swiss-Prot (74) and the Conserved Domain Database (75) using Delta-BLAST (76) via the NCBI web interface.
Mutations to the pertactin gene were identified in assembled genomes by alignment to a curated set of previously reported wild-type and deficient alleles (13) using BLASTn (77) with cutoffs of minimum sequence identity of 100% and query coverage of >99%. Alleles of fimH (fim3) were assigned in a similar manner.
Accession number(s).
The complete genome sequences determined in this study have been deposited at DDBJ/EMBL/GenBank under the accession numbers CP011245, CP012135, CP013867, CP016887, CP019869, and CP022361. The versions described in this paper are the first versions. Raw sequencing data have been deposited under the accession numbers listed in Table S1 in the supplemental material, organized under a BioProject with accession number PRJNA279196.
Supplementary Material
ACKNOWLEDGMENTS
We thank Christine Miner (Centers for Disease Control and Prevention) for technical assistance, Bruce Meade (U.S. Food and Drug Administration), Elizabeth Levin (University of Michigan), the California Department of Public Health Laboratory, the Minnesota Public Health Laboratory Division, Wadsworth Center New York State Department of Health, the Colorado Department of Public Health and Environment, and the Connecticut Department of Public Health for contributing isolates, and Nabsys for conducting HD mapping of B199 for genome repeat assembly and confirmation.
This work was supported by internal funds.
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Use of trade names and commercial sources is for identification only and does not imply endorsement by the Centers for Disease Control and Prevention, the Public Health Service, or the U.S. Department of Health and Human Services.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00869-17.
REFERENCES
- 1.Bowden KE, Williams MM, Cassiday PK, Milton A, Pawloski L, Harrison M, Martin SW, Meyer S, Qin X, DeBolt C, Tasslimi A, Syed N, Sorrell R, Tran M, Hiatt B, Tondella ML. 2014. Molecular epidemiology of the pertussis epidemic in Washington State in 2012. J Clin Microbiol 52:3549–3557. doi: 10.1128/JCM.01189-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Winter K, Harriman K, Zipprich J, Schechter R, Talarico J, Watt J, Chavez G. 2012. California pertussis epidemic, 2010. J Pediatr 161:1091–1096. doi: 10.1016/j.jpeds.2012.05.041. [DOI] [PubMed] [Google Scholar]
- 3.Clark TA. 2014. Changing pertussis epidemiology: everything old is new again. J Infect Dis 209:978–981. doi: 10.1093/infdis/jiu001. [DOI] [PubMed] [Google Scholar]
- 4.Ausiello CM, Cassone A. 2014. Acellular pertussis vaccines and pertussis resurgence: revise or replace? mBio 5:e01339-. doi: 10.1128/mBio.01339-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Warfel JM, Edwards KM. 2015. Pertussis vaccines and the challenge of inducing durable immunity. Curr Opin Immunol 35:48–54. doi: 10.1016/j.coi.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 6.Klein NP, Bartlett J, Rowhani-Rahbar A, Fireman B, Baxter R. 2012. Waning protection after fifth dose of acellular pertussis vaccine in children. N Engl J Med 367:1012–1019. doi: 10.1056/NEJMoa1200850. [DOI] [PubMed] [Google Scholar]
- 7.Misegades LK, Winter K, Harriman K, Talarico J, Messonnier NE, Clark TA, Martin SW. 2012. Association of childhood pertussis with receipt of 5 doses of pertussis vaccine by time since last vaccine dose, California, 2010. JAMA 308:2126–2132. doi: 10.1001/jama.2012.14939. [DOI] [PubMed] [Google Scholar]
