

ABSTRACT
The QseEF histidine kinase/response regulator system modulates expression of enterohemorrhagic Escherichia coli (EHEC) and Salmonella enterica serovar Typhimurium virulence genes in response to the host neurotransmitters epinephrine and norepinephrine. qseG, which encodes an outer membrane lipoprotein, is cotranscribed with qseEF in these enteric pathogens, but there is little knowledge of its role in virulence. Here, we found that in EHEC QseG interacts with the type III secretion system (T3SS) gate protein SepL and modulates the kinetics of attaching and effacing (AE) lesion formation on tissue-cultured cells. Moreover, an EHEC ΔqseG mutant had reduced intestinal colonization in an infant rabbit model. Additionally, in Citrobacter rodentium, an AE lesion-forming pathogen like EHEC, QseG is required for full virulence in a mouse model. In S. Typhimurium, we found that QseG regulates the phase switch between the two flagellin types, FliC and FljB. In an S. Typhimurium ΔqseG mutant, the phase-variable promoter for fljB is preferentially switched into the “on” position, leading to overproduction of this phase two flagellin. In infection of tissue-cultured cells, the S. Typhimurium ΔqseG mutant provokes increased inflammatory cytokine production versus the wild type; in vivo, in a murine infection model, the ΔqseG strain caused a more severe inflammatory response and was attenuated versus the wild-type strain. Collectively, our findings demonstrate that QseG is important for full virulence in several enteric pathogens and controls flagellar phase variation in S. Typhimurium, and they highlight both the complexity and conservation of the regulatory networks that control the virulence of enteric pathogens.
KEYWORDS: EHEC, QseG, Salmonella, flagella, type III secretion
INTRODUCTION
The gastrointestinal tract is a diverse ecosystem with many physiologically distinct niches. To mount a productive infection, bacterial pathogens that infect the gut must be able to sense these niches and deploy their virulence regime in the appropriate environment. Aberrant deployment of virulence factors may lead to wasted energy or detection by the host immune system. To accomplish this task, many bacterial pathogens have evolved highly complex regulatory networks to control expression of their virulence factors. Understanding these complex regulatory systems will lead to a greater understanding of disease progression (1).
Two-component systems are a common strategy used by bacterial pathogens to regulate virulence genes in response to environmental cues (2). Both enterohemorrhagic Escherichia coli (EHEC) and Salmonella enterica serovar Typhimurium use the QseEF two-component system to sense host hormones and regulate expression of virulence genes. The sensor kinase QseE senses epinephrine, norepinephrine, phosphate, and sulfate and phosphorylates its cognate regulator, QseF. In EHEC, QseF induces transcription of espFu (3, 4), a type III secretion system (T3SS) effector gene that interacts with host proteins to activate actin polymerization and initiate formation of attaching and effacing (AE) lesions (5–7). In S. Typhimurium, QseEF signaling enhances expression of the SPI-1 pathogenicity island (8), which encodes a T3SS and is necessary for host cell invasion (9). In a mouse model of systemic infection, an S. Typhimurium strain lacking qseE was significantly attenuated compared to the wild type (WT) (8). Although conserved across the two species, the QseEF system has undergone species-specific specialization to regulate virulence factors unique to each pathogen.
The qseG gene is located between qseE and qseF on the chromosome and is cotranscribed with the two-component system. Homologs of qseG are present in many of the Enterobacteriaceae; however, all previous work investigating QseG function has been carried out in EHEC. QseG is an outer membrane lipoprotein that is transported through the general secretory pathway (Sec) or the twin-arginine translocation (Tat) system (known to transport folded proteins [10, 11]) (4). QseG is predicted to contain alpha-helices but lacks significant homology to other proteins in the database. In an EHEC qseG deletion (ΔqseG) mutant, expression of qseE is increased, suggesting that QseG modulates expression of the two-component system. QseG is not required for secretion of T3SS components or effectors but impacts the translocation of the translocated intimin receptor (Tir) (4). Tir is the first effector translocated into the host cell via the T3SS (12). It interacts with intimin on the bacterial membrane to promote tight adherence and interacts with host proteins to promote actin polymerization and AE lesion formation (13). Because QseG impacts the translocation of Tir, and potentially other effectors, a strain lacking qseG does not form AE lesions efficiently (4). These data suggest that qseG is important for EHEC virulence; moreover, since qseG, along with qseEF, is conserved among many of the pathogenic Enterobacteriacae, it may play an important role in regulating virulence across many pathogens.
