

ABSTRACT
CD8+ T cells are the major effector cells that protect against malaria liver-stage infection, forming clusters around Plasmodium-infected hepatocytes and eliminating parasites after a prolonged interaction with these hepatocytes. We aimed to investigate the roles of specific and nonspecific CD8+ T cells in cluster formation and protective immunity. To this end, we used Plasmodium berghei ANKA expressing ovalbumin as well as CD8+ T cells from transgenic mice expressing a T cell receptor specific for ovalbumin (OT-I) and CD8+ T cells specific for an unrelated antigen, respectively. While antigen-specific CD8+ T cells were essential for cluster formation, both antigen-specific and nonspecific CD8+ T cells joined the clusters. However, nonspecific CD8+ T cells did not significantly contribute to protective immunity. In the livers of infected mice, specific CD8+ T cells expressed high levels of CD25, compatible with a local, activated effector phenotype. In vivo imaging of the liver revealed that specific CD8+ T cells interact with CD11c+ cells around infected hepatocytes. The depletion of CD11c+ cells virtually eliminated the clusters in the liver, leading to a significant decrease in protection. These experiments reveal an essential role of hepatic CD11c+ dendritic cells and presumably macrophages in the formation of CD8+ T cell clusters around Plasmodium-infected hepatocytes. Once cluster formation is triggered by parasite-specific CD8+ T cells, specific and unrelated activated CD8+ T cells join the clusters in a chemokine- and dendritic cell-dependent manner. Nonspecific CD8+ T cells seem to play a limited role in protective immunity against Plasmodium parasites.
KEYWORDS: CD8 T cells, dendritic cells, imaging, liver, malaria
INTRODUCTION
Malaria is a major infectious disease, with 212 million cases and 429,000 malaria-induced deaths in 2015 (1). In the life cycle of Plasmodium parasites, sporozoites are injected into the skin via infectious bites from mosquitoes and specifically arrest in the liver, where they invade hepatocytes (2). In the liver stage, parasites multiply and mature inside infected hepatocytes, generating thousands of merozoites that eventually lyse the hepatocytes and are released into circulation to initiate the blood-stage infection and cause malaria (3). Liver infection takes approximately 2 days in rodent malaria models and 7 to 10 days in Plasmodium falciparum-infected humans, and it represents the bottleneck of parasite burden in mammalian infections, making it an attractive target for vaccine development. Immunization with radiation-attenuated sporozoites or genetically attenuated parasites or sporozoite infection under a chloroquine shield can induce sterile immunity against sporozoite challenge (4). CD8+ T cells are the major effector cells that mediate this protective immunity by recognizing Plasmodium antigens in association with major histocompatibility complex (MHC) class I (MHC-I) molecules on infected hepatocytes (4–6). The effector mechanisms responsible for the elimination of intrahepatic parasites by antigen-specific CD8+ T cells remain controversial, however, and previous studies suggested that effector molecules of CD8+ T cells, such as gamma interferon (IFN-γ), tumor necrosis factor alpha (TNF-α), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), perforin, and Fas ligand, are involved in a multifactorial, redundant manner, with their contributions also varying depending on the parasite and host species (7, 8). In addition, although dendritic cells, Kupffer cells, and liver sinusoidal endothelial cells (LSECs) have been shown to express MHC class I and class II as well as costimulatory molecules and are able to cross-present antigens to CD8+ T cells (6, 9), the role of these cells in the activation of malaria-specific CD8+ T cells in the liver is not clearly understood.
Previous studies using CD8+ T cells that have a defined specificity for Plasmodium antigens showed that very high numbers of antigen-specific CD8+ T cells are required for sterile protection against liver-stage malaria (10). The percentage of antigen-specific memory CD8+ T cells required for sterile protection is on the order of 1 to 2% of CD8+ T cells in BALB/c mice, and this requirement is even higher in C57BL/6 mice (11, 12). Intravital imaging of malaria-specific CD8+ T cells revealed that effector CD8+ T cells are recruited to the liver after sporozoite infection by chemokine-mediated mechanisms, where they form clusters around infected hepatocytes and where parasites are eliminated following a prolonged interaction between infected hepatocytes and CD8+ T cells (12, 13). Activated CD8+ T cells of an unrelated specificity are also recruited to the clusters (13). Upon an infectious mosquito bite, it is likely that in addition to CD8+ T cells that are specific for liver-stage malaria antigens, those that are specific for other antigens, including mosquito antigens, are also primed. In addition, other infectious diseases are also common in regions where malaria is endemic, and it is important to consider the influence of activated CD8+ T cells that are not specific for Plasmodium antigens on protective immunity against malaria parasites (10). However, it is not clear whether these nonspecific CD8+ T cells, which are recruited to the clusters around infected hepatocytes, participate in the elimination of parasites from the liver.
In this study, we used Plasmodium berghei ANKA expressing the model antigen ovalbumin (OVA) epitope, as well as green fluorescent protein (GFP), here referred to as PbA-gfpOVA, to evaluate the role of CD8+ T cells with an unrelated specificity in the protective immune response against liver-stage malaria. Using this strategy, we found that protection was dependent on specific CD8+ T cells, while those of an unrelated specificity were barely involved in protection. In addition to CD8+ T cells that were recruited to clusters around infected hepatocytes, dendritic cells were recruited to these clusters in the liver and played pivotal roles in cluster development around infected hepatocytes.
RESULTS
Clusters around infected hepatocytes include both antigen-specific and nonspecific CD8+ T cells.
