

ABSTRACT
Coxiella burnetii is an intracellular pathogen that replicates in a lysosome-derived vacuole. A determinant necessary for C. burnetii virulence is the Dot/Icm type IVB secretion system (T4SS). The Dot/Icm system delivers more than 100 proteins, called type IV effectors (T4Es), across the vacuolar membrane into the host cell cytosol. Several T4Es have been shown to be important for vacuolar biogenesis. Here, transposon (Tn) insertion sequencing technology (INSeq) was used to identify C. burnetii Nine Mile phase II mutants in an arrayed library, which facilitated the identification and clonal isolation of mutants deficient in 70 different T4E proteins. These effector mutants were screened in HeLa cells for deficiencies in Coxiella-containing vacuole (CCV) biogenesis. This screen identified and validated seven new T4Es that were important for vacuole biogenesis. Loss-of-function mutations in cbu0414 (coxH1), cbu0513, cbu0978 (cem3), cbu1387 (cem6), cbu1524 (caeA), cbu1752, or cbu2028 resulted in a small-vacuole phenotype. These seven mutant strains produced small CCVs in all cells tested, which included macrophage-like cells. The cbu2028::Tn mutant, though unable to develop large CCVs, had intracellular replication rates similar to the rate of the parental strain of C. burnetii, whereas the other six effector mutants defective in CCV biogenesis displayed significant reductions in intracellular replication. Vacuoles created by the cbu0513::Tn mutant did not accumulate lipidated microtubule-associated protein 1A/1B light chain 3 (LC3-II), suggesting a failure in fusion of the CCV with autophagosomes. These seven T4E proteins add to the growing repertoire of C. burnetii factors that contribute to CCV biogenesis.
KEYWORDS: type IV secretion, vacuole biogenesis, bacterial effector proteins, lysosome
INTRODUCTION
Coxiella burnetii is a Gram-negative intracellular pathogen that causes a zoonotic disease in humans called Q fever (1). Ruminants such as cattle and sheep that are infected with C. burnetii are the most common source contributing to human infections (2). Q fever is most frequently exhibited as an acute flu-like illness; however, chronic illness is observed and can be fatal, typically as a form of bacterial endocarditis, especially in immunocompromised individuals (3). C. burnetii is typically transmitted by an aerosol route, and the infectious dose can be fewer than 10 bacteria (4). The infectious form of C. burnetii is a small-cell variant that is resistant to environmental stress and develops during late stages of intracellular replication. C. burnetii is capable of infecting a wide range of cell types, including many epithelial and fibroblast-like continuous cell lines, making it amenable to cell culture infection assays (5). Upon entry into a host cell, the bacteria develop into metabolically active large-cell variants that are resistant to lysosomal destruction (5, 6). During animal infections C. burnetii will undergo phase variation. Phase I bacteria contain a complex O-polysaccharide surface antigen, whereas phase II bacteria typically have alterations in O-polysaccharide that make them more susceptible to killing by antimicrobial factors in serum (7). The C. burnetii Nine Mile phase II clone 4 (NMII) strain, RSA493, is a laboratory-passaged isolate that contains a chromosomal deletion that eliminates several genes important for O-antigen biosynthesis (4, 8). This strain is unable to cause disease in animals but is fully virulent for most cells grown in tissue culture. Thus, the NMII strain is a reference strain used to study intracellular replication of C. burnetii.
After C. burnetii is internalized by a host cell, the bacteria are contained in a vacuole that is remodeled by endocytic maturation, which results in lysosomal fusion (9). This lysosome-derived Coxiella-containing vacuole (CCV) is highly fusogenic and frequently occupies a significant proportion of the area within a host cell, providing the bacteria with a spacious environment in which to replicate, a feature that is easily observed by microscopy (10). The CCV contains host proteins that reside in lysosomes, such as lysosomal-associated membrane protein 1 (LAMP-1), and expansion of the CCV involves continuous fusion with mature autophagosomes, which results in the accumulation of microtubule-associated protein 1A/1B light chain 3 (LC3) in the lumen of the vacuole (11, 12).
An essential determinant of pathogenicity is the C. burnetii Dot/Icm type IVB secretion system (T4SS). Fusion of the early CCV with lysosomes stimulates C. burnetii gene expression and increases metabolic activity in the bacterial cell, which upregulates expression of the T4SS. The T4SS is necessary for both intracellular replication and biogenesis of the mature CCV, presumably because bacterial effector proteins delivered by the T4SS control cellular processes important for subverting host membrane transport to the CCV (10, 13). This is supported by the observation that some type IV effector (T4E) mutants display defects in CCV biogenesis (14–17). The primary function of the C. burnetii T4SS is to deliver bacterial effector proteins across the vacuolar membrane into the host cytosol during infection. Collectively, these effector proteins modulate host pathways to facilitate biogenesis of a vacuole that supports C. burnetii replication (18). The T4E repertoire of C. burnetii is estimated to be as large as 130 different proteins, and recent studies have provided insight into how a subset of effector proteins manipulate host cell functions; however, there are many more T4Es whose functions we know little about (19, 20).
Axenic cultivation of C. burnetii led to advances in genetic manipulation so that both random and targeted mutagenesis is now possible (21, 22). Plasmid complementation studies have also developed to validate mutants isolated in forward genetic screens (23, 24). Thus, the ability to genetically manipulate C. burnetii has made it possible to identify isogenic mutants deficient in T4E proteins and to evaluate the contribution of these T4Es to CCV biogenesis and host virulence (14–17).
In this study, we report an expanded C. burnetii library of arrayed transposon (Tn) insertion mutants. An insertion sequencing technology (INSeq) pipeline mapped over 2,000 transposon insertion sites in this library, and single clones containing Tn insertions in genes encoding predicted T4E proteins were identified using this information. A sublibrary of T4E mutants was then screened to identify additional effector proteins that contribute to CCV biogenesis. Thus, this targeted approach has further expanded the repertoire of C. burnetii effector proteins that contribute to CCV biogenesis.
RESULTS
INSeq mapping of insertion sites in an arrayed C. burnetii transposon mutant library.
Previously, transposon insertion mutants of the Coxiella burnetii Nine Mile phase II strain, RSA493, were arrayed in 3,850 wells using 96-well plates. Bacteria from individual wells in the library were screened after host cell infection using a visual assay to identify individual wells likely to contain mutants defective in genes important for biogenesis of the CCV (16). Although several T4E mutants were identified using this screening approach, it was likely that, for a few reasons, this screen missed several T4E proteins that are important for CCV biogenesis. With only 3,850 mutants in the original library, the collection could be missing several effector mutants. Also, mutants with minor vacuole biogenesis defects are easy to miss when a visual screen involving over 3,000 isolated transposon insertion mutants is conducted. Last, the difficulty inherent in clonal selection of C. burnetii mutants from an agar plate after transposon mutagenesis sometimes results in the inadvertent coisolation of an adjacent microcolony, which results in a single well of the library containing two independent mutants. This is problematic because if two different mutants occupy the same well in an arrayed mutant library and if one of the mutants does not display a CCV biogenesis defect, robust replication will be the dominant phenotype observed upon visual inspection of the host cell infected with C. burnetii isolated from that well. To overcome potential limitations, we redesigned the approach to increase the number of total mutants available for screening and used INSeq technology to determine the sites of transposon insertions in the library and to map the location of each mutant to a specific well in the arrayed library, and this information was used to generate a sublibrary of validated clones that were defective in a single effector gene.