Here, we show that qseG is required for full virulence in animal models of EHEC; Citrobacter rodentium, a related AE pathogen; and S. Typhimurium infection. We show that although QseG is important for full virulence in animal models of both EHEC and S. Typhimurium infection, its mechanisms of action have diverged. In S. Typhimurium, QseG plays a role in modulating flagellar phase variation—a system that is missing from EHEC. This phase variation regulation leads to decreased inflammation at systemic sites during S. Typhimurium infection. The qseEFG operon is found in the genomes of many Enterobacteriaceae, and it is likely that QseG plays an important role in other species, as well. Our work highlights the complexity of the regulatory networks that bacterial pathogens employ, as well as the fact that proteins common to different pathogens can adapt to play species-specific roles.
RESULTS
QseG faces the periplasm and interacts with SepL.
QseG contains a predicted lipid attachment site, and previous work using cell fractionation demonstrated that QseG is located in the EHEC outer membrane (4). To determine whether QseG is exposed to the outside of the cell or instead faces the periplasm, a proteinase K protection assay was performed (Fig. 1A). Cells were treated with the membrane-impermeable protease proteinase K, and the effects on QseG and OmpA, as a control, were assessed by Western blotting. Levels of OmpA, which is exposed to the outside of the cell, were diminished by proteinase K treatment. In contrast, the levels of QseG were unchanged by proteinase K treatment, suggesting it is not exposed to the outside of the cell and therefore likely faces the periplasm.
FIG 1.

QseG interacts with SepL. (A) Proteinase K protection assays of OmpA and QseG. WT EHEC cells were treated with proteinase K, and then proteins were analyzed by Western blotting. A known outer membrane protein, OmpA, was included as a control. LPS, lipopolysaccharide. (B) Yeast two-hybrid assays to probe possible protein-protein interactions between QseG and QseG, EscC, EspA, SepD, SepL and Tir. Briefly, the yeast reporter strain L40 was transformed with all combinations of the proteins in both the bait (pLEX-ADE) and the library (pVP16) vectors. Dual transformants were selected on yeast minimal medium lacking leucine and tryptophan and then assayed for protein-protein interactions using the integrated lacZ and HIS3 reporters. Association of the two-protein fusions was determined by growth on yeast minimal medium lacking histidine. (C) Pulldown assays with SepL-HA and QseG. A plasmid expressing an HA-tagged sepL allele was transformed into EHEC ΔsepL and ΔsepL ΔqseG strains. SepL was captured using HA antisera, and the precipitate was probed for QseG by Western blotting using QseG antisera.
QseG is not involved in the expression of the locus of enterocyte effacement (LEE) or espFu genes or in the assembly of the T3SS but impacts the translocation of Tir into host cells (4). We hypothesized that QseG, because of its cellular location, may physically interact with components of the T3SS apparatus and thus have a role in modulating the T3SS. Using a yeast two-hybrid assay, QseG was tested for its ability to interact with various components of the T3SS thought to be present in the periplasm. Of the proteins screened, only SepL was identified as a potential binding partner for QseG (Fig. 1B). SepL, along with SepD, is required for secretion of EspA, EspB, and EspD (14, 15), the structural components of the T3SS, known as the translocon. Furthermore, they are responsible for regulating the switch from secretion of the translocon to secretion of effector proteins (14, 16). SepL directly binds to Tir to prevent its early secretion (17), but the signal that initiates the switch to begin secretion of effectors is not fully defined. Coimmunoprecipitation was employed to confirm the interaction of QseG with SepL. A hemagglutinin (HA)-tagged allele of sepL was expressed in a ΔsepL or ΔsepL ΔqseG strain of EHEC, and a pulldown was performed using an anti-HA antibody. When both SepL-HA and QseG were present, QseG was detected in the precipitated sample via Western blotting using an anti-QseG antibody (Fig. 1C), confirming the SepL-QseG interaction. The direct interaction of QseG with SepL in EHEC suggests that QseG may play a role in regulation of the T3SS and in the switch from translocon to effector secretion. Because Tir translocation is a key event in AE lesion formation, we investigated AE lesion formation by the ΔqseG mutant using live-cell imaging. The ΔqseG mutant exhibited delayed lesion formation and was almost abrogated in its ability to form AE lesions on epithelial cells compared to the WT. The ΔqseG mutant did not have any growth defects compared to the WT (18), and previous AE lesion assays were congruent with the live-cell image, showing that the ΔqseG mutant is defective for AE lesion formation and that this defect can be complemented (4). Taken together, these data suggest that differences in Tir translocation impact the kinetics of pedestal formation (Fig. 2; see Movies S1 and S2 in the supplemental material).