To examine the role of antigen-specific and nonspecific CD8+ T cells in the clearance of malaria liver infection, we utilized PbA-gfpOVA that expresses the model antigen OVA epitope as well as OT-I CD8+ T cells that recognize OVA and 2C CD8+ T cells that recognize an unrelated antigen, Ld (Fig. 1A). OT-I and 2C cells were activated in vitro and adoptively transferred into C57BL/6 mice. Mice were then infected with PbA-gfpOVA sporozoites, and imaging of the liver was performed 44 h later by using two-photon microscopy (14). Both OT-I and 2C cells joined clusters around hepatocytes infected with PbA-gfpOVA, as previously reported using a different parasite, mouse, and antigen (Fig. 1B; see also Video S1 in the supplemental material) (12, 13). OT-I cells formed clusters when they were transferred alone or with 2C cells, while 2C cells were unable to form clusters alone, indicating that the recognition of the OVA epitope by OT-I cells is required for the initiation of cluster formation (Fig. 1C). Once clusters were formed, similar numbers of OT-I and 2C cells were present in the clusters, following a positive linear correlation (Fig. 1D). The velocities of OT-I and 2C cells were similar, and those inside clusters were lower than those outside (Fig. 1E), suggesting that both OT-I and 2C cells were affected by the microenvironment inside the clusters. Overall, the data showed that OT-I and 2C cells were recruited to the clusters by similar mechanisms after the initiation of cluster formation mediated by OT-I cells.
FIG 1.

Both antigen-specific and nonspecific CD8+ T cells join clusters around infected hepatocytes. (A) Experimental design of two-photon imaging of antigen-specific (GFP/OT-I) and nonspecific (DsRed/2C) CD8+ T cells in the liver after sporozoite infection. (B) Time-lapse two-photon images of OT-I cells (green) and 2C cells (red) in a cluster formed in the liver. The yellow-dashed circle shows the arbitrary border of a cluster. Arrowheads indicate OT-I and 2C cells outside the cluster moving toward the cluster. (C) Numbers of clusters in 132-mm2 image fields from the livers of mice treated with OT-I, 2C, or both OT-I and 2C cells (n = 3 mice/group). Each dot represents data for one mouse. *, P < 0.05; ns, not significant (by one-way ANOVA followed by Sidak's multiple-comparison tests). (D) Numbers of OT-I and 2C cells located inside each cluster. Each dot represents data for one cluster (P < 0.0001 by Spearman's rank correlation coefficient). (E) Velocities of OT-I and 2C cells inside and outside clusters. Each dot represents data for one CD8+ T cell. Data are derived from five experiments with similar results. *, P < 0.05; ns, not significant (by two-way ANOVA followed by Sidak's multiple-comparison tests).
Essential role of antigen-specific CD8+ T cells in cluster formation and protection.
To investigate whether the recruitment of parasite-specific and nonspecific CD8+ T cells to the liver as well as to clusters after sporozoite infection is dependent on chemokine receptor signaling, OT-I and 2C cells were independently treated or not treated with pertussis toxin (PTX), an inhibitor of G-protein-coupled signaling, and transferred into C57BL/6 mice. In uninfected mice, the numbers of OT-I and 2C cells in the liver declined after PTX treatment, while those of cotransferred but untreated cells were not affected (Fig. 2A). Next, mice were infected with PbA-gfpOVA sporozoites after T cell transfer, and the numbers of OT-I and 2C cells in the liver and clusters were determined 44 h after infection. In infected mice, both OT-I and 2C cell numbers increased nearly 10-fold compared with the numbers in uninfected mice (Fig. 2B). When OT-I cells were treated with PTX, the recruitment of both OT-I and 2C cells to the infected liver declined, while treatment of 2C cells did not affect the recruitment of OT-I cells (Fig. 2B). The treatment of OT-I cells with PTX inhibited the formation of all T cell clusters, while that of 2C cells inhibited only their recruitment to the clusters of OT-I cells (Fig. 2C). These results demonstrate a basal-level accumulation of activated CD8+ T cells in the livers of noninfected mice. This accumulation strongly increases after hepatic infection in the presence of activated parasite-specific CD8+ T cells, which triggers cluster formation by specific and nonspecific cells. Both basal accumulation and accumulation in clusters after triggering by specific CD8+ T cells are dependent on G-protein signaling, presumably via the recruitment and/or retention of cells through chemokine receptors.
FIG 2.
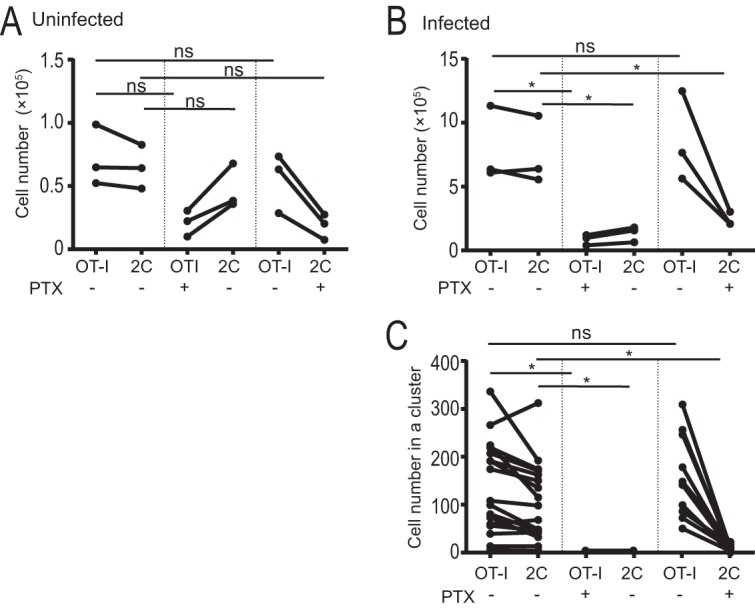
Antigen-specific CD8+ T cells initiate recruitment of activated CD8+ T cells. (A and B) Activated OT-I and 2C cells were incubated with (+) or without (−) PTX and transferred to C57BL/6 mice. The mice were uninfected (A) or infected (B) with PbA-gfpOVA sporozoites, and cell numbers in the liver were determined by flow cytometry. Each dot represents data for one mouse. (C) After infection with PbA-gfpOVA, numbers of OT-I and 2C cells in individual clusters were determined by using two-photon microscopy. Each dot represents data for one cluster. The values for OT-I and 2C cells in the same mouse (A and B) or in the same cluster (C) are connected by a line. Data are representative of data from three experiments with similar results. *, P < 0.05; ns, not significant (by two-way ANOVA followed by Sidak's multiple-comparison tests).
