

ABSTRACT
To understand the role of class I major histocompatibility complex (MHC-I) and class II MHC (MHC-II) antigen presentation pathways in host defense against Coxiella burnetii infection, we examined whether MHC-I or MHC-II deficiency in mice would significantly influence their susceptibility to virulent C. burnetii Nine Mile phase I (NMI) infection. The results indicate that NMI infection induced more severe disease in both MHC-I-deficient and MHC-II-deficient mice than in wild-type (WT) mice, while only MHC-I-deficient mice developed a severe persistent infection and were unable to control bacterial replication. These results suggest that both MHC-I-restricted CD8+ T cells and MHC-II-restricted CD4+ T cells contribute to host defense against primary C. burnetii infection, while MHC-I-restricted CD8+ T cells appear to play a more critical role in controlling bacterial replication. Additionally, although NMI infection induced more severe disease in TAP1-deficient mice than in their WT counterparts, TAP1 deficiency in mice did not significantly influence their ability to eliminate C. burnetii. This suggests that C. burnetii antigen presentation to CD8+ T cells by the MHC-I classical pathway may depend only partially on TAP1. Furthermore, granzyme B deficiency in mice did not significantly alter their susceptibility to C. burnetii infection, but perforin-deficient mice were unable to control host inflammatory responses during primary C. burnetii infection. These results suggest that perforin, but not granzyme B, is required for C. burnetii antigen-specific cytotoxic CD8+ T cells to control primary C. burnetii infection.
KEYWORDS: CTL, Coxiella burnetii, MHC-I, MHC-II, TAP, antigen presentation, granzyme, immune response, perforin
INTRODUCTION
Coxiella burnetii is an obligate intracellular Gram-negative bacterium that causes a worldwide zoonotic disease, Q fever, in humans. It is typically spread via aerosols produced by infected domestic animals, such as cattle, sheep, and goats (1). This makes C. burnetii an occupational hazard for people who closely work with livestock (2, 3). Q fever usually manifests as an acute illness but occasionally can develop into a severe chronic infection. Acute Q fever is characterized by high fever, atypical pneumonia, headache, and hepatitis, but it normally resolves within 2 to 3 weeks with or without antibiotic treatment. In contrast, chronic Q fever typically manifests as culture-negative endocarditis, most often in patients with preexisting heart damage and patients who are immunocompromised (4–7). This form of infection occurs in fewer than 5% of infected individuals but can have devastating consequences. Treatment involves a dual regimen of antibiotics over a period of 18 months to 3 years and typically requires surgical intervention to remove infected tissue (5). Even with treatment, chronic infection is fatal in 26 to 60% of cases (8). A recent outbreak in the Netherlands from 2007 to 2010 resulted in more than 3,500 reported human infections (9), highlighting that this worldwide zoonotic pathogen remains a significant threat to public health. Although the existing formalin-inactivated phase I whole-cell vaccine (Q-Vax) confers complete protection against clinical Q fever, widespread use of this vaccine is limited by its high incidence of adverse reactions, especially in individuals with prior immunity to C. burnetii (10, 11). Therefore, it is necessary to develop a safe and effective vaccine for prevention of human Q fever.
Understanding the immunological basis of host defense against C. burnetii infection is critical for developing a safe and effective new-generation vaccine against Q fever. Recently, efforts have been devoted to understanding the mechanisms of host immune responses to primary C. burnetii infection in mouse models. Andoh et al. have demonstrated that T cells and gamma interferon (IFN-γ) are essential for clearance of primary C. burnetii infection (12, 13). Additionally, it has been reported that either CD4+ or CD8+ T cells are able to control a primary pulmonary C. burnetii infection, suggesting that CD4+ or CD8+ T cells are required for host defense against a primary C. burnetii infection (14). These studies indicate that T cell-mediated immunity may be the essential mechanism for host defense against primary C. burnetii infection. However, the detailed role of CD4+ and CD8+ T cells in host defense against primary C. burnetii infection remains unclear.
The major histocompatibility complex (MHC) is a set of cell surface proteins essential for the acquired immune system to recognize foreign molecules in vertebrates, with its main function being to bind antigens derived from pathogens and display them on the cell surface for recognition by the appropriate T cells. MHC-I presents endogenous peptides to CD8+ T cells by the classical pathway via the endoplasmic reticulum (ER) and the transporter associated with antigen processing (TAP) complex (15). The classical TAP-dependent MHC-I pathway has been demonstrated to be critical in host defense against Mycobacterium tuberculosis (16). In addition, mice lacking β2-microglobulin (β2m), a component of the MHC-I chain, cannot stably present antigens on the surfaces of their cells. Recent studies using β2m knockout (KO) mice have shown that MHC-I plays an important role in host defense against several intracellular bacterial pathogens, including M. tuberculosis (17, 18), Listeria monocytogenes (15), and Chlamydia (19). The evidence that M. tuberculosis infection induces severe disease and a significant reduction of CD8+ T cells in MHC-I-deficient β2 m−/− mice indicates that MHC-I-restricted antigen-specific CD8+ T cells are crucial for host defense against M. tuberculosis infection (17, 18). In contrast, MHC-II molecules are present only on the surfaces of antigen-presenting cells, such as dendritic cells, B cells, and macrophages. MHC-II presents antigens from phagocytosed compounds to CD4+ T cells, which is critical for the expansion and function of CD4+ T cells during host immune responses (20). MHC-II is particularly important to sustain a long-term immune response during vaccination. Experimental vaccines against malaria, influenza, and Helicobacter pylori have been reported to require the MHC-II antigen presentation pathway (21–23). Despite previous studies demonstrating that both CD4+ and CD8+ T cells are involved in host defense against C. burnetii infection, the role of MHC-I and -II antigen presentation pathways in host defense against C. burnetii infection has not been investigated.
In this study, we examined the roles of MHC-I and MHC-II during primary C. burnetii infection in mice. Our results indicate that both MHC-II-restricted CD4+ T cells and MHC-I-restricted CD8+ T cells contribute to host defense against primary C. burnetii infection, while MHC-I-restricted CD8+ T cells appear to play a critical role in controlling bacterial replication.
RESULTS
Both MHC-I and -II molecules are important for host defense against primary C. burnetii infection.