Additional transposon insertion mutants were generated and arrayed to expand the library to contain more than 5,000 mutants. In order to identify the transposon insertion location in these mutants, INSeq was employed (Fig. 1A). All of the mutants were grown individually in axenic culture to expand the entire library. A total of 5,240 wells, or 85.2%, contained viable mutants and could be expanded in acidified citrate cysteine medium (ACCM-2) with antibiotic selection. Each mutant was then distributed into a subset of 24 pools in a unique pattern, as described previously (25, 26), and transposon-adjacent DNA was isolated by random shearing and PCR amplification (27). Illumina sequencing of these pools was used to determine the insertion site for each mutant in the arrayed library as described previously (25, 26). These data identified with high confidence the location of 2,342 insertion mutants in the library (Fig. 1B).
FIG 1.
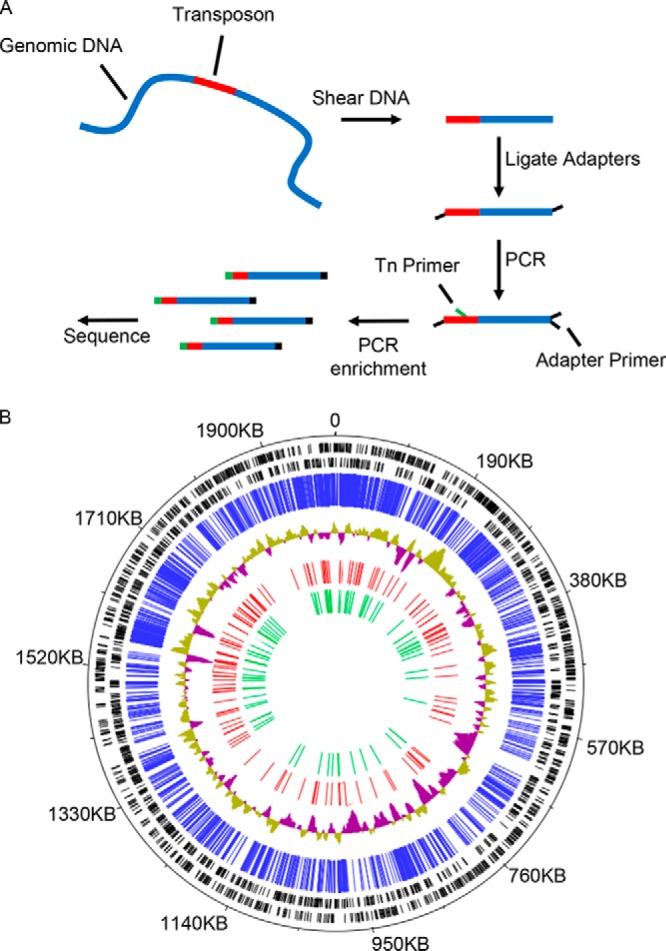
Sequencing and mapping of transposon mutant library by insertion sequencing. (A) A graphical depiction of the preparation of DNA for sequencing and mapping of the C. burnetii transposon insertion mutant library. (B) C. burnetii RSA493 genome annotated with the location of transposon insertion sites that were mapped by INSeq analysis. The black bars indicate the locations of predicted open reading frames encoded on forward and reverse strands. The blue bars indicate the location of each transposon insertion site in the mutant library that was identified and mapped by INSeq analysis. The blue and green topology graph depicts G-C content of the chromosomal DNA. The red bars indicate the relative locations of all predicted T4E genes. The green bars indicate the location of transposon insertion sites in predicted effector genes where the mutants were isolated and validated to construct the T4E mutant sublibrary.
Assembly of a T4E mutant sublibrary.
The INSeq data identified 260 insertions in genes encoding T4E proteins (Fig. 1B; see also Table S1 in the supplemental material). Multiple independent insertion mutants were identified for several T4E genes (Table S1). For example, there were 18 independent insertions in the gene encoding the T4E protein Cig2. In total, transposon insertion mutations were identified in 70 different genes encoding predicted T4E proteins. Importantly, several T4E mutants mapped to wells where more than one transposon insertion had mapped. This could indicate that either a single mutant with multiple transposon insertions was present in the well or that more than one independent mutant was in the well. Thus, to generate a sublibrary of C. burnetii mutants deficient in individual effector proteins, bacteria from a single well where a T4E mutant of interest had been mapped were grown on ACCM-2 agarose plates to obtain single colonies. Isolated clones were then analyzed using PCR to identify mutants that had a single insertion in the effector gene of interest. This analysis led to the generation of a sublibrary containing mutants deficient in 70 different T4E proteins. This sublibrary contained a relatively small number of mutants compared to the number in the original arrayed library, and each well of the sublibrary was validated to ensure that a single mutant was present. Thus, this sublibrary was predicted to be useful for screening T4E mutants for Dot/Icm-dependent phenotypes.
A visual screen identifies C. burnetii T4E mutants deficient in CCV biogenesis.
Vacuoles supporting intracellular replication of the parental NMII strain of C. burnetii are large and easy to visualize using microscopy at 5 days after infection of HeLa cells (Fig. 2A). Previously, we used a microscopy-based assay to screen an arrayed library of C. burnetii mutants in HeLa cells to identify genes required for CCV biogenesis and identified four T4E genes as being important for vacuole biogenesis (16). Transposon insertions in the genes cig2, cig57, coxCC8, and cbu1754 resulted in mutants that generated obvious CCV biogenesis defects. To determine if the T4E mutant sublibrary could be a useful tool in uncovering T4E proteins contributing to Dot/Icm-dependent phenotypes, we repeated the CCV biogenesis screen using the individual mutants in this collection.
FIG 2.