We next asked whether 2C cells that were recruited to clusters had any role in protection against malaria parasites in the liver. OT-I and/or 2C cells were transferred into C57BL/6 mice that were then infected with PbA-gfpOVA sporozoites, and the hepatic parasite burden was determined. The transfer of OT-I cells reduced the parasite burden in the liver, while that of 2C cells alone did not have any significant effect (Fig. 3A). We next mixed different doses of 2C cells with OT-I cells and cotransferred them into mice infected with PbA-gfpOVA sporozoites. The addition of 2C cells did not significantly contribute to the reduction in the parasite burden by OT-I cells when they were transferred into mice at the same dose (2.5 × 106 cells) (Fig. 3B) or at a dose that was 5 times higher (15.0 × 106 cells) (Fig. 3A). We concluded that, under our experimental conditions, protective immunity against liver-stage infection depends on Plasmodium-specific CD8+ T cells and that nonspecific activated CD8+ T cells do not significantly contribute to protection, although they join the cellular clusters initiated by specific CD8+ T cells around infected hepatocytes.
FIG 3.
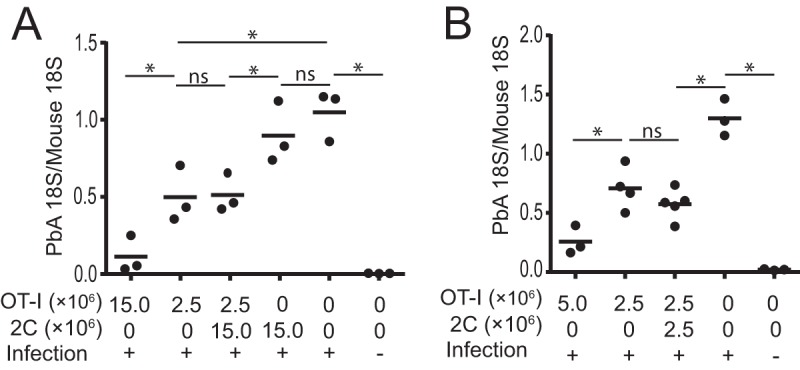
Antigen-nonspecific CD8+ T cells contribute little to protective immunity. C57BL/6 mice were treated with or without activated OT-I cells (2.5 × 106 to 15.0 × 106 cells) or 2C cells (2.5 × 106 to 15.0 × 106 cells) and infected (+) or not infected (−) with PbA-gfpOVA sporozoites, and after 44 h, the parasite burden in the liver was determined by real-time PCR. Numbers of transferred OT-I and 2C cells are indicated. Each dot represents data for one mouse. Panels A and B show data from two representative experiments from a total of nine replicates. *, P < 0.05; ns, not significant (by one-way ANOVA followed by Sidak's multiple-comparison tests).
Activation status of specific CD8+ T cells in infected livers.
Since both specific and nonspecific cells were recruited to the OT-I-dependent clusters around infected hepatocytes, we examined the numbers of these cells in the livers of infected mice. The numbers of OT-I and 2C cells increased dramatically in the livers but not the spleens of mice infected with PbA-gfpOVA (Fig. 4B and C). The number of host CD8+ T cells was not elevated in the liver, suggesting that activated OT-I and 2C cells were attracted to the microenvironment of the infected liver. To examine the activation status of OT-I and 2C cells that were recruited to the liver, we examined the expression of CD25 and CD69 on CD8+ T cells (Fig. 4). Interestingly, the expression level of CD25 on OT-I cells was significantly higher than that on 2C cells in the infected liver, although the CD69 expression level was similar (data not shown). This high level of CD25 expression was not seen in OT-I cells in the spleen, suggesting that OT-I cells in the infected liver were activated.
FIG 4.

Accumulation and activation of CD8+ T cells in the livers of infected mice. C57BL/6 mice were treated with activated GFP/OT-I and DsRed/2C cells and infected or not infected with PbA-gfpOVA, and single-cell suspensions from the livers were then stained for CD8 and CD25 44 h after infection. (A) After gating for CD8+ cells and for GFP versus DsRed, CD25 (orange) and isotype control (gray) profiles of OT-I (CD8+ GFP+), 2C (CD8+ DsRed+), and host CD8+ (CD8+ GFP− DsRed−) cells were determined. Numbers in the flow cytometry profiles indicate the proportions (percentages) of the population. (B and C) Numbers and CD25 expression levels of OT-I, 2C, and host CD8+ T cells in the livers (B) and spleens (C) of uninfected and infected mice. The expression levels of CD25 are shown as differences in the mean fluorescence intensity of anti-CD25 antibody staining and isotype controls (ΔMFI). Each dot represents data for one mouse. Data are representative of data from two experiments with similar results. *, P < 0.05; ns, not significant (by two-way ANOVA followed by Sidak's multiple-comparison tests).