- 8.Komatsu E, Yamaguchi F, Abe A, Weiss AA, Watanabe M. 2010. Synergic effect of genotype changes in pertussis toxin and pertactin on adaptation to an acellular pertussis vaccine in the murine intranasal challenge model. Clin Vaccine Immunol 17:807–812. doi: 10.1128/CVI.00449-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bottero D, Gaillard ME, Fingermann M, Weltman G, Fernandez J, Sisti F, Graieb A, Roberts R, Rico O, Rios G, Regueira M, Binsztein N, Hozbor D. 2007. Pulsed-field gel electrophoresis, pertactin, pertussis toxin S1 subunit polymorphisms, and surfaceome analysis of vaccine and clinical Bordetella pertussis strains. Clin Vaccine Immunol 14:1490–1498. doi: 10.1128/CVI.00177-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bart MJ, Harris SR, Advani A, Arakawa Y, Bottero D, Bouchez V, Cassiday PK, Chiang CS, Dalby T, Fry NK, Gaillard ME, van Gent M, Guiso N, Hallander HO, Harvill ET, He Q, van der Heide HG, Heuvelman K, Hozbor DF, Kamachi K, Karataev GI, Lan R, Lutynska A, Maharjan RP, Mertsola J, Miyamura T, Octavia S, Preston A, Quail MA, Sintchenko V, Stefanelli P, Tondella ML, Tsang RS, Xu Y, Yao SM, Zhang S, Parkhill J, Mooi FR. 2014. Global population structure and evolution of Bordetella pertussis and their relationship with vaccination. mBio 5:e01074-. doi: 10.1128/mBio.01074-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Gent M, Bart MJ, van der Heide HG, Heuvelman KJ, Mooi FR. 2012. Small mutations in Bordetella pertussis are associated with selective sweeps. PLoS One 7:e46407. doi: 10.1371/journal.pone.0046407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bowden KE, Weigand MR, Peng Y, Cassiday PK, Sammons S, Knipe K, Rowe LA, Loparev V, Sheth M, Weening K, Tondella ML, Williams MM. 2016. Genome structural diversity among 31 Bordetella pertussis isolates from two recent U.S. whooping cough statewide epidemics. mSphere 1:e00036-. doi: 10.1128/mSphere.00036-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pawloski LC, Queenan AM, Cassiday PK, Lynch AS, Harrison MJ, Shang W, Williams MM, Bowden KE, Burgos-Rivera B, Qin X, Messonnier N, Tondella ML. 2014. Prevalence and molecular characterization of pertactin-deficient Bordetella pertussis in the United States. Clin Vaccine Immunol 21:119–125. doi: 10.1128/CVI.00717-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lam C, Octavia S, Ricafort L, Sintchenko V, Gilbert GL, Wood N, McIntyre P, Marshall H, Guiso N, Keil AD, Lawrence A, Robson J, Hogg G, Lan R. 2014. Rapid increase in pertactin-deficient Bordetella pertussis isolates, Australia. Emerg Infect Dis 20:626–633. doi: 10.3201/eid2004.131478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin SW, Pawloski L, Williams M, Weening K, DeBolt C, Qin X, Reynolds L, Kenyon C, Giambrone G, Kudish K, Miller L, Selvage D, Lee A, Skoff TH, Kamiya H, Cassiday PK, Tondella ML, Clark TA. 2015. Pertactin-negative Bordetella pertussis strains: evidence for a possible selective advantage. Clin Infect Dis 60:223–227. doi: 10.1093/cid/ciu788. [DOI] [PubMed] [Google Scholar]
- 16.Breakwell L, Kelso P, Finley C, Schoenfeld S, Goode B, Misegades LK, Martin SW, Acosta AM. 2016. Pertussis vaccine effectiveness in the setting of pertactin-deficient pertussis. Pediatrics 137(5):e20153973. doi: 10.1542/peds.2015-3973. [DOI] [PubMed] [Google Scholar]
- 17.Kallonen T, Grondahl-Yli-Hannuksela K, Elomaa A, Lutynska A, Fry NK, Mertsola J, He Q. 2011. Differences in the genomic content of Bordetella pertussis isolates before and after introduction of pertussis vaccines in four European countries. Infect Genet Evol 11:2034–2042. doi: 10.1016/j.meegid.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 18.Octavia S, Maharjan RP, Sintchenko V, Stevenson G, Reeves PR, Gilbert GL, Lan R. 2011. Insight into evolution of Bordetella pertussis from comparative genomic analysis: evidence of vaccine-driven selection. Mol Biol Evol 28:707–715. doi: 10.1093/molbev/msq245. [DOI] [PubMed] [Google Scholar]