FIG 2.

AE lesion timing and dynamics. Time lapse microscopy of Lifeact::GFP-expressing HeLa cells being infected with mCherry-expressing WT and ΔqseG EHEC. The time elapsed after imaging was begun is shown at the top. EHEC pedestal formation (arrows) can be observed as yellow areas where actin polymerization (green puncta) colocalized with attached EHEC bacteria (red).
QseG is required for robust colonization and full virulence in in vivo models of EHEC infection.
QseG impacts Tir translocation and is required for efficient pedestal formation (4). To test if this defect in T3SS activity translates into attenuated colonization and virulence in vivo, we used two different small-animal models of EHEC infection. We infected infant rabbits with WT or a ΔqseG mutant of the EHEC strain 86-24 and measured the bacterial load in the colon 5 days postinfection. Significantly fewer CFU were recovered from rabbits infected with ΔqseG than from those infected with the WT (Fig. 3A), demonstrating that QseG contributes to efficient colonization in this model. We also tested the role of QseG in vivo using C. rodentium, a natural pathogen of mice that is closely related to EHEC. The C. rodentium genome also harbors the LEE (19), and many of the regulatory pathways that control its expression are conserved between C. rodentium and EHEC. Importantly, while EHEC's virulence in mice can be solely attributed to Shiga toxin (20), C. rodentium infection in mice exhibits many of the hallmarks of EHEC disease in humans, including AE lesion formation, colonic hyperplasia, and diarrhea (21, 22), making it a useful model for studying LEE regulation in vivo. Mice infected with a ΔqseG mutant survived significantly longer than those infected with WT C. rodentium (Fig. 3B). ΔqseG-infected mice also displayed attenuated disease symptoms compared to WT-infected mice. The ΔqseG-infected mice had decreased colon weights (consistent with decreased colonic hyperplasia [16, 23, 24]) (Fig. 3C), and their stool was formed in normal pellets while WT-infected mice displayed unformed, liquid diarrheal feces. (Fig. 3D). The increased survival and attenuation of disease symptoms where qseG was absent indicates that the gene is required for full virulence in vivo.
FIG 3.

QseG is necessary for EHEC and C. rodentium virulence. (A) EHEC infection in infant rabbits. Three-day-old rabbits were inoculated intragastrically with ∼5 × 108 CFU of WT or ΔqseG EHEC. At 5 days postinfection, the colonic contents were plated on LB containing streptomycin to determine EHEC CFU. **, P < 0.01. (B) C. rodentium murine infections. C3H/HeJ mice (3.5 weeks old) were gavaged with 109 CFU of WT or ΔqseG C. rodentium or PBS. Survival was monitored daily (n = 10). Statistical significance was determined via Kaplan Meyer test. (C) Colon weights. Mice (n = 5) were infected as described for panel B. At day 6 postinfection, the mice were sacrificed, and their colons were collected, washed, and weighed. *, P < 0.05. (D) Colons collected as described for panel C were examined for gross pathology differences.
Absence of qseG leads to overproduction of the S. Typhimurium type II flagellin.
The QseEF two-component system regulates type III secretion in both EHEC and S. Typhimurium and is required for full virulence in both pathogens (4, 8, 25, 26). We hypothesized that qseG modulates virulence in Salmonella, as well, and studying its role in the pathogen could help uncover its mechanism of action. A sequence alignment of the qseEFG operons of EHEC and S. Typhimurium revealed that the sequence similarity of qseG was lower at both the nucleotide and protein levels (59% and 88%, respectively) than those of the surrounding qseE (71% and 94%) and qseF (81% and 98%) genes (Fig. 4A). This suggests that QseG has undergone greater adaptation than the QseEF two-component system with which it is cotranscribed. We know that the QseEF regulon differs between EHEC and S. Typhimurium, controlling virulence factors specific to each species, and therefore, we hypothesized that the role of QseG may differ between the two pathogens, as well.