Discovery of seven effector genes important for CCV biogenesis. Mutants containing transposon insertions in 70 different C. burnetii T4E genes, constituting a majority of the effector repertoire, were analyzed using a visual screen to detect vacuole biogenesis defects. HeLa cells were infected with the mutant strains at an MOI of 100, and vacuole phenotypes were determined after 5 days of infection. Mutants with vacuole biogenesis defects were complemented by either expression of the wild-type gene of interest in single copy at the Tn7 insertion site (cbu1387::Tn) or by expression of the wild-type gene on a multicopy plasmid. The mutant strains shown in panel A displayed CCVs that were smaller than CCVs in cells infected with the parental NMII strain (wild type). Infected cells were stained with an anti-Coxiella antibody (red), an anti-LAMP-1 antibody (green), and Hoechst dye (blue) prior to acquisition of the representative images. (B) The areas of the CCVs were quantified by averaging the areas of vacuoles of all infected cells from images at ×600 magnification across 10 random fields of view. *, P < 0.01 (by Student's t test).
The CCV biogenesis screen of the T4E mutant sublibrary independently confirmed that the cig2::Tn, cig57::Tn, coxCC8::Tn, and cbu1754::Tn mutants produced small vacuoles. The cbu0937::Tn mutant also produced small vacuoles, which validated screens conducted by other groups that identified Cbu0937 as being an effector important for CCV biogenesis (17, 19) (Table S1). Importantly, this screen identified seven additional T4E genes as potentially being important for CCV biogenesis. The mutants cbu0414::Tn, cbu0513::Tn, cbu0978::Tn, cbu1387::Tn, cbu1524::Tn, cbu1752::Tn, and cbu2028::Tn all displayed a small-vacuole phenotype in the visual screen (Fig. 2A). To better assess the defect in CCV biogenesis, we calculated the average size of the vacuoles generated by each T4E mutant and normalized these data to the size of the vacuoles created by the parental strain of C. burnetii (Fig. 2B). The vacuoles created by these T4E mutants were all significantly smaller than the vacuoles created by the parental strain. To demonstrate that the defect in vacuole size for each mutant was the result of the transposon insertion in the corresponding T4E gene, we complemented the defect by reintroducing the wild-type allele of each T4E gene either in trans on a plasmid (for the mutants cbu0414::Tn, cbu0513::Tn, cbu0978::Tn, cbu1524::Tn, cbu1752::Tn, and cbu2028::Tn) or in single copy through integration at an unlinked Tn7 site on the chromosome (for the cbu1387::Tn mutant) (Fig. 2A and B). Thus, the screening of this newly created sublibrary was successful in identifying T4E genes that contribute to Dot/Icm-dependent phenotypes that had been missed using previous approaches.
Infections at a high multiplicity of infection (MOI) can skew the size of CCVs because homotypic fusion of vacuoles formed by multiple C. burnetii bacteria that were internalized independently will enhance vacuole size before intracellular replication is initiated. Thus, infections were repeated at an MOI of 1 to examine the size of vacuoles created in cells infected with a single bacterium. For these studies, HeLa cells were infected with the parental C. burnetii strain as a positive control and with the type IV-deficient icmL::Tn mutant as a negative control in parallel to infections with the seven mutant strains identified in this study (Fig. S1A). Measurements of CCV area demonstrated that the cbu0414::Tn, cbu0513::Tn, cbu0978::Tn, cbu1387::Tn, cbu1524::Tn, cbu1752::Tn, and cbu2028::Tn mutants created CCVs that were smaller than the CCVs formed by the parental strain of C. burnetii but larger than the vacuoles containing the icmL::Tn mutant (Fig. S1B). Thus, these data confirm that these newly identified effector mutants have a defect in vacuole biogenesis but do not have a CCV biogenesis defect that is equivalent to that of a Dot/Icm-deficient strain.
Although the initial screen of the effector mutant sublibrary reidentified several effector mutants shown previously to have CCV biogenesis defects, there were several effector mutants reported to have CCV biogenesis defects that were not identified in the sublibrary screen. To make certain that a defect was not missed due to use of a high MOI in the initial screen, we reexamined these other effector mutants in a low-MOI assay. In this low-MOI assay, no obvious vacuole biogenesis defects were observed for the mutants cbu0041::Tn, cbu0425::Tn, cbu0388::Tn, cbu0069::Tn, and cbu1665::Tn (Fig. S1), which were mutants deficient for effectors reported previously as being important for vacuole biogenesis and intracellular replication (15, 17). Thus, the seven effector mutants identified in this screen have a vacuole biogenesis defect that is more severe than that of several other effectors reported previously to be involved in this process.
Newly identified T4E proteins that are important for CCV biogenesis contribute to intracellular replication of C. burnetii.
Several previously identified T4E mutants with CCV biogenesis defects are also attenuated for intracellular replication (16). To determine if the newly identified T4E mutants with CCV biogenesis defects have corresponding reductions in intracellular replication, we measured genome equivalents (GE) over 7 days of intracellular infection for each mutant. The T4E mutants cbu0414::Tn, cbu0513::Tn, cbu0978::Tn, cbu1387::Tn, cbu1524::Tn, and cbu1752::Tn had significantly reduced levels of intracellular replication in HeLa cells, and this reduction was complemented upon introduction of the wild-type allele of the mutated gene (Fig. 3A to F). Even though the cbu2028::Tn mutant had a defect in vacuole size, no measurable intracellular replication defect was observed in HeLa cells (Fig. 3G). This suggests that the cbu2028 gene is important for Dot/Icm-dependent vacuole expansion but that the CCV in which this mutant resides remains highly permissive for intracellular replication.
FIG 3.
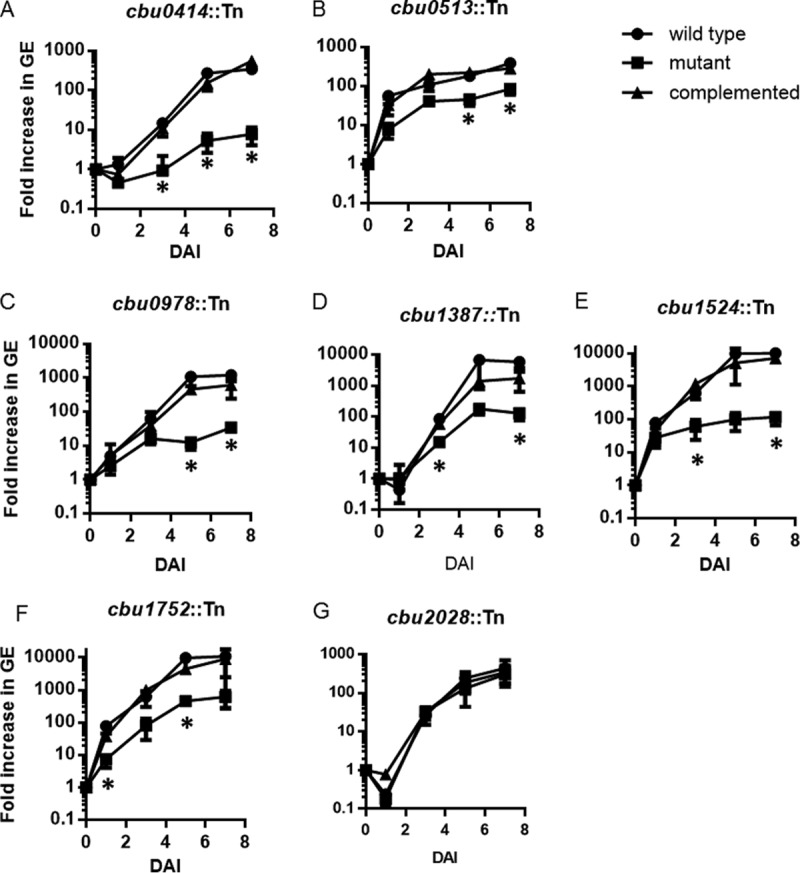
Intracellular replication kinetics of T4E mutants that displayed a small-CCV phenotype. Intracellular replication over 7 days was determined by measuring fold increase in genomic equivalents at the indicated times relative to the level at first day of infection (time zero on the x axis). Each experiment was conducted independently with the parental NMII strain (wild type), the indicated transposon insertion mutant strain, and the complemented mutant strain analyzed in parallel. Panels A to G show the data for mutant strains as indicated. *, P < 0.01, for values compared to those of the wild-type strain (two-tailed Student's t test). DAI, days after inoculation.