- 19.Sealey KL, Harris SR, Fry NK, Hurst LD, Gorringe AR, Parkhill J, Preston A. 2015. Genomic analysis of isolates from the United Kingdom 2012 pertussis outbreak reveals that vaccine antigen genes are unusually fast evolving. J Infect Dis 212:294–301. doi: 10.1093/infdis/jiu665. [DOI] [PubMed] [Google Scholar]
- 20.Xu Y, Liu B, Grondahl-Yli-Hannuksila K, Tan Y, Feng L, Kallonen T, Wang L, Peng D, He Q, Wang L, Zhang S. 2015. Whole-genome sequencing reveals the effect of vaccination on the evolution of Bordetella pertussis. Sci Rep 5:12888. doi: 10.1038/srep12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cummings CA, Bootsma HJ, Relman DA, Miller JF. 2006. Species- and strain-specific control of a complex, flexible regulon by Bordetella BvgAS. J Bacteriol 188:1775–1785. doi: 10.1128/JB.188.5.1775-1785.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Decker KB, James TD, Stibitz S, Hinton DM. 2012. The Bordetella pertussis model of exquisite gene control by the global transcription factor BvgA. Microbiology 158:1665–1676. doi: 10.1099/mic.0.058941-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheller EV, Cotter PA. 2015. Bordetella filamentous hemagglutinin and fimbriae: critical adhesins with unrealized vaccine potential. Pathog Dis 73:ftv079. doi: 10.1093/femspd/ftv079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villarino Romero R, Osicka R, Sebo P. 2014. Filamentous hemagglutinin of Bordetella pertussis: a key adhesin with immunomodulatory properties? Future Microbiol 9:1339–1360. doi: 10.2217/fmb.14.77. [DOI] [PubMed] [Google Scholar]
- 25.Cattelan N, Dubey P, Arnal L, Yantorno OM, Deora R. 2016. Bordetella biofilms: a lifestyle leading to persistent infections. Pathog Dis 74:ftv108. doi: 10.1093/femspd/ftv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serra DO, Conover MS, Arnal L, Sloan GP, Rodriguez ME, Yantorno OM, Deora R. 2011. FHA-mediated cell-substrate and cell-cell adhesions are critical for Bordetella pertussis biofilm formation on abiotic surfaces and in the mouse nose and the trachea. PLoS One 6:e28811. doi: 10.1371/journal.pone.0028811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henderson MW, Inatsuka CS, Sheets AJ, Williams CL, Benaron DJ, Donato GM, Gray MC, Hewlett EL, Cotter PA. 2012. Contribution of Bordetella filamentous hemagglutinin and adenylate cyclase toxin to suppression and evasion of interleukin-17-mediated inflammation. Infect Immun 80:2061–2075. doi: 10.1128/IAI.00148-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inatsuka CS, Julio SM, Cotter PA. 2005. Bordetella filamentous hemagglutinin plays a critical role in immunomodulation, suggesting a mechanism for host specificity. Proc Natl Acad Sci U S A 102:18578–18583. doi: 10.1073/pnas.0507910102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abramson T, Kedem H, Relman DA. 2008. Modulation of the NF-kappaB pathway by Bordetella pertussis filamentous hemagglutinin. PLoS One 3:e3825. doi: 10.1371/journal.pone.0003825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asgarian-Omran H, Amirzargar AA, Zeerleder S, Mahdavi M, van Mierlo G, Solati S, Jeddi-Tehrani M, Rabbani H, Aarden L, Shokri F. 2015. Interaction of Bordetella pertussis filamentous hemagglutinin with human TLR2: identification of the TLR2-binding domain. APMIS 123:156–162. doi: 10.1111/apm.12332. [DOI] [PubMed] [Google Scholar]
- 31.Dieterich C, Relman DA. 2011. Modulation of the host interferon response and ISGylation pathway by B. pertussis filamentous hemagglutinin. PLoS One 6:e27535. doi: 10.1371/journal.pone.0027535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elahi S, Holmstrom J, Gerdts V. 2007. The benefits of using diverse animal models for studying pertussis. Trends Microbiol 15:462–468. doi: 10.1016/j.tim.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 33.van der Ark AA, Hozbor DF, Boog CJ, Metz B, van den Dobbelsteen GP, van Els CA. 2012. Resurgence of pertussis calls for re-evaluation of pertussis animal models. Expert Rev Vaccines 11:1121–1137. doi: 10.1586/erv.12.83. [DOI] [PubMed] [Google Scholar]