Since OT-I cells in the liver express CD25 at higher levels than do 2C cells in infected mice, we hypothesized that they were activated via the recognition of the OVA epitope in the hepatic environment. To visualize interactions among dendritic cells, specific CD8+ T cells, and infected cells in the liver, OT-I cells were transferred into CD11c-eYFP mice, which were subsequently infected with PbA-gfpOVA sporozoites. In hepatic myeloid cells, CD11c+ cells consisted of CD11c+ F4/80− dendritic cells and CD11c+ F4/80+ cells that are likely to be a subpopulation of Kupffer cells (15). Intravital imaging showed that CD11c+ cells were present in clusters around infected hepatocytes and that OT-I cells around the infected hepatocytes were in close contact with them (Fig. 5A and B; see also Videos S2 and S3 in the supplemental material). This pervasive presence of CD11c+ cells in clusters and their close association with CD8+ T cells suggest that they might play a role in cluster formation. To examine the role of CD11c+ cells in the formation of clusters of OT-I cells, OT-I cells were transferred into CD11c-DTR chimeric mice, which were then infected with PbA-gfpOVA sporozoites. In the liver of chimeric mice in which OT-I cells were transferred and infected with PbA-OVA, the numbers of CD11c+ MHC-II+ dendritic cells increased compared to those that did not receive OT-I cells (Fig. 5C). Treatment of these mice with diphtheria toxin (DTX) resulted in a dramatic reduction in the number of CD11c+ cells in both the liver and spleen (Fig. 5C). After the depletion of CD11c+ cells, the numbers of CD8+ T cells and OT-I cells in the liver were severely diminished, while those in the spleen were not affected (Fig. 5D). Imaging of the liver showed that the number of OT-I clusters was severely reduced after the depletion of CD11c+ cells (Fig. 5E). This reduction in the number of clusters was not due to the direct toxic effect of DTX on the parasites, since the parasite burden in the livers of mice without OT-I cells was not affected by DTX treatment (Fig. 5F). Furthermore, the expression of CD25 on OT-I cells in the liver after infection was diminished in mice depleted of CD11c+ cells (Fig. 5G), suggesting that CD25 expression on OT-I cells in the livers of PbA-gfpOVA-infected mice was, at least in part, dependent on CD11c+ cells in the liver. Therefore, we examined whether the protective effect of OT-I cells against PbA-gfpOVA sporozoite infection was altered by DTX treatment (Fig. 5H). The depletion of CD11c+ cells resulted in reduced protection elicited by OT-I cells, which correlated with the numbers of OT-I cells in the liver.
FIG 5.

Dendritic cells play a pivotal role in the formation of CD8+ T cell clusters. (A and B) CD11c-eYFP mice were transferred with activated DsRed/OT-I cells and infected with PbA-gfpOVA sporozoites, and liver imaging was performed by using two-photon microscopy. (A) Time-lapse images of OT-I cells (red cells, indicated by small yellow arrowheads) associated with CD11c+ cells (green) around infected hepatocytes (yellow, indicated by large white arrowheads). (B) Images of CD11c-eYFP cells (green) in a cluster (left) and overlay image of OT-I (red) and CD11c+ cells (green) in the same cluster (right). (C to H) CD11c-DTR chimeric mice were transferred (+) or not transferred (−) with activated OT-I cells; infected with PbA-gfpOVA sporozoites, except for one group in panel G; and treated (+) or not treated (−) with DTX. (C) Liver and spleen cells were stained and analyzed by using flow cytometry. The proportions (percentages) of MHC-II+ CD11c+ dendritic cells from CD45.2+ liver (top) and spleen (bottom) cells were determined (left), and their absolute numbers were calculated (right). (D) Total numbers of suspended cells (left) and OT-I cells (right) in the liver (top) and spleen (bottom) were determined. (E) Livers were examined under a two-photon microscope 44 h after infection, and the numbers of clusters in a 132-mm2 area were determined. (F) Parasite burdens in mice treated with higher (0.1 μg) and lower (0.05 μg) doses of DTX without OT-I transfer were determined by real-time PCR. (G and H) Proportions (percentages) of CD25+ cells of OT-I cells in liver cell suspensions (G) and parasite burdens in the liver (H) were determined 44 h after infection (+) or without infection (−). Each dot represents data for one mouse. Data are representative of data from three experiments with similar results. Results are presented as means ± SD. *, P < 0.05; ns, not significant (by unpaired t tests [D, right, and E] or by one-way ANOVA followed by Sidak's multiple-comparison tests [C, D, left, and F to H]).