Newly identified T4E proteins are important for CCV biogenesis and intracellular replication in both macrophages and epithelial cells.
Vacuole biogenesis and intracellular replication of the newly identified T4E mutants were analyzed in multiple cell lines to make certain the phenotypes displayed by these mutants were not specific for the HeLa cell model of infection. Specifically, phenotypes for the T4E mutants were evaluated using Vero cells (an epithelial cell line derived from African green monkey), CHO cells (an epithelial cell line derived from Chinese hamster), J774 cells (a macrophage-like cell line derived from mice), and THP-1 cells (a macrophage-like cell line derived from humans). In all of the cell lines tested, the vacuoles containing the T4E mutants were significantly smaller than the vacuoles created by the NMII parental strain (Fig. 4B, D, F, and H). Likewise, intracellular replication of all the T4E mutants was reduced in all of the cell lines tested, as measured by fluorescence of the mCherry protein encoded on the transposon (Fig. 4A, C, E, and G). Because the cbu2028::Tn mutant displayed a minor defect in intracellular replication in the fluorescence-based assays but did not show reduced replication in HeLa cells, as determined by measuring genome equivalents, we further analyzed intracellular replication of the cbu2028::Tn mutant in these other cell lines using a GE assay (Fig. S2). Similar to what was observed in HeLa cells, the GE assay showed that there was no significant reduction in the intracellular replication of the cbu2028::Tn mutant in these other cell lines, confirming that Cbu2028 is a T4E that contributes to CCV expansion but is not required for bacterial intracellular replication. In summary, these data indicate that the T4E proteins identified in this screen have a broad and conserved role in promoting CCV biogenesis in both phagocytic and nonphagocytic mammalian host cells.
FIG 4.
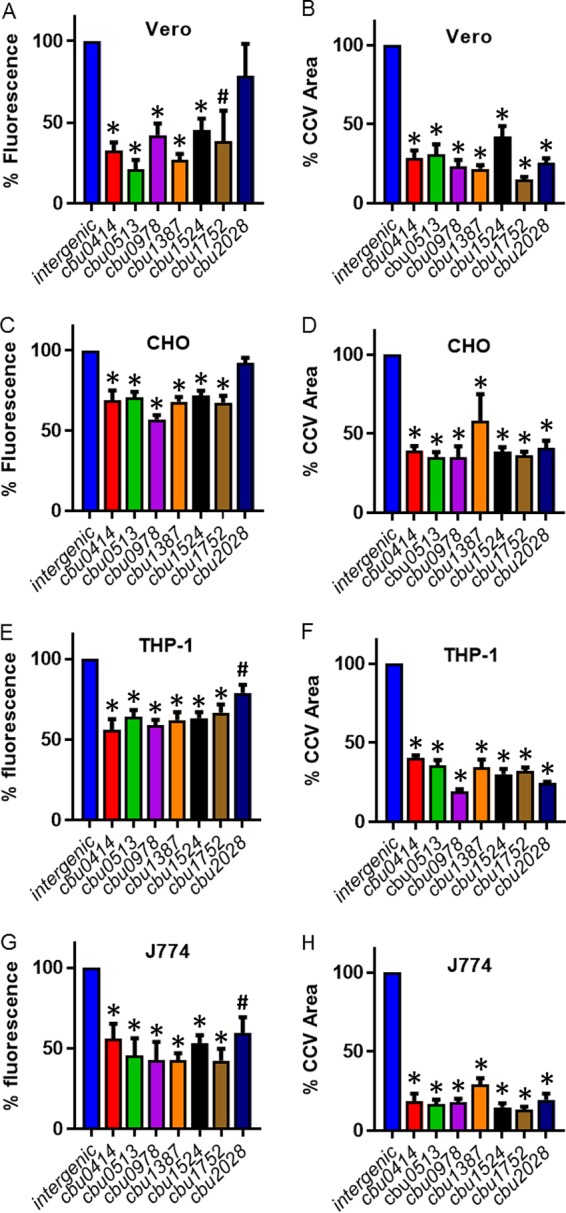
Vacuole biogenesis and intracellular replication phenotypes for the newly identified T4E mutants are conserved in both macrophages and epithelial cells. Intracellular replication was measured by fluorescence from the mCherry fluorescent protein that is encoded on the transposon. A control strain having the transposon inserted in an intergenic region was used as the positive control because this strain does not have a replication defect or vacuole biogenesis defect compared to the parental NMII strain. The infections were conducted in 96-well plates at an MOI of 20, and total fluorescence was measured using a plate reader. The measurements were taken at 5 days postinfection, and the readings from 10 replicate wells were averaged. Results from independent infections are shown in panels A, C, E, and G for the indicated cell lines. To determine the size of the CCVs, the areas of vacuoles of all infected cells in low-magnification images across 10 randomly selected fields of view were averaged at 5 days postinfection, and results are shown in panels B, D, F, and H for the indicated cell lines. CCVs were visualized after cells were stained with anti-Coxiella and anti-LAMP-1 antibodies, and overlapping areas for both anti-Coxiella and anti-LAMP-1 were measured. *, P < 0.005; #, P < 0.05, for results relative to those with the wild type (two-tailed Student's t test).
Subcellular localization of T4E proteins expressed ectopically.