To understand the roles of MHC-I and -II molecules in host defense against primary C. burnetii infection, we examined whether MHC-I or -II deficiency in mice would significantly influence their susceptibility to virulent C. burnetii Nine Mile phase I (NMI) intraperitoneal (i.p.) infection. The severity of disease induced by C. burnetii infection in wild-type (WT), MHC-I-deficient (β2m KO), and MHC-II-deficient mice was evaluated by comparing body weight loss, splenomegaly, bacterial burden, and pathological changes in the spleen on different days postinfection (dpi). Compared to WT mice, β2m KO (day 8, P = 0.0154; day 11, P = 0.0242) and MHC-II-deficient (day 8, P = 0.0478) mice had transitional body weight loss during early stages of infection (Fig. 1A). These observations suggest that both MHC-I and -II molecules are required for protecting C. burnetii-infected hosts from developing disease during early stages of infection. As shown in Fig. 1B, both β2m KO (P = 0.0082) and MHC-II-deficient (P = 0.0189) mice showed significantly higher splenomegaly than WT mice at 14 dpi, while splenomegaly was similar in WT and MHC-II-deficient mice (P = 0.9931) but was significantly lower than that in β2m KO mice (P = 0.0031) at 28 dpi. Interestingly, C. burnetii genomic copies in the spleen were similar in WT and MHC-II-deficient mice (P = 0.8511), but significantly more C. burnetii genomic copies were detected in the spleens from β2m KO mice at 7 (P = 0.0027) and 28 (P = 0.0006) dpi (Fig. 1C). These results indicate that C. burnetii can induce a persistent bacterial infection in β2m KO mice, suggesting MHC-I-restricted CD8+ T cells may play a critical role in clearance of bacteria during C. burnetii primary infection. Histopathological differences were observed in the spleens from WT and β2m KO or MHC-II-deficient mice. As shown in Fig. 1D, moderate to large multifocal accumulations of macrophages (encircled by a black border) were present in red pulp of spleens from both β2m KO and MHC-II-deficient mice, but few small accumulations of macrophages (encircled by black border) were observed in red pulp of spleens from WT mice at 14 and 28 dpi. The histology score in β2m KO or MHC-II-deficient mice was significantly higher than that in WT mice at both 14 (P < 0.0001) and 28 (P < 0.0001 and P = 0.0010, respectively) dpi (Fig. 1E). These results suggest that both MHC-I and -II molecules are required for regulating host inflammatory responses during C. burnetii primary infection. Collectively, these data demonstrate that both MHC class I and class II molecules are required, but MHC-I molecules appear to play a more critical role in host defense against primary C. burnetii infection.
FIG 1.

MHC-I-deficient β2m mice have more severe infection with NMI. Deficient and WT mice were infected i.p. with 1 × 107 NMI organisms. Disease severity was assessed at 7, 14, and 28 dpi. (A) Relative body weight (current body weight/day 0 body weight) was measured throughout infection to monitor disease severity. (B) Splenomegaly was determined as a ratio of spleen weight to body weight. (C) Bacterial burden in the spleen was determined by real-time PCR and reported as log10 C. burnetii com1 gene copy numbers. (D) Spleen sections from wild-type and β2m KO mice at day 14 (top panels) and day 28 (bottom panels). (E) Spleens (1 section per mouse) from both groups were scored for histiocytic inflammation in red pulp of spleen using the following scale: 0, none (no accumulations of macrophages); 1, few small accumulations of macrophages; 2, few small to moderate accumulations of macrophages; 3, large numbers of moderate to large accumulations of macrophages. Much of the increase in volumes of spleens in all groups was a result of extramedullary hematopoiesis. The data presented for each group are the average with standard deviation for four mice. Results were analyzed by one-way (B, C, and E) or two-way (A) ANOVA with Tukey's multiple-comparison test for statistical significance. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Presentation of C. burnetii antigens by the MHC-I classical pathway depends partially on TAP1.
The observation that C. burnetii induced more severe disease and persistent bacterial infection in β2m KO mice than in their WT counterparts suggests that a functional MHC-I antigen presentation pathway may be crucial for host defense against primary C. burnetii infection. Considering that TAP is an important component for presentation of endogenous peptides to CD8+ T cells by the MHC-I classical pathway (18), TAP may be involved modulating disease and bacterial clearance in β2m KO mice infected with C. burnetii. We utilized TAP1 knockout mice to examine whether MHC-I classical pathway deficiency in mice would significantly influence their susceptibility to C. burnetii infection. As shown in Fig. 2A, compared to WT mice, TAP1− mice