DISCUSSION
CD8+ T cells are major effector cells in the protective immune response against malaria liver-stage infection. Both parasite-specific and nonspecific activated CD8+ T cells form clusters around infected hepatocytes during the protective immune response (12, 13). We used OVA as a model antigen as well as OT-I and 2C T cell receptor (TCR) transgenic T cells as antigen-specific and nonspecific CD8+ T cells, respectively, to study the mechanisms underlying the formation of clusters of CD8+ T cells and their role in the protective immune response against OVA-expressing malaria parasites in the liver stage. We activated OT-I and 2C cells in vitro prior to their transfer into mice, since naive CD8+ T cells did not accumulate in the liver and were not protective against PbA-gfpOVA infection (data not shown) (16). The initiation of cluster formation was tightly regulated by the recognition of antigens by specific CD8+ T cells, suggesting that the recruitment of activated CD8+ T cells to infected hepatocytes is initiated by the recognition of parasite antigens by activated malaria antigen-specific CD8+ T cells. However, activated nonspecific CD8+ T cells joined these clusters in a manner independent of TCR recognition. The expression of chemokine receptors or adhesion molecules on activated CD8+ T cells may be sufficient for their recruitment to clusters of CD8+ T cells, as shown previously (13, 17), although the chemokine that is required for the migration of CD8+ T cells to the liver during malaria infection has not been identified. However, under our experimental conditions, the nonspecific CD8+ T cells in these clusters did not significantly contribute to protection, indicating that TCR-mediated signaling is required for directing the effector function of CD8+ T cells toward infected hepatocytes. In regions where malaria is endemic, the population is constantly exposed to various infectious microbes and may possess circulating effector T cells that are not specific for malaria antigens. In addition, mosquito antigen-specific CD8+ T cells may also be induced upon an infectious bite. Our study suggests that these nonspecific T cells may join the cluster but will not directly contribute to protective immunity at the liver stage of Plasmodium infection. TCR sharing of bystander CD8+ T cells does not appear to take place in liver-stage infection (18). It will be intriguing to determine whether the recruitment of nonspecific activated T cells with different profiles of anti-liver-stage cytokine secretion (19–21) to clusters enhances the elimination of infected cells.
CD11c+ cells in hepatic myeloid cells consist of at least two subpopulations, CD11c+ F4/80− and CD11c+ F4/80+ cells, which are mostly dendritic cells and a subpopulation of Kupffer cells, respectively (15). We found that the number of CD11c+ cells increased in mice in which OT-I cells were transferred and infected with PbA-OVA and accumulated in the clusters of activated CD8+ T cells that were formed around infected hepatocytes. The recruitment of CD11c+ cells may be triggered by infected hepatocytes, which recognize Plasmodium parasites via receptors for pathogen-associated molecular patterns in a type I interferon (IFN)-dependent, and, thus, activated CD8+ T cell-independent, manner (22, 23). However, since cluster formation is dependent on specific CD8+ T cells, it is more likely that OT-I cells recruit CD11c+ cells following the recognition of infected hepatocytes. When CD11c+ cells were depleted by DTX treatment in CD11c-DTR chimeric mice, the formation of CD8+ T cell clusters was strongly reduced, implying that CD11c+ cells in the liver play a pivotal role in the orchestration of cluster formation. The depletion of CD11c+ cells significantly decreased the protection conferred by the transfer of OT-I cells, correlating with the decrease in OT-I cell numbers in the liver, suggesting that CD11c+ dendritic cells in the clusters have pivotal roles for the effector function of these specific CD8+ T cells in protection. Alternatively, there is the possibility of a direct protective role of CD11c+ cells in the clusters via cytokine secretion or phagocytosis, because targeting of CD11c+ cells with DTX depletes a majority of CD11c+ dendritic cells and a significant proportion of CD11c+ F4/80+ Kupffer cells (15). However, CD11c− F4/80+ Kupffer cells should remain intact in DTX-treated mice, and we believe that this possibility is less likely.
In our imaging study, we observed that OT-I cells that approached clusters around infected hepatocytes were closely associated with CD11c+ cells. These results suggest that CD11c+ cells that were recruited to infected hepatocytes form a microenvironment that attracts activated CD8+ T cells, increasing the opportunity for antigen-specific CD8+ T cells to recognize and eliminate infected hepatocytes. CD11c+ dendritic cells are known to cross-present antigens to specific CD8+ T cells when in close contact. In the livers of mice infected with radiation-attenuated Plasmodium sporozoites, CD8α+ dendritic cells present liver-stage antigens to activate effector CD8+ T cells (24). However, it was reported previously that effector CD8+ cells do not require bone marrow-derived antigen-presenting cells for protection at the liver stage of Plasmodium yoelii infection (25). Furthermore, dendritic cells use the endosome-cytosol pathway to cross-present Plasmodium antigens to CD8+ T cells, and this endosomal pathway of antigen presentation has been shown to be dispensable for protective immunity against infected hepatocytes mediated by activated specific CD8+ T cells (26). Thus, we believe that this pathway is unlikely to be essential for protection in the liver, although it may have an ancillary role. An alternative possibility is that specific CD8+ T cells directly receive activation signals upon the recognition of the antigens presented on infected hepatocytes via their TCR. In this scenario, CD11c+ cells that are recruited to infected hepatocytes do not directly activate specific CD8+ T cells but may attract activated CD8+ T cells to infected hepatocytes in an antigen-nonspecific manner via the expression of adhesion molecules and the secretion of chemokines.
We observed clusters of CD8+ T cells 44 h after infection with sporozoites, representing a relatively late period of liver-stage infection. At this late time, CD8+ T cell clusters were heterogeneous, ranging from the collection of a few CD8+ T cells around infected hepatocytes to large clusters of CD8+ T cells that accumulated after the elimination of parasites. Our study demonstrated the critical role of dendritic cells in the formation of these CD8+ T cell clusters and their impact on protection. In addition, we showed that Plasmodium-specific CD8+ T cells within the clusters are likely involved in the elimination of infected cells. It is well known that the activation of innate immunity is critical for the induction of adaptive immune responses (27). Our study highlights the important role of innate immune cells in inducing the effector mechanisms of activated CD8+ T cells during the liver stage of Plasmodium infection. Further study will reveal the molecular mechanisms underlying the interactions between dendritic cells and effector CD8+ T cells that play essential roles in the elimination of liver-stage Plasmodium parasites.