Ectopic expression of the newly identified T4E proteins in mammalian host cells was conducted to determine if they have the capacity to localize to specific subcellular organelles. We did not include the proteins Cbu0414, Cbu0513, and Cbu1524 in this analysis because previous studies have examined the localizations of these T4E proteins following ectopic production in mammalian cells (10, 17). Cbu0414 and Cbu513 were reported to localize to the host cell cytosol, and Cbu1524 was reported to localize to the host cell nucleus (10, 17). Cbu1461 was used as a control effector reported previously to localize to the host cytosol (17). HeLa cells infected persistently with the parental NMII strain of C. burnetii were transfected with plasmids encoding effector proteins with three copies of a FLAG epitope (3×FLAG) tag: Cbu0978, Cbu1387, Cbu1752, Cbu2028, or Cbu1461. Cells were fixed, and the 3×FLAG-tagged proteins were stained using an anti-FLAG antibody. Immunofluorescence microscopy revealed that the ectopically produced effector proteins did not display strong localization to a discrete organelle in the cell but were enriched around the vacuole containing C. burnetii (Fig. 5). This suggests that these T4E proteins may interact with factors near the CCV membrane but do not have predicted transmembrane domains that mediate intimate attachment to this organelle.
FIG 5.

Subcellular localization of ectopically expressed type IV effectors in infected HeLa cells. HeLa 229 cells persistently infected with the NMII strain were transfected with plasmids encoding the 3×FLAG-tagged effector protein Cbu0978, Cbu1387, Cbu1752, Cbu2028, or Cbu1461. Cells were fixed 24 h after transfection and stained with anti-FLAG antibodies to view the epitope-tagged effector proteins (green), with anti-LAMP-1 to identify the CCV membrane (red), and with Hoechst to identify bacterial and host DNA (blue).
Vacuoles containing the cbu0513::Tn mutant arrest prior to fusion with autophagosomes.
The host protein LC3 has been shown to localize to mature vacuoles that support intracellular replication of C. burnetii, which is an indicator that fusion has occurred between the CCV and autophagosomes (11, 12, 28). The effector protein Cig2 is one of the factors that is critical for fusion of autophagosomes with the CCV (16, 29). To better characterize the CCV biogenesis defect displayed by the newly identified T4E mutants, we analyzed LC3 localization to the vacuoles containing these mutants. HeLa cells infected with either the parental strain NMII, cbu0414::Tn, cbu0513::Tn, cbu0978::Tn, cbu1387::Tn, cbu1524::Tn, cbu1752::Tn, or cbu2028::Tn were fixed after 4 days of infection, and immunofluorescence microscopy was used to examine localization of LC3 to the CCVs. Roughly 30% of the vacuoles containing the parental NMII strain stained positive for LC3 at this time (Fig. 6B). With the exception of the cbu0513 mutant-containing vacuoles, LC3 localization was observed on the vacuoles formed by the other effector mutants. Vacuoles containing the cbu0513::Tn mutant did not display LC3 localization; however, complementation of the cbu0513::Tn mutant with a plasmid encoding the wild-type cbu0513 allele restored LC3 localization to the CCV (Fig. 6C). This confirms that the LC3 localization defect displayed by this mutant was the result of cbu0513 inactivation. Thus, the defect in CCV biogenesis resulting from a deficiency in the effector protein Cbu0513 arrests vacuole maturation prior to fusion with autophagosomes, whereas the remaining T4E mutants have a vacuole biogenesis defect that does not affect fusion of the CCV with autophagosomes.
FIG 6.

LC3 localization to vacuoles containing C. burnetii transposon insertion mutants. HeLa cells were infected with C. burnetii or the indicated transposon insertion mutant and stained 4 days after infection with anti-LC3-II (green) and anti-Coxiella (red) antibodies. (A) Representative micrographs showing the presence or absence of LC3-II on vacuoles containing C. burnetii. (B) Quantification of the percentage of CCVs that stained positive for LC3-II. (C) Complementation of the cbu0513::Tn mutant restored LC3-II localization on the CCV. *, P < 0.05; **, P < 0.01 (Student's t test).
DISCUSSION
INSeq technology was used to map transposon insertion sites in an arrayed library of C. burnetii mutants. This technology allowed us to determine the locations of individual mutants in the arrayed library on a large scale. Results demonstrate that mapping data obtained using this technology can be leveraged to isolate a large subclass of C. burnetii mutants for focused genetic screens. We applied this approach to isolate a collection of clonally isolated and validated mutants deficient in different T4E proteins.
This T4E mutant sublibrary was created to screen for effectors that could be important for phenotypes that require the Dot/Icm secretion system. To establish proof of principle, the sublibrary was screened for T4E mutants that had CCV biogenesis defects, which allowed us to compare results using the sublibrary to our previous results where we screened the entire nonannotated library for mutants defective in CCV biogenesis (16). Screening the T4E mutant sublibrary identified effectors previously shown to be involved in CCV biogenesis. Importantly, seven additional effectors that were not previously identified as being important for CCV biogenesis were identified during this analysis.
There are multiple reasons why screening of the T4E mutant sublibrary led to the identification of additional effectors that participate in CCV biogenesis. Unlike using a large screen containing over 2,000 mutants, focusing on a smaller subset of mutants reduced the throughput of the screen, which made it easier to assess the phenotypes observed for each mutant using a visual approach. Additionally, the mapping data indicated that several effector mutants were in wells that contained additional independent mutants, and because these other mutants did not display a CCV biogenesis defect, the phenotype of the effector mutant was masked.
HeLa cells were used for the initial screen to identify mutants with aberrant CCV biogenesis phenotypes (Fig. 2A). The seven mutants with CCV biogenesis defects displayed similar phenotypes in several other cell lines, including macrophage-like cells from both human and mouse (Fig. 4). Thus, these seven effectors are of fundamental importance for CCV biogenesis irrespective of the cells being infected. This suggests that these effectors are subverting cellular processes essential for biogenesis of the CCV.
The cbu0414, cbu0978, cbu1387, and cbu2028 genes encode hypothetical proteins of unknown function and have been shown to be translocated in a T4SS-dependent manner (17, 30–32). The protein encoded by cbu0513 is annotated as a fructose-1,6-bisphosphatase but has also been shown to be a T4SS substrate (17). Thus, it is possible that the Cbu0513 protein has enzymatic activity that alters host metabolic function after it is translocated into the cytosol during infection. The effector protein Cbu1524 was named CaeA (Coxiella antiapoptosis effector A) because it was shown to inhibit host apoptosis when produced ectopically in mammalian cells (33, 34). The finding that a loss-of-function mutation in cbu1524 interferes with CCV biogenesis suggests that inhibition of host apoptosis might be important for intracellular replication, which could explain why C. burnetii encodes multiple effectors that disrupt host cell apoptosis (33, 34).