- 34.Gates I, DuVall M, Ju H, Tondella ML, Pawloski L, Pertussis Working G. 2017. Development of a qualitative assay for screening of Bordetella pertussis isolates for pertussis toxin production. PLoS One 12:e0175326. doi: 10.1371/journal.pone.0175326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bart MJ, van der Heide HG, Zeddeman A, Heuvelman K, van Gent M, Mooi FR. 2015. Complete genome sequences of 11 Bordetella pertussis strains representing the pandemic ptxP3 lineage. Genome Announc 3(6):e01394-. doi: 10.1128/genomeA.01394-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weigand MR, Peng Y, Loparev V, Batra D, Bowden KE, Burroughs M, Cassiday PK, Davis JK, Johnson T, Juieng P, Knipe K, Mathis MH, Pruitt AM, Rowe L, Sheth M, Tondella ML, Williams MM. 2017. The history of Bordetella pertussis genome evolution includes structural rearrangement. J Bacteriol 199:e00806-16. doi: 10.1128/JB.00806-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weigand MR, Peng Y, Loparev V, Batra D, Burroughs M, Johnson T, Juieng P, Rowe LA, Tondella ML, Williams MM. 2016. Complete genome sequences of Bordetella pertussis vaccine reference strains 134 and 10536. Genome Announc 4(5):e00979-16. doi: 10.1128/genomeA.00979-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mascher T. 2013. Signaling diversity and evolution of extracytoplasmic function (ECF) sigma factors. Curr Opin Microbiol 16:148–155. doi: 10.1016/j.mib.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Deora R. 2004. Multiple mechanisms of bipA gene regulation by the Bordetella BvgAS phosphorelay system. Trends Microbiol 12:63–65. doi: 10.1016/j.tim.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Williams CL, Boucher PE, Stibitz S, Cotter PA. 2005. BvgA functions as both an activator and a repressor to control Bvg phase expression of bipA in Bordetella pertussis. Mol Microbiol 56:175–188. doi: 10.1111/j.1365-2958.2004.04526.x. [DOI] [PubMed] [Google Scholar]
- 41.Sundin GW, Weigand MR. 2007. The microbiology of mutability. FEMS Microbiol Lett 277:11–20. doi: 10.1111/j.1574-6968.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- 42.Caro V, Hot D, Guigon G, Hubans C, Arrive M, Soubigou G, Renauld-Mongenie G, Antoine R, Locht C, Lemoine Y, Guiso N. 2006. Temporal analysis of French Bordetella pertussis isolates by comparative whole-genome hybridization. Microbes Infect 8:2228–2235. doi: 10.1016/j.micinf.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 43.Dalet K, Weber C, Guillemot L, Njamkepo E, Guiso N. 2004. Characterization of adenylate cyclase-hemolysin gene duplication in a Bordetella pertussis isolate. Infect Immun 72:4874–4877. doi: 10.1128/IAI.72.8.4874-4877.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weigand MR, Peng Y, Loparev V, Johnson T, Juieng P, Gairola S, Kumar R, Shaligram U, Gowrishankar R, Moura H, Rees J, Schieltz DM, Williamson Y, Woolfitt A, Barr J, Tondella ML, Williams MM. 2016. Complete genome sequences of four Bordetella pertussis vaccine reference strains from Serum Institute of India. Genome Announc 4(6):e01404-. doi: 10.1128/genomeA.01404-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Williams MM, Sen K, Weigand MR, Skoff TH, Cunningham VA, Halse TA, Tondella ML, CDC Pertussis Working Group. 2016. Bordetella pertussis strain lacking pertactin and pertussis toxin. Emerg Infect Dis 22:319–322. doi: 10.3201/eid2202.151332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henderson IR, Owen P, Nataro JP. 1999. Molecular switches–the ON and OFF of bacterial phase variation. Mol Microbiol 33:919–932. doi: 10.1046/j.1365-2958.1999.01555.x. [DOI] [PubMed] [Google Scholar]