had transitional body weight loss at 8 dpi. In addition, although splenomegaly (P = 0.0111) (Fig. 2B) and C. burnetii genomic copies in the spleens (P = 0.0161) (Fig. 2C) from TAP1− mice were significantly higher than in WT mice at 14 dpi, there was no significant difference in either splenomegaly (P = 0.2717) or C. burnetii genomic copies in the spleens (P = 0.6155) between TAP1− and WT mice at 28 dpi. These results indicate that C. burnetii induced more severe infection in TAP1− mice than in their WT counterparts during the early stage of infection, but TAP1 deficiency in mice may not significantly influence their ability to eliminate C. burnetii at later stages of infection. These results suggest that C. burnetii antigen presentation by the MHC-I classical pathway may depend only partially on TAP1 and that TAP-independent pathways may also be involved in presenting antigen to CD8+ T cells during C. burnetii primary infection.
FIG 2.
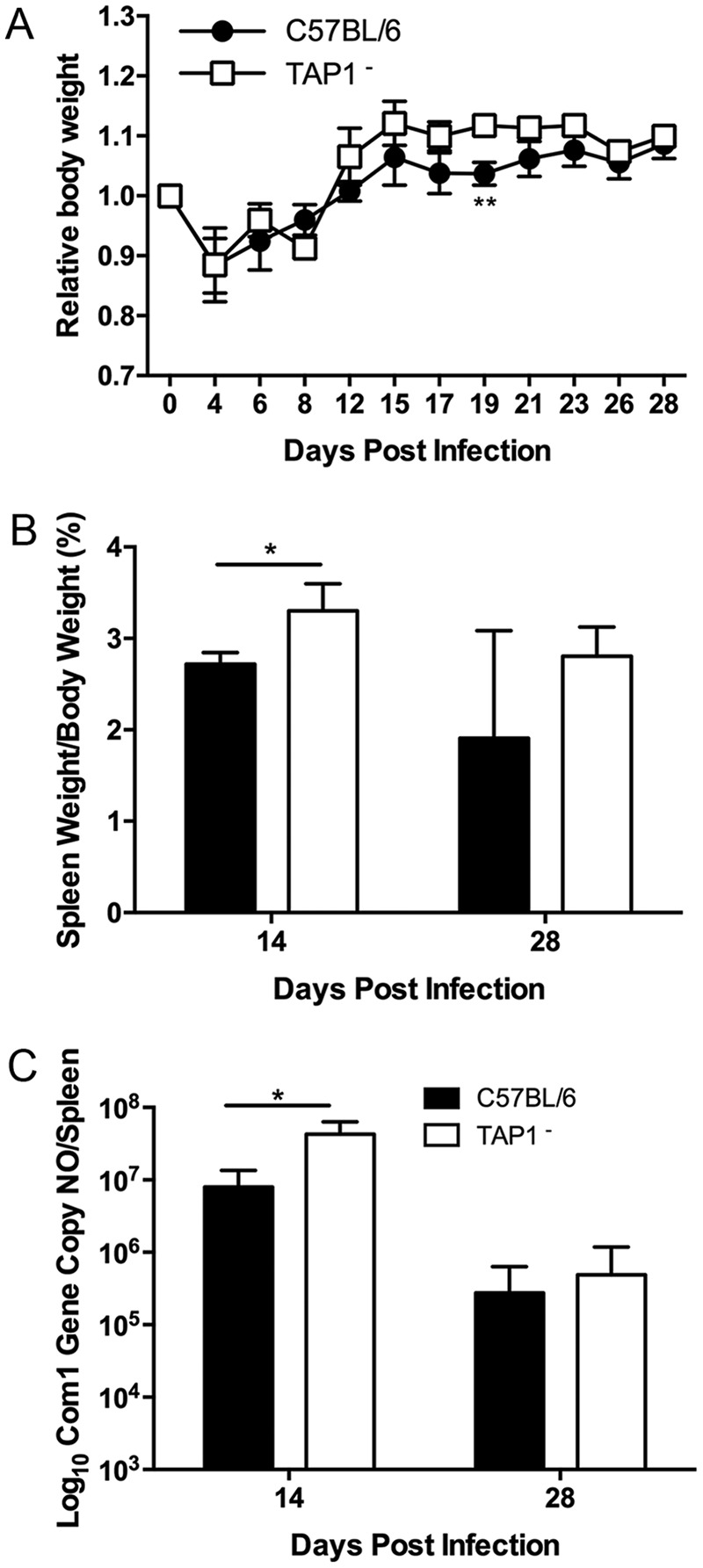
Antigen presentation of NMI by MHC-I is partially dependent on TAP1. Deficient and WT mice were infected i.p. with 1 × 107 NMI organisms. Disease severity was assessed at 14 and 28 dpi. (A) Relative body weight was determined throughout infection for monitoring disease severity. (B) Splenomegaly was determined as a ratio of spleen weight to body weight. (C) Bacterial burden in the spleen was determined by real-time PCR and reported as log10 C. burnetii com1 gene copy numbers. These data indicate that TAP1-deficient mice had more severe disease following infection with NMI at 14 dpi. The data presented for each group are the average with standard deviation for four mice. Results were analyzed by t test (B and C) or two-way ANOVA with Sidak's multiple-comparison test (A) for statistical significance. *, P < 0.05; **, P < 0.01.
C. burnetii infection induces elevated levels of IFN-γ in β2m- and TAP1-deficient mice.
The observation that C. burnetii NMI induces a lethal disease in IFN-γ-deficient mice (12, 13) suggests that IFN-γ plays a critical role in host defense against C. burnetii infection. Since antigen-specific CD8+ T cells can produce IFN-γ during host immune responses to microbial infections, the defects in TAP-dependent MHC-I-restricted CD8+ T cells may result in lower levels of IFN-γ during primary C. burnetii infection. To test this hypothesis, we analyzed the levels of IFN-γ in serum samples from NMI-infected WT, β2m KO, and TAP1-deficient mice at 14 dpi. Surprisingly, significantly higher levels of IFN-γ were detected in sera from NMI-infected β2m KO (P = 00182) (Fig. 3A) and TAP1− (P = 0.0016) (Fig. 3B) mice than in sera from WT mice at 14 dpi. These results suggest that the fact that C. burnetii infection induced more severe disease in β2m KO and TAP1− mice may not be due to the reduction of IFN-γ-producing CD8+ T cells.
FIG 3.
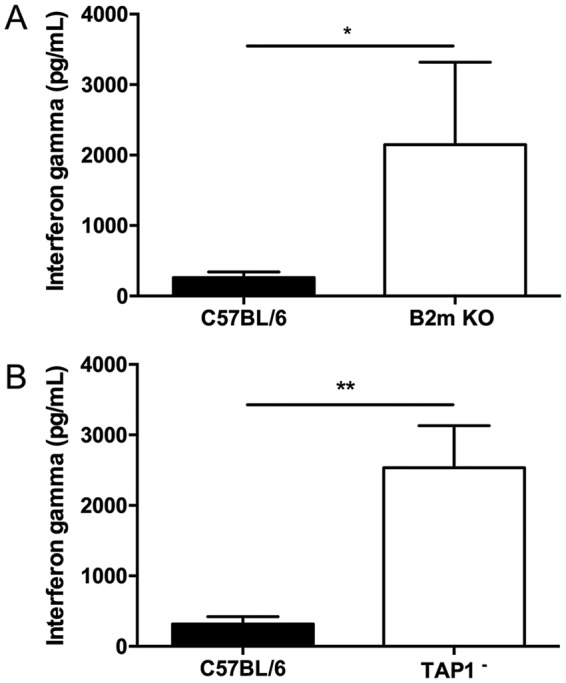
MHC-I-deficient β2m and TAP1-deficient mice have higher levels of serum IFN-γ following infection with NMI. Serum was collected at 14 dpi, and levels of IFN-γ were measured by ELISA. (A) Serum IFN-γ levels from β2m KO mice at 14 dpi. (B) Serum IFN-γ levels from TAP1− mice 14 dpi. These data indicate that serum levels of IFN-γ were higher in both β2m and TAP1 knockout mice following infection with NMI. The data presented for each group are the average with standard deviation for four serum samples. Results were analyzed by t test for statistical significance. *, P < 0.05; **, P < 0.01.
IFN-γ-producing CD8+ T cells are involved in controlling bacterial burden during primary C. burnetii infection.
To determine whether IFN-γ producing CD8+ T cells play an important role in host defense against primary C. burnetii infection, we examined whether there were differences between WT CD8+ T cells and IFN-γ-deficient CD8+ T cells in their ability to protect against C. burnetii NMI infection when adoptively transferred into recombination activating gene 1 (Rag1)-deficient mice. As shown in Fig. 4A, both WT CD8+ T cell recipients (day 12, P = 0.0005; day 14, P = 0.0002) and IFN-γ-deficient CD8+ T cell recipients (day 12, P = 0.0007; day 14, P < 0.0001) showed significantly higher body weights than control Rag1 KO mice beginning at 12 dpi. In addition, there was no significant difference in splenomegaly among the different groups of mice (P = 0.9149, Fig. 4B). However, although C. burnetii genomic copies in the spleens were similar in Rag1 KO and IFN-γ-deficient CD8+ T cell recipient mice (P = 0.4548), fewer C. burnetii genomic copies were detected in the spleens from WT CD8+ T cell recipient mice at 14 dpi (P = 0.0572) (Fig. 4C). These results indicate that IFN-γ-producing CD8+ T cells may not be important for protecting against C. burnetii infection-induced disease, but they may play a role in controlling bacterial replication during C. burnetii infection.
FIG 4.