MATERIALS AND METHODS
Animals.
OT-I transgenic mice expressing a TCR specific for OVA257–264/Kb were obtained from H. Kosaka (Osaka University, Osaka, Japan) (28). B6.SJL and OT-I mice were interbred, and the offspring were intercrossed to obtain CD45.1+ OT-I mice. In addition, 2C mice (29) (RBRC00123) were provided by RIKEN BRC through the National Bio-Resource Project of the Ministry of Education, Culture, Sports, Science, and Technology, Japan. DsRed.T3 (30), CD11c-DTR (31), and CD11c-eYFP (32) transgenic mice were purchased from The Jackson Laboratory (Bar Harbor, ME). B6-EGFP (GFP) and C57BL/6 mice were purchased from SLC (Shizuoka, Japan). OT-I mice were interbred with DsRed.T3 and GFP transgenic mice to generate DsRed/OT-I and GFP/OT-I mice, respectively, while 2C mice were crossed with DsRed.T3 mice to generate DsRed/2C mice. Mice were maintained in the Laboratory Animal Core for Animal Research at Nagasaki University and used at the age of 8 to 14 weeks. The animal experiments presented here were approved by the Institutional Animal Care and Use Committee of Nagasaki University and were conducted according to guidelines for animal experimentation at Nagasaki University.
T cell transfer, parasite infection, and imaging.
For the preparation of activated OT-I cells, cells were harvested from the spleens and inguinal lymph nodes of OT-I mice, pulsed with the OVA257–264 peptide (2 μg/ml) for 4 h, washed, and cultured for 3 days. For 2C cells, cells were prepared from DsRed/2C mice and pulsed with the 2C SIYRYYGL peptide (5 μg/ml) (33). Mice received activated OT-I or 2C cells (5 × 106 to 10 × 106 cells) intravenously through the tail vein.
Activated OT-I and 2C CD8+ T cells were treated with PTX (100 ng/ml; Sigma, St. Louis, MO) for 3 h at 37°C and washed three times with phosphate-buffered saline (PBS) before transfer to mice. Each type of cell (OT-I and 2C, PTX-treated and untreated) was transferred separately into the recipient mouse.
PbA-gfpOVA sporozoites were collected from the salivary glands of infected Anopheles stephensi mosquitoes. One day after cell transfer, mice were intravenously infected with PbA-gfpOVA sporozoites (1 × 104 sporozoites). Next, 44 h after infection, the liver was imaged by using an inverted TCS SP5 two-photon microscope equipped with an OPO laser (Leica Microsystems, Wetzlar, Germany) with a 25× water immersion objective, as described previously (12, 14). Analysis of two-photon imaging data was performed by using Imaris 7.6.5 software (Bitplane, Zurich, Switzerland).
Liver cell suspension.
The liver cell suspension was prepared as described previously, with modifications (34). Briefly, the isolated liver was crushed with a syringe plunger in a petri dish placed on ice in 5 ml PBS. The cell suspension was centrifuged, and the pellet was suspended in a solution of 33% Percoll in PBS and centrifuged at 800 × g for 30 min at 20°C. Parenchymal cells and debris on the top were removed by using a glass pipette, and the pellet was suspended in Gey's solution to lyse red blood cells, centrifuged, washed with PBS, resuspended in PBS, and stained with fluorochrome-conjugated antibodies.
Flow cytometry.
Cells were blocked with anti-CD16/CD32 (2.4G2) and incubated with antibodies specific for CD8α and CD25 (eBioscience, San Diego, CA), as described previously (12). All samples were analyzed by using a BD FACSCanto II or BD LSRFortessa X-20 instrument (BD Biosciences, San Jose, CA). Data were analyzed by using FlowJo software v10.2 (TreeStar, Ashland, OR). The difference in the mean fluorescence intensity (ΔMFI) was used to determine the difference between experimental staining and the isotype control.
Real-time PCR to determine parasite burden.
Total liver RNA was isolated by using Isogen II (Nippon Gene, Tokyo, Japan) 44 h after infection with 10,000 PbA-gfpOVA sporozoites. RNA was treated with DNase (TaKaRa, Kusatsu, Japan) according to the manufacturer's protocol and separated on a 1% agarose gel to verify its integrity, using the ratio of 28S to 18S rRNA. cDNA was prepared from 2 μg of RNA and amplified by PCR using primer pairs targeted to the 18S rRNA sequence of Plasmodium yoelii (35) or mouse (36) in a mixture containing SYBR green (Applied Biosystems, Foster City, CA). Samples were amplified by using an ABI Prism 7900HT automatic real-time PCR system (Applied Biosystems). The threshold cycle of each PCR was converted into a DNA equivalent using standard curves made by amplifying 10-fold dilutions of a plasmid bearing the relevant target sequences. The liver parasite burden was determined as the ratio of the cDNA equivalent measured for P. yoelii 18S rRNA to that for mouse 18S.
Depletion of CD11c+ cells.
C57BL/6 mice were lethally irradiated (900 rads) and intravenously received bone marrow cells from CD11c-DTR mice (1.0 × 107 cells) on the following day to generate bone marrow chimeric (CD11c-DTR chimeric) mice. Mice were maintained for more than 2 months before infection to allow the reconstitution of the hematopoietic cells. To deplete dendritic cells, CD11c-DTR chimeric mice were intraperitoneally injected with 0.5 μg/mouse DTX in 1 ml PBS 2 h after infection with sporozoites.