The cbu1752 gene appears to be located on a region of the chromosome that contains multiple effectors important for CCV biogenesis. In addition to cbu1752, the genes cbu1751 (cig57) and cbu1754 have been shown to be important for CCV biogenesis and intracellular replication (16). The similar phenotype displayed by these effector mutants and chromosomal linkage could indicate that these effector proteins work in concert to subvert a host cell pathway. The Cig57 protein encoded by cbu1751 has been shown to bind the host FCHO proteins to subvert clathrin-dependent vesicular transport (35). The effector protein CvpA has also been shown to subvert clathrin-dependent vesicular transport (14). Thus, C. burnetii has evolved multiple effectors to subvert this host pathway, which raises the possibility that cbu1752 also contributes to this pathway.
Ectopic expression of the T4E proteins in mammalian cells infected with C. burnetii showed enrichment of the effector near the CCV, which is consistent with these proteins participating in biogenesis of this unique organelle. In addition to subverting clathrin-dependent vesicular transport, the CCV fuses with host autophagosomes. The effector protein Cig2 was shown to be important for fusion of the CCV with autophagosomes and to promote homotypic fusion of independently derived CCVs in a single infected cell CCV (16, 29, 36). None of the effectors identified in this screen phenocopied a cig2 mutant strain, which displays a unique multivacuole phenotype but does not have a pronounced reduction in intracellular replication. However, similar to vacuoles containing a cig2::Tn mutant, the vacuoles containing the cbu0513::Tn mutant did not stain positive for the autophagy protein LC3. The cbu0513::Tn mutant had a reduction in intracellular replication, which indicates a severe defect in CCV biogenesis. Thus, these data suggest that the effector encoded by cbu0513 is important for an early stage in CCV biogenesis that precedes fusion of the vacuole with autophagosomes.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
C. burnetii Nine Mile phase II (NMII) strain, RSA493 (clone 4), was grown in a modified acidified citrate cysteine medium (ACCM-2) at 37°C in 5% CO2 and 2.5% O2 (22, 37). Mutant strains were grown with 3 μg/ml of chloramphenicol, and strains harboring complementation constructs were grown with 375 μg/ml kanamycin. Coxiella burnetii genome equivalents were quantified by quantitative PCR (qPCR) using primers specific to the dotA gene (38). Transposon mutagenesis of C. burnetii was carried out as previously described (16). Plasmid complementation was achieved by cloning the C. burnetii T4E genes into a pJB-Kan expression plasmid or a pMiniTn7T plasmid (13, 37).
Cell culture.
HeLa 229 cells, CHO cells, and Vero cells were grown in Dulbecco minimal Eagle's medium (DMEM) containing 5 or 10% fetal bovine serum (FBS) at 37°C in 5% CO2. J774.1 cells and THP-1 cells were grown in RPMI 1640 medium containing 5 or 10% fetal bovine serum at 37°C in 5% CO2.
Combinatorial mapping of individual insertion strains from arrayed library.
Each of the 5,240 insertion mutants was individually grown in 1 ml of ACCM-2 for 7 days. Every culture was then pelleted by centrifugation and resuspended in 100 μl of H2O and individually added into single wells of 96-well plates. Each resuspended bacterial mutant strain was then pipetted into a unique combination of 10 or 12 of a total of 24 pools using an EpMotion liquid handling robot (Eppendorf, Hamburg, Germany) for automated pipetting, as previously described (25, 26).
High-throughput insertion mapping by deep sequencing.
The genomic DNA of all of the mutants in each of the 24 generated pools was isolated as previously described (25, 26). Preparation of the DNA to be submitted for Illumina sequencing was done according to a modified version of a previously published protocol (27). Briefly, genomic DNA was sheared into fragments approximately 200 bp in length using a focused ultrasonicator set to a duty cycle of 10%, intensity of 5%, and number of cycles/burst set at 200 for 180 s (Covaris, Woburn, MA). Partially complementary adapters containing pool-specific adapters were ligated to the ends of the DNA fragments. Fragments containing transposon insertions were amplified by PCR using transposon-specific and adapter-specific primers (see Table S2 in the supplemental material). Two rounds of PCR, first for 10 cycles and then for 20 cycles using the products from the first PCR as the template for the second, were conducted. DNA fragments were size selected by cutting DNA in a range of 200 to 300 bp from a 2% agarose gel. The fragments were sequenced by Illumina sequencing, and the insertion site for each mutant was determined in parallel by INSeq as described previously (25, 26). The C. burnetii NMII strain, RSA493 version 3, in the NCBI database (accession number NC_002971.3) was used as the reference genome.
CCV biogenesis assay.
HeLa 229 cells, Vero cells, CHO cells, J774.1 cells, or THP-1 cells were plated in 24-well plates with 10-mm glass coverslips at a density of 5 × 104 cells per well 24 h prior to infection. Wild-type and transposon insertion mutant C. burnetii strains were grown to stationary phase in ACCM-2 and quantified by qPCR as described above. The bacteria were resuspended in DMEM containing 5% FBS for an MOI of 100, placed in a sonicator bath for 12 min to prevent bacterial aggregates, and added to the cells. Cells were washed with phosphate-buffered saline (PBS) at 4 h postinfection before the addition of fresh DMEM with 5% FBS. The infection was carried out for 120 h, at which point cells were fixed with 4% paraformaldehyde and then blocked and permeabilized in PBS containing 2% bovine serum albumin (BSA) and 0.05% saponin. Bacterial and host DNA was stained with Hoechst 33342 (Invitrogen), C. burnetii was stained with rabbit anti-C. burnetii polyclonal antibody at a concentration of 1:10,000, and the CCVs were stained using anti-LAMP-1 monoclonal H4A3 (Developmental Studies Hybridoma Bank) at a concentration of 1:1,000 in blocking buffer. Alexa Fluor 488 and 568 (Invitrogen) were the secondary antibodies used at 1:2,000 dilutions in blocking buffer, and the coverslips were subsequently mounted on slides using ProLong Gold (Invitrogen). The vacuoles were then visually assessed for vacuole biogenesis relative to that of the wild-type control. Digital images were acquired with a Nikon Eclipse TE2000-S inverted fluorescence microscope using a 60× objective lens and a Photometrics CoolSNAP EZ camera controlled by SlideBook (version 6.0) imaging software. To measure vacuole size by microscopy, images were acquired for 10 random fields of view using a 60× objective. SlideBook was used to determine the area of each CCV in these images. Staining for LAMP-1 was used to visualize the membrane surrounding each CCV in the image. An anti-Coxiella antibody was used to identify bacteria in each LAMP-1-positive CCV. Data for THP-1 cells were derived from images in which the sizes of 311 CCVs were measured, which resulted in an average of 39 CCVs analyzed for each strain and roughly 4 CCVs for each image acquired. Data for CHO cells were derived from images in which the sizes of 573 CCVs were measured, which resulted in an average of 72 CCVs analyzed for each strain and roughly 7 CCVs for each image acquired. Data for Vero cells were derived from images in which the sizes of 534 CCVs were measured, which resulted in an average of 67 CCVs analyzed for each strain and roughly 7 CCVs for each image acquired. Data for J774 cells were derived from images in which the sizes of 196 CCVs were measured, which resulted in an average of 25 CCVs analyzed for each strain and roughly 3 CCVs for each image acquired. Data for HeLa cells were derived from images in which the sizes of 776 CCVs were measured, which resulted in an average of 52 CCVs analyzed for each strain and roughly 5 CCVs for each image acquired.