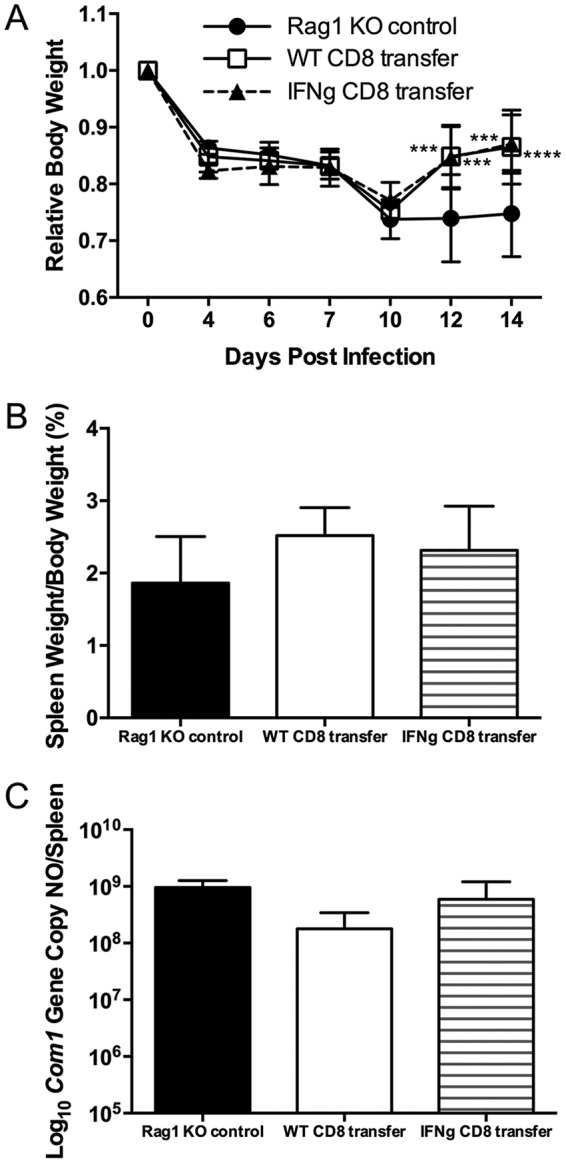
Adoptive transfer of wild-type but not IFN-γ-deficient CD8+ T cells lessens bacterial burden in Rag1 knockout mice following NMI infection. Rag1 knockout mice received 5 × 106 CD8+ T cells from WT or IFN-γ knockout mice at 24 h prior to i.p. infection with 1 × 107 NMI organisms. Disease severity was assessed at 14 dpi. (A) Relative body weight was determined throughout infection to monitor disease severity. (B) Splenomegaly was determined as a ratio of spleen weight to body weight. (C) Bacterial burden in the spleen was determined by real-time PCR and reported as log10 C. burnetii com1 gene copy numbers. These data indicate that adoptive transfer of WT, but not IFN-γ-deficient, CD8+ T cells helps to control bacterial burden in mice following NMI infection. The data presented for each group are the average with standard deviation for four mice. Results were analyzed by one-way (B and C) or two-way (A) ANOVA with Tukey's multiple-comparison test for statistical significance. ***, P < 0.001; ****, P < 0.0001.
C. burnetii infection induces more severe disease in granzyme B-deficient mice.
Granzyme B is a serine protease that is secreted by cytotoxic T lymphocytes (CTLs) along with the pore-forming protein perforin to induce apoptosis in target cells. To determine whether C. burnetii antigen-specific CTLs play an important role in host defense against primary C. burnetii infection, we investigated whether granzyme B deficiency in mice would significantly alter their ability to resist C. burnetii infection. As shown in Fig. 5A, granzyme B KO mice had significantly lower body weights than their WT counterparts throughout the NMI infection (day 15, P = 0.0189; day 19, P = 0.0096; day 23, P = 0.0296; day 26, P = 0.0402). This result indicates that C. burnetii infection induces a more severe disease in granzyme B-deficient mice, suggesting that CTLs may play an important role in protecting mice from development of severe disease. However, there was no significant difference in splenomegaly (day 14, P = 0.0881; day 28, P = 0.8862) (Fig. 5B) or C. burnetii genomic copies in the spleens (day 14, P = 0.2644; day 28, P = 0.5468) (Fig. 5C) between WT and granzyme B KO mice at 14 and 28 dpi. These results indicate that granzyme B may not play a critical role in controlling host inflammatory responses or bacterial replication during primary C. burnetii infection. Considering that granzymes have redundant roles during host immune responses, it is possible that other granzymes could overcome the granzyme B deficiency in mice, leading to control of C. burnetii infection in granzyme B KO mice.
FIG 5.
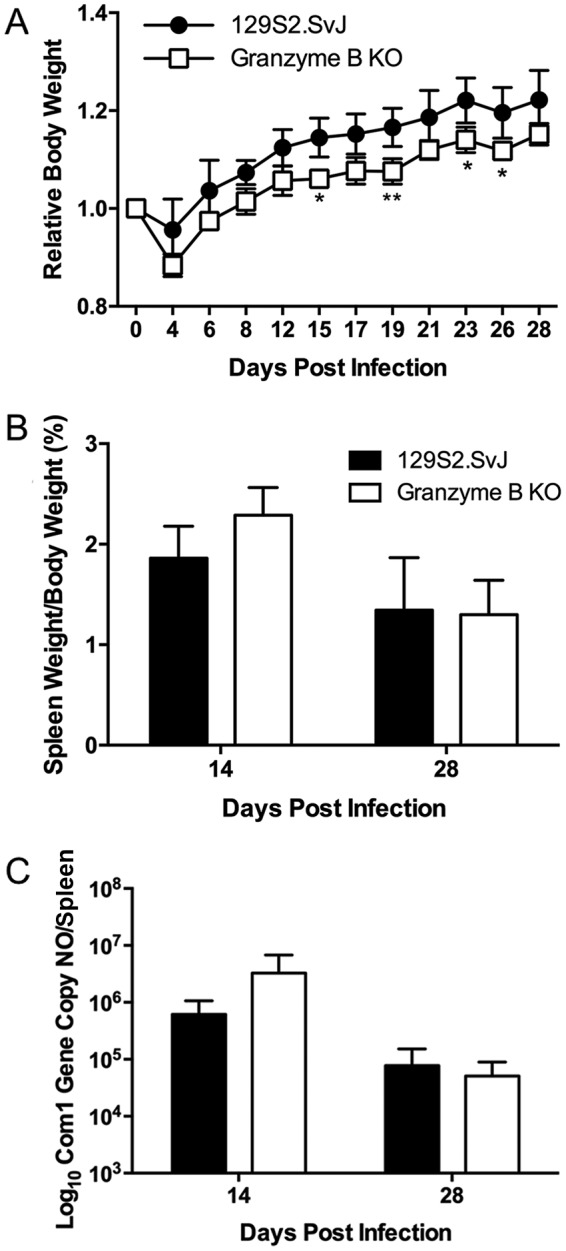
Granzyme B KO mice have more severe disease following infection with NMI. WT and granzyme B knockout mice were infected i.p. with 1 × 107 NMI organisms, and disease severity was assessed at 14 and 28 dpi. (A) Relative body weight was determined throughout infection to monitor disease severity. (B) Splenomegaly was determined as a ratio of spleen weight to body weight. (C) Bacterial burden in the spleen was determined by real-time PCR and reported as log10 C. burnetii com1 gene copy numbers. The data presented for each group are the average with standard deviation for four mice. Results were analyzed by t test (B and C) or two-way ANOVA with Sidak's multiple-comparison test (A) for statistical significance. *, P < 0.05; **, P < 0.01.