Statistical analysis.
In comparisons between two groups, unpaired two-tailed Student's t tests were used. In comparisons of three or more groups, an overall difference was first determined by one-way or two-way analysis of variance (ANOVA) followed by Sidak's multiple-comparison tests. Statistical analysis was performed by using GraphPad Prism version 6 (GraphPad Software, San Diego, CA, USA). Correlations were analyzed by using Spearman's rank correlation coefficient. Results are presented as means ± standard deviations (SD).
Supplementary Material
ACKNOWLEDGMENTS
We thank Nobuka Kawamoto and Mikiyo Yoshida for their technical assistance.
This work was supported by grants-in-aid for scientific research from the Japan Society for the Promotion of Science (25113717 and 15K15124) to K.Y.
We have no financial conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00717-17.
REFERENCES
- 1.WHO. 2017. World malaria report 2016. WHO, Geneva, Switzerland. [Google Scholar]
- 2.Tavares J, Costa DM, Teixeira AR, Cordeiro-da-Silva A, Amino R. 2017. In vivo imaging of pathogen homing to the host tissues. Methods 127:37–44. doi: 10.1016/j.ymeth.2017.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Menard R, Tavares J, Cockburn I, Markus M, Zavala F, Amino R. 2013. Looking under the skin: the first steps in malarial infection and immunity. Nat Rev Microbiol 11:701–712. doi: 10.1038/nrmicro3111. [DOI] [PubMed] [Google Scholar]
- 4.Hafalla JC, Silvie O, Matuschewski K. 2011. Cell biology and immunology of malaria. Immunol Rev 240:297–316. doi: 10.1111/j.1600-065X.2010.00988.x. [DOI] [PubMed] [Google Scholar]
- 5.Schofield L, Villaquiran J, Ferreira A, Schellekens H, Nussenzweig R, Nussenzweig V. 1987. γ interferon, CD8+ T cells and antibodies required for immunity to malaria sporozoites. Nature 330:664–666. doi: 10.1038/330664a0. [DOI] [PubMed] [Google Scholar]
- 6.Frevert U, Nacer A. 2013. Immunobiology of Plasmodium in liver and brain. Parasite Immunol 35:267–282. doi: 10.1111/pim.12039. [DOI] [PubMed] [Google Scholar]
- 7.Morrot A, Zavala F. 2004. Effector and memory CD8+ T cells as seen in immunity to malaria. Immunol Rev 201:291–303. doi: 10.1111/j.0105-2896.2004.00175.x. [DOI] [PubMed] [Google Scholar]
- 8.Butler NS, Schmidt NW, Harty JT. 2010. Differential effector pathways regulate memory CD8 T cell immunity against Plasmodium berghei versus P. yoelii sporozoites. J Immunol 184:2528–2538. doi: 10.4049/jimmunol.0903529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomson AW, Knolle PA. 2010. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol 10:753–766. doi: 10.1038/nri2858. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt NW, Harty JT. 2011. Cutting edge: attrition of Plasmodium-specific memory CD8 T cells results in decreased protection that is rescued by booster immunization. J Immunol 186:3836–3840. doi: 10.4049/jimmunol.1003949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Braeckel-Budimir N, Harty JT. 2014. CD8 T-cell-mediated protection against liver-stage malaria: lessons from a mouse model. Front Microbiol 5:272. doi: 10.3389/fmicb.2014.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimura K, Kimura D, Matsushima Y, Miyakoda M, Honma K, Yuda M, Yui K. 2013. CD8+ T cells specific for a malaria cytoplasmic antigen form clusters around infected hepatocytes and are protective at the liver stage of infection. Infect Immun 81:3825–3834. doi: 10.1128/IAI.00570-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cockburn IA, Amino R, Kelemen RK, Kuo SC, Tse SW, Radtke A, Mac-Daniel L, Ganusov VV, Zavala F, Menard R. 2013. In vivo imaging of CD8+ T cell-mediated elimination of malaria liver stages. Proc Natl Acad Sci U S A 110:9090–9095. doi: 10.1073/pnas.1303858110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akbari M, Kimura K, Houts JT, Yui K. 2016. Intravital imaging of the immune responses during liver-stage malaria infection: an improved approach for fixing the liver. Parasitol Int 65:502–505. doi: 10.1016/j.parint.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 15.David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB, Sousa Pereira RV, Marchesi SC, Alvarenga DM, Nakagaki BN, Araujo AM, Dos Reis DS, Rocha RM, Marques PE, Lee WY, Deniset J, Liew PX, Rubino S, Cox L, Pinho V, Cunha TM, Fernandes GR, Oliveira AG, Teixeira MM, Kubes P, Menezes GB. 2016. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology 151:1176–1191. doi: 10.1053/j.gastro.2016.08.024. [DOI] [PubMed] [Google Scholar]
- 16.John B, Crispe IN. 2004. Passive and active mechanisms trap activated CD8+ T cells in the liver. J Immunol 172:5222–5229. doi: 10.4049/jimmunol.172.9.5222. [DOI] [PubMed] [Google Scholar]