- 47.Orsi RH, Bowen BM, Wiedmann M. 2010. Homopolymeric tracts represent a general regulatory mechanism in prokaryotes. BMC Genomics 11:102. doi: 10.1186/1471-2164-11-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gogol EB, Cummings CA, Burns RC, Relman DA. 2007. Phase variation and microevolution at homopolymeric tracts in Bordetella pertussis. BMC Genomics 8:122. doi: 10.1186/1471-2164-8-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vaughan TE, Pratt CB, Sealey K, Preston A, Fry NK, Gorringe AR. 2014. Plasticity of fimbrial genotype and serotype within populations of Bordetella pertussis: analysis by paired flow cytometry and genome sequencing. Microbiology 160:2030–2044. doi: 10.1099/mic.0.079251-0. [DOI] [PubMed] [Google Scholar]
- 50.Stibitz S, Aaronson W, Monack D, Falkow S. 1989. Phase variation in Bordetella pertussis by frameshift mutation in a gene for a novel two-component system. Nature 338:266–269. doi: 10.1038/338266a0. [DOI] [PubMed] [Google Scholar]
- 51.Melvin JA, Scheller EV, Noel CR, Cotter PA. 2015. New insight into filamentous hemagglutinin secretion reveals a role for full-length FhaB in Bordetella virulence. mBio 6(4):e01189-. doi: 10.1128/mBio.01189-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ahuja U, Shokeen B, Cheng N, Cho Y, Blum C, Coppola G, Miller JF. 2016. Differential regulation of type III secretion and virulence genes in Bordetella pertussis and Bordetella bronchiseptica by a secreted anti-sigma factor. Proc Natl Acad Sci U S A 113:2341–2348. doi: 10.1073/pnas.1600320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mattoo S, Yuk MH, Huang LL, Miller JF. 2004. Regulation of type III secretion in Bordetella. Mol Microbiol 52:1201–1214. doi: 10.1111/j.1365-2958.2004.04053.x. [DOI] [PubMed] [Google Scholar]
- 54.Stockbauer KE, Fuchslocher B, Miller JF, Cotter PA. 2001. Identification and characterization of BipA, a Bordetella Bvg-intermediate phase protein. Mol Microbiol 39:65–78. doi: 10.1046/j.1365-2958.2001.02191.x. [DOI] [PubMed] [Google Scholar]
- 55.Mattoo S, Cherry JD. 2005. Molecular pathogenesis, epidemiology, and clinical manifestations of respiratory infections due to Bordetella pertussis and other Bordetella subspecies. Clin Microbiol Rev 18:326–382. doi: 10.1128/CMR.18.2.326-382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cassiday PK, Skoff TH, Jawahir S, Tondella ML. 2016. Changes in predominance of pulsed-field gel electrophoresis profiles of Bordetella pertussis isolates, United States, 2000–2012. Emerg Infect Dis 22:442–448. doi: 10.3201/eid2203.151136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parkhill J, Sebaihia M, Preston A, Murphy LD, Thomson N, Harris DE, Holden MT, Churcher CM, Bentley SD, Mungall KL, Cerdeno-Tarraga AM, Temple L, James K, Harris B, Quail MA, Achtman M, Atkin R, Baker S, Basham D, Bason N, Cherevach I, Chillingworth T, Collins M, Cronin A, Davis P, Doggett J, Feltwell T, Goble A, Hamlin N, Hauser H, Holroyd S, Jagels K, Leather S, Moule S, Norberczak H, O'Neil S, Ormond D, Price C, Rabbinowitsch E, Rutter S, Sanders M, Saunders D, Seeger K, Sharp S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Unwin L, Whitehead S, Barrell BG, Maskell DJ. 2003. Comparative analysis of the genome sequences of Bordetella pertussis, Bordetella parapertussis and Bordetella bronchiseptica. Nat Genet 35:32–40. doi: 10.1038/ng1227. [DOI] [PubMed] [Google Scholar]