C. burnetii replication assays.
HeLa 229 cells were plated in 24-well plates at a density of 5 × 104 cells per well at 24 h prior to infection. Wild-type and transposon insertion mutant C. burnetii bacteria were grown to stationary phase in ACCM-2 and quantified by qPCR as described above. Bacteria were pelleted, resuspended in DMEM to the experimental concentration (MOI of 20), and placed in a liquid sonicator for 12 min. The medium was aspirated from the cells, and the medium containing the bacteria for infection was added to each well. Cells were washed with PBS at 4 h postinfection before the addition of fresh DMEM with 5% FBS. A sample was taken at this time for the day 0 data and then again at 24, 72, 120, and 168 h postinfection for the day 1, 3, 5, and 7 data, respectively. Genomic DNA was extracted from these samples using an Illustra Bacteria GenomicPrep Mini Prep kit (GE Healthcare, Piscataway, NJ). Genomic equivalents were quantified by qPCR as described above.
CHO cells, Vero cells, THP-1 cells, or J774.1 cells were plated in 96-well plates at a density of 1 × 104 cells per well. Cells were infected and washed in the same manner as described above for HeLa cells. Fluorescence was read using a Tecan Infinite M1000 plate reader (Tecan, Mannedorf, Switzerland) and was measured as fold growth from day 0 after the background autofluorescence of uninfected cells was subtracted.
Effector subcellular localization experiments.
To examine the localization of ectopically produced 3×FLAG-tagged proteins, HeLa 229 cells persistently infected with wild-type C. burnetii were plated in 24-well plates at a density of 5 × 104 cells per well with 10-mm glass coverslips. Cells were transfected with plasmid DNA using TransIT LT-1 transfection reagent (Mirus, Madison, WI). The T4E genes were expressed on pcDNA4/TO plasmids expressing a 3×FLAG epitope tag. At 24 h after transfection cells were fixed with 4% paraformaldehyde and then blocked and permeabilized in PBS containing 2% BSA and 0.05% saponin. Bacterial and host DNA was stained with Hoechst 33342 (Invitrogen), C. burnetii was stained with rabbit anti-C. burnetii polyclonal antibody at a concentration of 1:10,000, and 3×FLAG epitope-tagged effector proteins were stained with anti-Flag M2 antibody (Sigma). Alexa Fluor 488 and 568 (Invitrogen) were the secondary antibodies used at 1:2,000 dilutions in blocking buffer, and the coverslips were subsequently mounted on slides using ProLong Gold (Invitrogen). Images were acquired as described above.
LC3 localization.
HeLa 229 cells were plated on 10-mm glass coverslips in 24-well plates at a density of 5 × 104 cells per well 24 h prior to infection. Cells were infected as previously described, and infection was allowed to proceed for 4 days. Cells were fixed and permeabilized with ice-cold methanol and then blocked with 2% BSA (Sigma). Bacterial and host DNA was stained with Hoechst 33342 (Invitrogen), C. burnetii was stained with rabbit anti-C. burnetii polyclonal antibody at a concentration of 1:5,000, and endogenous LC3-II was stained with mouse anti-LC3-II antibody at a 1:200 concentration (Nanotools, Teningen, Germany). Secondary antibodies used were anti-mouse 488 wavelength and anti-rabbit 568 wavelength at 1:2,000 concentration. The coverslips were subsequently mounted on slides using ProLong Gold (Invitrogen). Images were acquired as described above. The percentage of infected cells with LC3-II on the CCV was quantified by counting CCVs in 10 randomly chosen fields of view, and it was visually determined if LC3-II was colocalized with the CCVs.
Supplementary Material
ACKNOWLEDGMENTS
We thank Shawna Reed for the cbu1387::Tn complemented strain and members of the Roy lab for their input and suggestions.
This work was funded by the National Institutes of Health (NIH) (F32 GM 108411 R01 AI 114760 and R35 GM118159). A.L.G. is supported by the Burroughs Wellcome Fund and the HHMI Faculty Scholars Program.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00758-17.
REFERENCES
- 1.Honarmand H. 2012. Q Fever: an old but still a poorly understood disease. Interdiscip Perspect Infect Dis 2012:131932. doi: 10.1155/2012/131932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eldin C, Melenotte C, Mediannikov O, Ghigo E, Million M, Edouard S, Mege JL, Maurin M, Raoult D. 2017. From Q fever to Coxiella burnetii infection: a paradigm change. Clin Microbiol Rev 30:115–190. doi: 10.1128/CMR.00045-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maurin M, Raoult D. 1999. Q fever. Clin Microbiol Rev 12:518–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moos A, Hackstadt T. 1987. Comparative virulence of intra- and interstrain lipopolysaccharide variants of Coxiella burnetii in the guinea pig model. Infect Immun 55:1144–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voth DE, Heinzen RA. 2007. Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell Microbiol 9:829–840. doi: 10.1111/j.1462-5822.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- 6.McCaul TF, Williams JC. 1981. Developmental cycle of Coxiella burnetii: structure and morphogenesis of vegetative and sporogenic differentiations. J Bacteriol 147:1063–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vishwanath S, Hackstadt T. 1988. Lipopolysaccharide phase variation determines the complement-mediated serum susceptibility of Coxiella burnetii. Infect Immun 56:40–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoover TA, Culp DW, Vodkin MH, Williams JC, Thompson HA. 2002. Chromosomal DNA deletions explain phenotypic characteristics of two antigenic variants, phase II and RSA 514 (crazy), of the Coxiella burnetii Nine Mile strain. Infect Immun 70:6726–6733. doi: 10.1128/IAI.70.12.6726-2733.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McDonough JA, Newton HJ, Roy CR. 2012. Coxiella burnetii secretion systems. Adv Exp Med Biol 984:171–197. doi: 10.1007/978-94-007-4315-1_9. [DOI] [PubMed] [Google Scholar]
- 10.Carey KL, Newton HJ, Luhrmann A, Roy CR. 2011. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog 7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gutierrez MG, Vazquez CL, Munafo DB, Zoppino FC, Beron W, Rabinovitch M, Colombo MI. 2005. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol 7:981–993. doi: 10.1111/j.1462-5822.2005.00527.x. [DOI] [PubMed] [Google Scholar]
- 12.Romano PS, Gutierrez MG, Beron W, Rabinovitch M, Colombo MI. 2007. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell Microbiol 9:891–909. doi: 10.1111/j.1462-5822.2006.00838.x. [DOI] [PubMed] [Google Scholar]