Perforin plays a role in host defense against primary C. burnetii infection.
Perforin is a pore-forming protein secreted by CTLs to induce apoptosis in target cells by allowing entry of granzymes and other cytotoxic components into the targeted cell. We utilized perforin-deficient mice to examine whether C. burnetii antigen-specific CTLs play an important role in host defense against primary C. burnetii infection. As shown in Fig. 6A, both WT and Prf1 KO mice showed transitional body weight loss from 4 to 12 dpi (day 8, P = 0.0062). Splenomegaly was significantly higher in Prf1 KO mice than in WT mice at 14 dpi (P = 0.0129) (Fig. 6B), although there was only a moderate increase in C. burnetii genomic copies in the spleens (P = 0.0726) (Fig. 6C). These results indicate that perforin may be important for functional CTLs to control C. burnetii infection in mice, suggesting that CTLs may play a role in controlling primary C. burnetii infection.
FIG 6.
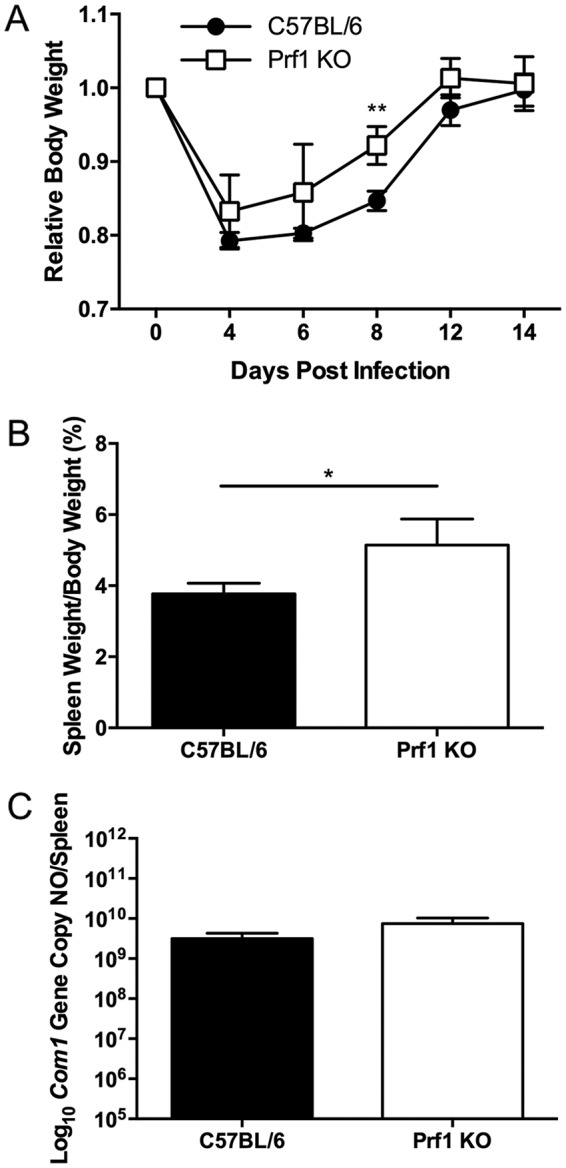
Perforin knockout mice have more severe disease following NMI infection than wild-type mice. WT and perforin knockout mice were infected i.p. with 5 × 107 NMI organisms, and disease severity was assessed 14 dpi. (A) Relative body weight was determined throughout infection to monitor disease severity. (B) Splenomegaly was determined as a ratio of spleen weight to body weight. (C) Bacterial burden in the spleen was determined by real-time PCR and reported as log10 C. burnetii com1 gene copy numbers. These data indicate that Prf1 KO mice had more severe splenomegaly than WT mice following NMI infection. The data presented for each group are the average with standard deviation for four mice. Results were analyzed by t test (B and C) or two-way ANOVA with Sidak's multiple-comparison test (A) for statistical significance. *, P < 0.05; **, P < 0.01.
DISCUSSION
The obligate intracellular lifestyle of C. burnetii makes it a potential target for T cells during primary infection. A previous study (13) demonstrated that T cells and IFN-γ are essential for controlling primary C. burnetii infection. Additionally, it has been shown that either CD4+ or CD8+ T cells alone were able to control primary pulmonary C. burnetii infection, suggesting that CD4+ or CD8+ T cells are required for host defense against primary C. burnetii infection (14). These studies suggest that T cell-mediated immunity may be the essential mechanism for host defense against primary C. burnetii infection. However, the role of MHC-I and -II antigen presentation pathways in host defense against C. burnetii infection remains unclear. C. burnetii infection in immunocompetent mice does not result in death or overt clinical signs; however, several recent studies (13, 24–26) demonstrated that C. burnetii infection reliably induces significant splenomegaly, and splenomegaly was correlated to bacterial burden in the spleen as measured by quantitative real-time PCR. In addition, recent studies (13, 14, 25, 27–30) demonstrated that both C. burnetii i.p. and respiratory infections cause similar systemic infections in mice and induce a significant and comparable splenomegaly at different time points postinfection. Thus, splenomegaly and bacterial burden in the spleen have been widely used as indicators for monitoring the severity of infection and evaluating the protective efficacy of vaccine-induced protection in both C. burnetii i.p. and respiratory infection mouse models. Since with i.p. infection it is easy to control the infectious dose and does not induce a large variation within the same group of mice, the model of C. burnetii i.p. infection of mice was used to examine whether MHC-I or -II deficiency in mice would significantly influence their susceptibility to C. burnetii infection in this study. Interestingly, although C. burnetii infection induced more severe disease in both β2m KO and MHC-II-deficient mice than in WT mice during early stages of infection, only β2m KO mice developed a severe persistent infection and were unable to control bacterial replication at late stages of infection. It is known that MHC-I-deficient β2m KO mice have a severely reduced CD8+ T cell population (17, 18), and MHC-II antigen presentation is critical for the expansion and function of CD4+ T cells (20). Our results indicate that both MHC-II-restricted CD4+ T cells and MHC-I-restricted CD8+ T cells contribute to host defense against primary C. burnetii infection, while MHC-I-restricted CD8+ T cells may play a more critical role in controlling bacterial replication. These results corroborate a previous study (14) that demonstrated that CD8+ T cell depletion in NMI-infected mice resulted in development of markedly worse disease, while CD4+ T cell depletion in NMI-infected mice showed disease severity similar to that in WT mice. These data support the hypothesis that CD8+ T cells play a critical role in host defense against primary C. burnetii infection.