- 58.Bouchez V, Caro V, Levillain E, Guigon G, Guiso N. 2008. Genomic content of Bordetella pertussis clinical isolates circulating in areas of intensive children vaccination. PLoS One 3:e2437. doi: 10.1371/journal.pone.0002437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lam C, Octavia S, Sintchenko V, Gilbert GL, Lan R. 2014. Investigating genome reduction of Bordetella pertussis using a multiplex PCR-based reverse line blot assay (mPCR/RLB). BMC Res Notes 7:727. doi: 10.1186/1756-0500-7-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heikkinen E, Kallonen T, Saarinen L, Sara R, King AJ, Mooi FR, Soini JT, Mertsola J, He Q. 2007. Comparative genomics of Bordetella pertussis reveals progressive gene loss in Finnish strains. PLoS One 2:e904. doi: 10.1371/journal.pone.0000904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Skoff TH, Baumbach J, Cieslak PR. 2015. Tracking pertussis and evaluating control measures through enhanced pertussis surveillance, emerging infections program, United States. Emerg Infect Dis 21:1568–1573. doi: 10.3201/eid2109.150023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hardwick TH, Cassiday P, Weyant RS, Bisgard KM, Sanden GN. 2002. Changes in predominance and diversity of genomic subtypes of Bordetella pertussis isolated in the United States, 1935 to 1999. Emerg Infect Dis 8:44–49. doi: 10.3201/eid0801.010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gautom RK. 1997. Rapid pulsed-field gel electrophoresis protocol for typing of Escherichia coli O157:H7 and other gram-negative organisms in 1 day. J Clin Microbiol 35:2977–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Swaminathan B, Barrett TJ, Hunter SB, Tauxe RV, CDC PulseNet Task Force. 2001. PulseNet: the molecular subtyping network for foodborne bacterial disease surveillance, United States. Emerg Infect Dis 7:382–389. doi: 10.3201/eid0703.017303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hulbert RR, Cotter PA. 2009. Laboratory maintenance of Bordetella pertussis. Curr Protoc Microbiol Chapter 4:Unit 4B.1. doi: 10.1002/9780471729259.mc04b01s15. [DOI] [PubMed] [Google Scholar]
- 66.Green MR, Sambrook J, Sambrook J. 2012. Molecular cloning: a laboratory manual, 4th ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 67.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, Clum A, Copeland A, Huddleston J, Eichler EE, Turner SW, Korlach J. 2013. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10:563–569. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 68.Chaisson MJ, Tesler G. 2012. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13:238. doi: 10.1186/1471-2105-13-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gardner SN, Slezak T, Hall BG. 2015. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31:2877–2878. doi: 10.1093/bioinformatics/btv271. [DOI] [PubMed] [Google Scholar]
- 70.Han MV, Zmasek CM. 2009. phyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics 10:356. doi: 10.1186/1471-2105-10-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Letunic I, Bork P. 2016. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44(W1):W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Powell S, Forslund K, Szklarczyk D, Trachana K, Roth A, Huerta-Cepas J, Gabaldon T, Rattei T, Creevey C, Kuhn M, Jensen LJ, von Mering C, Bork P. 2014. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res 42:D231–D239. doi: 10.1093/nar/gkt1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.UniProt Consortium. 2015. UniProt: a hub for protein information. Nucleic Acids Res 43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH. 2015. CDD: NCBI's conserved domain database. Nucleic Acids Res 43:D222–D226. doi: 10.1093/nar/gku1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boratyn GM, Schaffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL. 2012. Domain enhanced lookup time accelerated BLAST. Biol Direct 7:12. doi: 10.1186/1745-6150-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bart MJ, Zeddeman A, van der Heide HG, Heuvelman K, van Gent M, Mooi FR. 2014. Complete genome sequences of Bordetella pertussis isolates B1917 and B1920, representing two predominant global lineages. Genome Announc 2(6):e01301-14. doi: 10.1128/genomeA.01301-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.