- 13.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2:e00175-. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Larson CL, Beare PA, Howe D, Heinzen RA. 2013. Coxiella burnetii effector protein subverts clathrin-mediated vesicular trafficking for pathogen vacuole biogenesis. Proc Natl Acad Sci U S A 110:E4770–E4779. doi: 10.1073/pnas.1309195110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez E, Cantet F, Fava L, Norville I, Bonazzi M. 2014. Identification of OmpA, a Coxiella burnetii protein involved in host cell invasion, by multi-phenotypic high-content screening. PLoS Pathog 10:e1004013. doi: 10.1371/journal.ppat.1004013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newton HJ, Kohler LJ, McDonough JA, Temoche-Diaz M, Crabill E, Hartland EL, Roy CR. 2014. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog 10:e1004286. doi: 10.1371/journal.ppat.1004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weber MM, Chen C, Rowin K, Mertens K, Galvan G, Zhi H, Dealing CM, Roman VA, Banga S, Tan Y, Luo ZQ, Samuel JE. 2013. Identification of C. burnetii type IV secretion substrates required for intracellular replication and Coxiella-containing vacuole formation. J Bacteriol 195:3914–3924. doi: 10.1128/JB.00071-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohler LJ, Roy CR. 2015. Biogenesis of the lysosome-derived vacuole containing Coxiella burnetii. Microbes Infect 17:766–771. doi: 10.1016/j.micinf.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larson CL, Martinez E, Beare PA, Jeffrey B, Heinzen RA, Bonazzi M. 2016. Right on Q: genetics begin to unravel Coxiella burnetii host cell interactions. Future Microbiol 11:919–939. doi: 10.2217/fmb-2016-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moffatt JH, Newton P, Newton HJ. 2015. Coxiella burnetii: turning hostility into a home. Cell Microbiol 17:621–631. doi: 10.1111/cmi.12432. [DOI] [PubMed] [Google Scholar]
- 21.Omsland A, Cockrell DC, Fischer ER, Heinzen RA. 2008. Sustained axenic metabolic activity by the obligate intracellular bacterium Coxiella burnetii. J Bacteriol 190:3203–3212. doi: 10.1128/JB.01911-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, Porcella SF, Heinzen RA. 2009. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci U S A 106:4430–4434. doi: 10.1073/pnas.0812074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beare PA. 2012. Genetic manipulation of Coxiella burnetii. Adv Exp Med Biol 984:249–271. doi: 10.1007/978-94-007-4315-1_13. [DOI] [PubMed] [Google Scholar]
- 24.Beare PA, Howe D, Cockrell DC, Omsland A, Hansen B, Heinzen RA. 2009. Characterization of a Coxiella burnetii ftsZ mutant generated by Himar1 transposon mutagenesis. J Bacteriol 191:1369–1381. doi: 10.1128/JB.01580-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, Gordon JI. 2009. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6:279–289. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodman AL, Wu M, Gordon JI. 2011. Identifying microbial fitness determinants by insertion sequencing using genome-wide transposon mutant libraries. Nat Protoc 6:1969–1980. doi: 10.1038/nprot.2011.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong SM, Gawronski JD, Lapointe D, Akerley BJ. 2011. High-throughput insertion tracking by deep sequencing for the analysis of bacterial pathogens. Methods Mol Biol 733:209–222. doi: 10.1007/978-1-61779-089-8_15. [DOI] [PubMed] [Google Scholar]
- 28.Beron W, Gutierrez MG, Rabinovitch M, Colombo MI. 2002. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun 70:5816–5821. doi: 10.1128/IAI.70.10.5816-5821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohler LJ, Reed SR, Sarraf SA, Arteaga DD, Newton HJ, Roy CR. 2016. Effector protein Cig2 decreases host tolerance of infection by directing constitutive fusion of autophagosomes with the Coxiella-containing vacuole. mBio 7:e01127-. doi: 10.1128/mBio.01127-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, Tan Y, Luo ZQ, Samuel JE. 2010. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc Natl Acad Sci U S A 107:21755–21760. doi: 10.1073/pnas.1010485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lifshitz Z, Burstein D, Peeri M, Zusman T, Schwartz K, Shuman HA, Pupko T, Segal G. 2013. Computational modeling and experimental validation of the Legionella and Coxiella virulence-related type-IVB secretion signal. Proc Natl Acad Sci U S A 110:E707–E715. doi: 10.1073/pnas.1215278110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lifshitz Z, Burstein D, Schwartz K, Shuman HA, Pupko T, Segal G. 2014. Identification of novel Coxiella burnetii Icm/Dot effectors and genetic analysis of their involvement in modulating a mitogen-activated protein kinase pathway. Infect Immun 82:3740–3752. doi: 10.1128/IAI.01729-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bisle S, Klingenbeck L, Borges V, Sobotta K, Schulze-Luehrmann J, Menge C, Heydel C, Gomes JP, Luhrmann A. 2016. The inhibition of the apoptosis pathway by the Coxiella burnetii effector protein CaeA requires the EK repetition motif, but is independent of survivin. Virulence 7:400–412. doi: 10.1080/21505594.2016.1139280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klingenbeck L, Eckart RA, Berens C, Luhrmann A. 2013. The Coxiella burnetii type IV secretion system substrate CaeB inhibits intrinsic apoptosis at the mitochondrial level. Cell Microbiol 15:675–687. doi: 10.1111/cmi.12066. [DOI] [PubMed] [Google Scholar]
- 35.Latomanski EA, Newton P, Khoo CA, Newton HJ. 2016. The effector Cig57 hijacks FCHO-mediated vesicular trafficking to facilitate Intracellular replication of Coxiella burnetii. PLoS Pathog 12:e1006101. doi: 10.1371/journal.ppat.1006101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez E, Allombert J, Cantet F, Lakhani A, Yandrapalli N, Neyret A, Norville IH, Favard C, Muriaux D, Bonazzi M. 2016. Coxiella burnetii effector CvpB modulates phosphoinositide metabolism for optimal vacuole development. Proc Natl Acad Sci U S A 113:E3260–E3269. doi: 10.1073/pnas.1522811113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Omsland A, Beare PA, Hill J, Cockrell DC, Howe D, Hansen B, Samuel JE, Heinzen RA. 2011. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl Environ Microbiol 77:3720–3725. doi: 10.1128/AEM.02826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coleman SA, Fischer ER, Howe D, Mead DJ, Heinzen RA. 2004. Temporal analysis of Coxiella burnetii morphological differentiation. J Bacteriol 186:7344–7352. doi: 10.1128/JB.186.21.7344-7352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.