- 17.Tse SW, Radtke AJ, Espinosa DA, Cockburn IA, Zavala F. 2014. The chemokine receptor CXCR6 is required for the maintenance of liver memory CD8+ T cells specific for infectious pathogens. J Infect Dis 210:1508–1516. doi: 10.1093/infdis/jiu281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chaudhri G, Quah BJ, Wang Y, Tan AH, Zhou J, Karupiah G, Parish CR. 2009. T cell receptor sharing by cytotoxic T lymphocytes facilitates efficient virus control. Proc Natl Acad Sci U S A 106:14984–14989. doi: 10.1073/pnas.0906554106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferreira A, Schofield L, Enea V, Schellekens H, van der Meide P, Collins WE, Nussenzweig RS, Nussenzweig V. 1986. Inhibition of development of exoerythrocytic forms of malaria parasites by γ-interferon. Science 232:881–884. doi: 10.1126/science.3085218. [DOI] [PubMed] [Google Scholar]
- 20.Nussler A, Pied S, Goma J, Renia L, Miltgen F, Grau GE, Mazier D. 1991. TNF inhibits malaria hepatic stages in vitro via synthesis of IL-6. Int Immunol 3:317–321. doi: 10.1093/intimm/3.4.317. [DOI] [PubMed] [Google Scholar]
- 21.Schmieg J, Yang G, Franck RW, Tsuji M. 2003. Superior protection against malaria and melanoma metastases by a C-glycoside analogue of the natural killer T cell ligand α-galactosylceramide. J Exp Med 198:1631–1641. doi: 10.1084/jem.20031192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liehl P, Zuzarte-Luis V, Chan J, Zillinger T, Baptista F, Carapau D, Konert M, Hanson KK, Carret C, Lassnig C, Muller M, Kalinke U, Saeed M, Chora AF, Golenbock DT, Strobl B, Prudencio M, Coelho LP, Kappe SH, Superti-Furga G, Pichlmair A, Vigario AM, Rice CM, Fitzgerald KA, Barchet W, Mota MM. 2014. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat Med 20:47–53. doi: 10.1038/nm.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller JL, Sack BK, Baldwin M, Vaughan AM, Kappe SH. 2014. Interferon-mediated innate immune responses against malaria parasite liver stages. Cell Rep 7:436–447. doi: 10.1016/j.celrep.2014.03.018. [DOI] [PubMed] [Google Scholar]
- 24.Jobe O, Donofrio G, Sun G, Liepinsh D, Schwenk R, Krzych U. 2009. Immunization with radiation-attenuated Plasmodium berghei sporozoites induces liver cCD8α+ DC that activate CD8+ T cells against liver-stage malaria. PLoS One 4:e5075. doi: 10.1371/journal.pone.0005075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakravarty S, Cockburn IA, Kuk S, Overstreet MG, Sacci JB, Zavala F. 2007. CD8+ T lymphocytes protective against malaria liver stages are primed in skin-draining lymph nodes. Nat Med 13:1035–1041. doi: 10.1038/nm1628. [DOI] [PubMed] [Google Scholar]
- 26.Cockburn IA, Tse SW, Radtke AJ, Srinivasan P, Chen YC, Sinnis P, Zavala F. 2011. Dendritic cells and hepatocytes use distinct pathways to process protective antigen from Plasmodium in vivo. PLoS Pathog 7:e1001318. doi: 10.1371/journal.ppat.1001318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iwasaki A, Medzhitov R. 2015. Control of adaptive immunity by the innate immune system. Nat Immunol 16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. 1994. T cell receptor antagonist peptides induce positive selection. Cell 76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 29.Sha WC, Nelson CA, Newberry RD, Kranz DM, Russell JH, Loh DY. 1988. Selective expression of an antigen receptor on CD8-bearing T lymphocytes in transgenic mice. Nature 335:271–274. doi: 10.1038/335271a0. [DOI] [PubMed] [Google Scholar]
- 30.Vintersten K, Monetti C, Gertsenstein M, Zhang P, Laszlo L, Biechele S, Nagy A. 2004. Mouse in red: red fluorescent protein expression in mouse ES cells, embryos, and adult animals. Genesis 40:241–246. doi: 10.1002/gene.20095. [DOI] [PubMed] [Google Scholar]
- 31.Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, Pamer EG, Littman DR, Lang RA. 2002. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity 17:211–220. doi: 10.1016/S1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lindquist RL, Shakhar G, Dudziak D, Wardemann H, Eisenreich T, Dustin ML, Nussenzweig MC. 2004. Visualizing dendritic cell networks in vivo. Nat Immunol 5:1243–1250. doi: 10.1038/ni1139. [DOI] [PubMed] [Google Scholar]
- 33.Udaka K, Tsomides TJ, Eisen HN. 1992. A naturally occurring peptide recognized by alloreactive CD8+ cytotoxic T lymphocytes in association with a class I MHC protein. Cell 69:989–998. doi: 10.1016/0092-8674(92)90617-L. [DOI] [PubMed] [Google Scholar]
- 34.Watarai H, Nakagawa R, Omori-Miyake M, Dashtsoodol N, Taniguchi M. 2008. Methods for detection, isolation and culture of mouse and human invariant NKT cells. Nat Protoc 3:70–78. doi: 10.1038/nprot.2007.515. [DOI] [PubMed] [Google Scholar]
- 35.Kawabata Y, Udono H, Honma K, Ueda M, Mukae H, Kadota J, Kohno S, Yui K. 2002. Merozoite surface protein 1-specific immune response is protective against exoerythrocytic forms of Plasmodium yoelii. Infect Immun 70:6075–6082. doi: 10.1128/IAI.70.11.6075-6082.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimura D, Miyakoda M, Kimura K, Honma K, Hara H, Yoshida H, Yui K. 2016. Interleukin-27-producing CD4+ T cells regulate protective immunity during malaria parasite infection. Immunity 44:672–682. doi: 10.1016/j.immuni.2016.02.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.