The observation that NMI infection induced more severe disease and persistent bacterial infection in MHC-I-deficient β2m KO mice than in their WT counterparts suggests that MHC-I-restricted CD8+ T cells may be crucial for host defense against primary C. burnetii infection. β2m is a component of several other antigen-presenting molecules, such as H2-M3, TL, Qa-1, Qa-2, and CD1d. The MHC-encoded class I, H2-M3, Qa-1, and Qa-2 molecules have all been shown to bind peptide antigens loaded in a TAP-dependent manner (17, 18). This leads us to hypothesize that TAP may be involved in the severe disease and defects in bacterial clearance in β2m KO mice infected with NMI. To determine whether the severe disease phenotype in β2m KO mice is due to defects in the MHC-I-restricted CD8+ T cell classical pathway, we examined whether TAP1 deficiency in mice would significantly influence their susceptibility to NMI infection. Our results demonstrated that NMI infection induced more severe disease in TAP1− mice than in their WT counterparts during the early stages of infection. However, TAP1 deficiency in mice did not significantly influence elimination of C. burnetii at the later stages of infection. These results suggest that C. burnetii antigen presentation by the MHC-I classical pathway may depend partially upon TAP1, but TAP-independent pathways may also be involved in antigen presentation to CD8+ T cells during C. burnetii primary infection. The classical TAP-dependent antigen presentation pathway has been demonstrated to play a critical role in host defense against M. tuberculosis (16). In contrast, lipid-based antigens can also be presented by the TAP-independent CD1 antigen presentation pathway (31–33). It has been shown that TAP-independent pathways also play important roles in host defense against Toxoplasma gondii infection (34). Although the current study did not rule out the possibility that CD1-restricted CD8+ T cells are involved in host defense against primary C. burnetii infection, our results indicate that C. burnetii antigen presentation by the MHC-I pathway depends only partially upon TAP1. It is possible that the CD1-restricted antigen presentation pathway may play a critical role in activating CD8+ T cells during C. burnetii primary infection. Cross presentation and the host autophagy system are additional forms of TAP-independent antigen processing, which could also play important roles in CD8+ T cell clearance of C. burnetii. Future study is needed to examine whether deficiencies in these pathways in mice would significantly increase their susceptibility to NMI infection.
The evidence that NMI infection induces lethal disease in IFN-γ-deficient mice (13) suggests that IFN-γ is crucial for host defense against C. burnetii infection. Since antigen-specific CD8+ T cells may function via production of IFN-γ in response to C. burnetii infection, we examined whether the defects in the TAP-dependent MHC-I-restricted CD8+ T cell pathway would reduce the levels of IFN-γ during primary C. burnetii infection. Interestingly, significantly higher levels of IFN-γ were detected in sera from NMI-infected β2m KO and TAP1− mice than in sera from WT mice. This result suggests that C. burnetii infection induced more severe disease in β2m KO and TAP1− mice independently of IFN-γ-producing CD8+ T cells. On the other hand, this result may be due to the lack of antigen-specific CD8+ T cells in NMI-infected β2m KO and TAP1− mice resulting in uncontrolled bacterial replication that can strongly activate CD4+ T cells or natural killer cells to produce higher levels of IFN-γ. To further determine whether CD8+ T cells play an important role via production of IFN-γ during primary C. burnetii infection, we examined whether there were differences between WT CD8+ T cells and IFN-γ-deficient CD8+ T cells in their ability to protect B and T cell-deficient Rag1 knockout mice against NMI infection following adoptive transfer. The results indicate that although the numbers of C. burnetii genomic copies in the spleens were similar in Rag1 KO and IFN-γ-deficient CD8+ T cell recipient mice, fewer C. burnetii genomic copies were detected in the spleens from WT CD8+ T cell recipient mice. These results suggest that while IFN-γ-producing CD8+ T cells may not be important for protecting against C. burnetii infection-induced inflammatory responses, they may be involved in controlling bacterial replication during C. burnetii infection. Collectively, these data provide additional evidence to support an important role for antigen-specific CD8+ T cells, via production of IFN-γ, in host defense against primary C. burnetii infection.
To determine whether the cytolytic function of CD8+ T cells is critical for host defense against primary C. burnetii infection, we first investigated whether granzyme B deficiency in mice decreases resistance to NMI infection. The observation that NMI infection induced more severe disease in granzyme B KO mice than in their WT counterparts indicates that cytotoxic CD8+ T cells may play a role in protecting mice from development of severe disease during C. burnetii infection. However, splenomegaly and bacterial burden in the spleens were similar in WT and granzyme B KO mice. This result suggests that granzyme B may not play a critical role in controlling host inflammatory responses or bacterial replication during primary C. burnetii infection. As granzymes play redundant roles in cytotoxic CD8+ T cells during host immune responses, it is possible that other granzymes could overcome the granzyme B deficiency in mice, resulting in control of NMI infection. Perforin is considered more critical for CTLs to induce apoptosis in target cells because without perforin, granzymes and other cytotoxic components cannot enter CTL-targeted cells. We next examined whether perforin deficiency in mice would influence their ability to control NMI infection. Interestingly, we found that splenomegaly in Prf1 KO mice was significantly higher than that in WT mice. These results indicate that perforin may be important for functional CTLs to control C. burnetii infection in mice and suggest that cytotoxic CD8+ T cells may play an important role in controlling primary C. burnetii infection.
In summary, this study provides novel evidence to support that both MHC-II-restricted CD4+ T cells and MHC-I-restricted CD8+ T cells are important for host defense against primary C. burnetii infection, while highlighting that MHC-I-restricted CD8+ T cells play a critical role in controlling bacterial replication and ultimately eliminating invading bacteria. Our results also suggest that MHC-I-restricted antigen-specific CD8+ T cells may play a critical role during primary C. burnetii infection via their ability to produce IFN-γ and induce cell death in C. burnetii-infected target cells. Current clinical treatment of acute and chronic Q fever patients relies on antibiotics, but in some cases C. burnetii can persistently infect host cells even after treatment with multiple antimicrobial drugs. Further understanding of how C. burnetii antigen-specific CD8+ T cells clear infection locally, in the lungs, as well as systemically could lead to the development of novel treatments to fully clear infection from both acute and chronic Q fever patients.
MATERIALS AND METHODS
C. burnetii strain.
C. burnetii Nine Mile phase I (NMI) clone 7 (RSA493) was grown in L929 cells and purified via density gradient centrifugation as previously described (35). All experiments using virulent NMI were performed in biosafety level 3 (BSL3) or animal biosafety level 3 (ABSL3) facilities at the University of Missouri Laboratory for Infectious Disease Research (MU-LIDR).
Animals.
Six-week-old female β2m KO, MHC II−, TAP1−, Rag1 KO, Ifng KO, granzyme B KO, Prf1 KO, and WT C57BL/6 or 129S2.SvJ mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Mice were housed in sterile microisolator cages in a conventional animal facility or in the ABSL3 facility at the MU-LIDR. All research involving animals was conducted in accordance with guidelines of, and all animal use protocols were approved by, the Animal Care and Use Committee at the University of Missouri. All C. burnetii NMI infection experiments were conducted in the ABSL3 facility at MU-LIDR.
CD8+ T cell adoptive transfer.
Spleens were harvested from either wild-type C57BL/6 or IFN-γ−/− mice. Briefly, spleens were removed and homogenized using a glass tissue homogenizer. The cell suspension was passed through a 70-μm nylon mesh, and red blood cells were lysed using ammonium chloride-potassium (ACK) lysis buffer for 5 min at room temperature. To purify CD8+ T cells, we used the CD8+ T cell isolation kit (Miltenyi) and LS columns according to manufacturer specifications. Purified T cells were adoptively transferred to Rag1 knockout mice at 5 × 106 cells i.p. at 24 h prior to infection with NMI.
C. burnetii infection.
Mice were infected with NMI via intraperitoneal (i.p.) injection of 1 × 107 NMI organisms per mouse in 0.4 ml phosphate-buffered saline (PBS). Mice were weighed every other day throughout the course of infection. At desired time points, mice were sacrificed and spleens were removed and weighed to calculate splenomegaly [(spleen weight/body weight) × 100]. Pieces of spleens were collected for measuring C. burnetii genomic copies by real-time PCR and examining histopathological changes.
Real-time PCR.
Spleen pieces were homogenized in 200 μl lysis buffer (1 M Tris, 0.5 M EDTA, 7 mg/ml glucose, 28 mg/ml lysozyme) with 10 μl of proteinase K (20 mg/ml). Samples were then incubated for 18 h at 60°C. Following incubation, 21 μl of 10% SDS was added to samples and left for 1 h at room temperature. DNA was then extracted using the High Pure PCR template preparation kit according to manufacturer specifications (Roche). The com1 gene copy number was quantified using a standard curve with SYBR green (Applied Biosciences) on an Applied Biosystems 7300 real-time PCR system. Recombinant plasmid DNA (com1 gene ligated into pBluescript vector) was used as standard to quantify com1 gene copy numbers.
ELISAs.
Serum IFN-γ levels were measured using the mouse IFN-γ enzyme-linked immunosorbent assay (ELISA) Ready-Set-Go kit (eBiosciences) according to manufacturer specifications. Plates were analyzed at 490 nm using the Tecan F50 plate reader and Magellan software.
Histological staining.
Spleens were fixed in 10% formalin for at least 72 h and processed by the Veterinary Medicine Diagnostic Laboratory (VMDL) at the University of Missouri. Samples were stained with hematoxylin and eosin and scored in a blind fashion by a trained pathologist.
Statistical analysis.
Statistical analysis was performed using Prism 5.0 (GraphPad Software Inc., San Diego, CA). Results were compared using an analysis of variance (ANOVA) with multiple comparisons or t test when appropriate, as stated in the figure legends. Differences were considered significant if the P value was ≤0.05.
ACKNOWLEDGMENTS
This study was supported by NIAID/NIH grant RO1AI083364 to G.Z. and DTRA funding grant HDTRA1-13-0003 subcontract to G.Z.
We thank the staff at the MU-LIDR for their assistance with these experiments. We also thank Nicholas Olivarez for critical reading and editing of the manuscript.
REFERENCES
- 1.Maurin M, Raoult D. 1999. Q fever. Clin Microbiol Rev 12:518–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitney EA, Massung RF, Candee AJ, Ailes EC, Myers LM, Patterson NE, Berkelman RL. 2009. Seroepidemiologic and occupational risk survey for Coxiella burnetii antibodies among US veterinarians. Clin Infect Dis 48:550–557. doi: 10.1086/596705. [DOI] [PubMed] [Google Scholar]
- 3.McQuiston JH, Holman RC, McCall CL, Childs JE, Swerdlow DL, Thompson HA. 2006. National surveillance and the epidemiology of human Q fever in the United States, 1978-2004. Am J Trop Med Hyg 75:36–40. [DOI] [PubMed] [Google Scholar]
- 4.Raoult D. 1993. Treatment of Q fever. Antimicrob Agents Chemother 37:1733–1736. doi: 10.1128/AAC.37.9.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raoult D, Houpikian P, Tissot Dupont H, Riss JM, Arditi-Djiane J, Brouqui P. 1999. Treatment of Q fever endocarditis: comparison of 2 regimens containing doxycycline and ofloxacin or hydroxychloroquine. Arch Intern Med 159:167–173. doi: 10.1001/archinte.159.2.167. [DOI] [PubMed] [Google Scholar]
- 6.Raoult D, Fenollar F, Stein A. 2002. Q fever during pregnancy. Arch Intern Med 162:701–704. doi: 10.1001/archinte.162.6.701. [DOI] [PubMed] [Google Scholar]
- 7.Wegdam-Blans MC, Vainas T, Van Sambeek MR, Cuypers PW, Tjhie HT, Van Straten AH, Teijink JA. 2011. Vascular complications of Q-fever infections. Eur J Vasc Endovasc Surg 42:384–392. doi: 10.1016/j.ejvs.2011.04.013. [DOI] [PubMed] [Google Scholar]
- 8.Million M, Thuny F, Richet H, Raoult D. 2010. Long-term outcome of Q fever endocarditis: a 26-year personal survey. Lancet Infect Dis 10:527–535. doi: 10.1016/S1473-3099(10)70135-3. [DOI] [PubMed] [Google Scholar]
- 9.Dijkstra F, Van Der Hoek W, Wijers N, Schimmer B, Rietveld A, Wijkmans CJ, Vellema P, Schneeberger PM. 2012. The 2007-2010 Q fever epidemic in The Netherlands: characteristics of notified acute Q fever patients and the association with dairy goat farming. FEMS Immunol Med Microbiol 64:3–12. doi: 10.1111/j.1574-695X.2011.00876.x. [DOI] [PubMed] [Google Scholar]
- 10.Bell M, Patel M, Sheridan J. 1997. Q fever vaccination in Queensland abattoirs. Commun Dis Intell 21:29–31. [DOI] [PubMed] [Google Scholar]
- 11.Marmion BP, Ormsbee RA, Kyrkou M, Wright J, Worswick DA, Izzo AA, Esterman A, Feery B, Shapiro RA. 1990. Vaccine prophylaxis of abattoir-associated Q fever: eight years' experience in Australian abattoirs. Epidemiol Infect 104:275–287. doi: 10.1017/S0950268800059458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andoh M, Naganawa T, Hotta A, Yamaguchi T, Fukushi H, Masegi T, Hirai K. 2003. SCID mouse model for lethal Q fever. Infect Immun 71:4717–4723. doi: 10.1128/IAI.71.8.4717-4723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andoh M, Zhang G, Russell-Lodrigue KE, Shive HR, Weeks BR, Samuel JE. 2007. T cells are essential for bacterial clearance, and gamma interferon, tumor necrosis factor alpha, and B cells are crucial for disease development in Coxiella burnetii infection in mice. Infect Immun 75:3245–3255. doi: 10.1128/IAI.01767-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Read AJ, Erickson S, Harmsen AG. 2010. Role of CD4+ and CD8+ T cells in clearance of primary pulmonary infection with Coxiella burnetii. Infect Immun 78:3019–3026. doi: 10.1128/IAI.00101-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanchard N, Shastri N. 2010. Cross-presentation of peptides from intracellular pathogens by MHC class I molecules. Ann N Y Acad Sci 1183:237–250. doi: 10.1111/j.1749-6632.2009.05135.x. [DOI] [PubMed] [Google Scholar]
- 16.Behar SM, Dascher CC, Grusby MJ, Wang C, Brenner MB. 1999. Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis. J Exp Med 189:1973–1980. doi: 10.1084/jem.189.12.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ladel CH, Flesch IE, Arnoldi J, Kaufmann SH. 1994. Studies with MHC-deficient knock-out mice reveal impact of both MHC I- and MHC II-dependent T cell responses on Listeria monocytogenes infection. J Immunol 153:3116–3122. [PubMed] [Google Scholar]
- 18.Abbas AK, Lichtman AH, Pillai S. 2011. Cellular and molecular immunology, 7th ed Saunders, Philadelphia, PA. [Google Scholar]
- 19.Gervassi AL, Probst P, Stamm WE, Marrazzo J, Grabstein KH, Alderson MR. 2003. Functional characterization of class Ia- and non-class Ia-restricted Chlamydia-reactive CD8+ T cell responses in humans. J Immunol 171:4278–4286. doi: 10.4049/jimmunol.171.8.4278. [DOI] [PubMed] [Google Scholar]
- 20.Takeda S, Rodewald H-R, Arakawa H, Bluethmann H, Shimizu T. 1996. MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity 5:217–228. doi: 10.1016/S1074-7613(00)80317-9. [DOI] [PubMed] [Google Scholar]
- 21.Dobano C, Rogers WO, Gowda K, Doolan DL. 2007. Targeting antigen to MHC class I and class II antigen presentation pathways for malaria DNA vaccines. Immunol Lett 111:92–102. doi: 10.1016/j.imlet.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 22.O E, Lee YT, Ko EJ, Kim KH, Lee YN, Song JM, Kwon YM, Kim MC, Perez DR, Kang SM. 2014. Roles of major histocompatibility complex class II in inducing protective immune responses to influenza vaccination. J Virol 88:7764–7775. doi: 10.1128/JVI.00748-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ermak TH, Giannasca PJ, Nichols R, Myers GA, Nedrud J, Weltzin R, Lee CK, Kleanthous H, Monath TP. 1998. Immunization of mice with urease vaccine affords protection against Helicobacter pylori infection in the absence of antibodies and is mediated by MHC class II-restricted responses. J Exp Med 188:2277–2288. doi: 10.1084/jem.188.12.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang G, Peng Y, Schoenlaub L, Elliott A, Mitchell W, Zhang Y. 2013. Formalin-inactivated Coxiella burnetii phase I vaccine-induced protection depends on B cells to produce protective IgM and IgG. Infect Immun 81:2112–2122. doi: 10.1128/IAI.00297-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang G, Russell-Lodrigue KE, Andoh M, Zhang Y, Hendrix LR, Samuel JE. 2007. Mechanisms of vaccine-induced protective immunity against Coxiella burnetii infection in BALB/c mice. J Immunol 179:8372–8380. doi: 10.4049/jimmunol.179.12.8372. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Zhang G, Hendrix LR, Tesh VL, Samuel JE. 2012. Coxiella burnetii induces apoptosis during early stage infection via a caspase-independent pathway in human monocytic THP-1 cells. PLoS One 7:e30841. doi: 10.1371/journal.pone.0030841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Elliott A, Peng Y, Zhang G. 2013. Coxiella burnetii interaction with neutrophils and macrophages in vitro and in SCID mice following aerosol infection. Infect Immun 81:4604–4614. doi: 10.1128/IAI.00973-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elliott A, Schoenlaub L, Freches D, Mitchell W, Zhang G. 2015. Neutrophils play an important role in protective immunity against Coxiella burnetii infection. Infect Immun 83:3104–3113. doi: 10.1128/IAI.00042-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peng Y, Schoenlaub L, Elliott A, Mitchell WJ, Zhang G. 2014. Characterization of a lipopolysaccharide-targeted monoclonal antibody and its variable fragments as candidates for prophylaxis against the obligate intracellular bacterial pathogen Coxiella burnetii. Infect Immun 82:4530–4541. doi: 10.1128/IAI.01695-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russell-Lodrigue KE, Andoh M, Poels MW, Shive HR, Weeks BR, Zhang GQ, Tersteeq C, Masegi T, Hotta A, Yamaguchi T, Fukushi H, Hirai K, McMurray DN, Samuel JE. 2009. Coxiella burnetii isolates cause genogroup-specific virulence in mouse and guinea pig models of acute Q fever. Infect Immun 77:5640–5650. doi: 10.1128/IAI.00851-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brigl M, Brenner MB. 2004. CD1: antigen presentation and T cell function. Annu Rev Immunol 22:817–890. doi: 10.1146/annurev.immunol.22.012703.104608. [DOI] [PubMed] [Google Scholar]
- 32.Rolph MS, Kaufmann SHE. 2000. Partially TAP-independent protection against Listeria monocytogenes by H2-M3-restricted CD8+ T cells. J Immunol 165:4575–4580. doi: 10.4049/jimmunol.165.8.4575. [DOI] [PubMed] [Google Scholar]
- 33.Seaman MS, Perarnau B, Lindahl KF, Lemonnier FA, Forman J. 1999. Response to Listeria monocytogenes in mice lacking MHC class Ia molecules. J Immunol 162:5429–5436. [PubMed] [Google Scholar]
- 34.Goldszmid RS, Bafica A, Jankovic D, Feng CG, Caspar P, Winkler-Pickett R, Trinchieri G, Sher A. 2007. TAP-1 indirectly regulates CD4+ T cell priming in Toxoplasma gondii infection by controlling NK cell IFN-gamma production. J Exp Med 204:2591–2602. doi: 10.1084/jem.20070634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ho T, Htwe KK, Yamasaki N, Zhang GQ, Ogawa M, Yamaguchi T, Fukushi H, Hirai K. 1995. Isolation of Coxiella burnetii from dairy cattle and ticks, and some characteristics of the isolates in Japan. Microbiol Immunol 39:663–671. doi: 10.1111/j.1348-0421.1995.tb03254.x. [DOI] [PubMed] [Google Scholar]