FIG 4.

QseG controls flagellin phase variation in S. Typhimurium. (A) Nucleotide and protein sequence similarities of qseE, qseF, and qseG between EHEC and S. Typhimurium. (B) S. Typhimurium (Stm) strains were grown statically in LB broth to late log phase. A standard amount of bovine serum albumin (BSA) was spiked into the cultures just prior to collection to serve as a concentration/loading control. The cells were pelleted, and proteins in the filtered, concentrated supernatants were separated via SDS-PAGE and stained using Coomassie blue. Presumed identifications of selected proteins based on molecular weight are indicated, and the band of interest that is overexpressed in the qseG mutant supernatant is indicated with an asterisk. Relative quantification of FljB and FliC levels from the pictured supernatants was performed via LC–MS-MS and is shown below. (C) WT, ΔqseG, and complemented strains were grown to an OD600 of 1.0 in LB broth plus 0.2% arabinose to induce the complementation plasmid. RNA extracted from these cells was probed via qPCR for expression levels of fliC and fljB. The levels of genes of interest were normalized to rpoA levels and expressed as a fold change over WT levels. The averages and standard deviations of three replicates are shown. (D) Using a qPCR-based assay, the orientation of the fljB promoter was assessed in S. Typhimurium grown as described in the legend to Fig. 3B. The data are displayed as the molar on/off ratio of the fljB promoter. The averages and standard deviations of three replicates are shown. Student's t test was used to determine statistical significance. ns, not significant.
We created a qseG deletion mutant in S. Typhimurium strain SL1344 and confirmed that, like the corresponding EHEC mutant (18), it displayed growth kinetics identical to those of WT S. Typhimurium in vitro (data not shown). We then analyzed the secreted proteome of WT versus ΔqseG S. Typhimurium by SDS-PAGE and Coomassie staining. The supernatant of a ΔinvA ΔspiB mutant, which is deficient in both the Salmonella T3SSs encoded in pathogenicity islands SPI-1 and SPI-2, was run as a control to identify proteins secreted through a T3SS. Compared to the WT, the ΔqseG mutant did not appear to have any change in the amounts of T3SS-secreted proteins. However, a large band appeared in the ΔqseG supernatant that was not observed in the WT supernatant (Fig. 4B). The unknown band was extracted and identified by mass spectrometry (MS) as FljB (Table 1), the S. Typhimurium phase II flagellin. Unlike E. coli, many strains of Salmonella possess two antigenically distinct alleles of the flagellin protein: fliC and fljB (27). fljB, the phase II flagellin gene, is under the control of an invertible promoter that in the on position leads to transcription of fljB and the repressor of fliC, fljA, producing reciprocal control of the two flagellin types (28–33). A relative quantification of the total secreted proteome was performed by mass spectrometry (Table 1), confirming that ΔqseG mutant supernatants contained nearly 50-fold higher FljB protein levels than WT supernatants (Fig. 4B). A corresponding decrease in FliC levels was observed, as expected, but no other significant changes in flagellum-associated proteins were observed. The secreted proteome analysis also confirmed that no significant, consistent change in SPI-1 secreted proteins was observed (Table 1).
TABLE 1.
Mass spectrometry analysis of secreted proteins in WT versus ΔqseG S. Typhimurium strain SL1344
| Protein | Fold change (WT/ΔqseG) |
|---|---|
| Flagella | |
| FliC | −5.63 |
| FljB | 46.70 |
| FliD | 1.04 |
| FlgK | −1.49 |
| FlgM | 1.18 |
| FlgL | −1.15 |
| FlgE | −1.10 |
| FliK | −1.11 |
| FlgF | 2.05 |
| FlgG | 1.38 |
| SPI-1 | |
| SipC | −1.13 |
| SipB | −2.90 |
| SipA | −5.42 |
| SipD | −1.05 |
| SopB | −1.50 |
| SopE | 1.05 |
| SopE2 | 1.3 |
| SopA | −13.50 |
QseG decreases fljB transcription by modulating its invertible promoter to favor the off position.
To test whether increased FljB secretion in the ΔqseG mutant was modulated at the transcriptional level, we measured fljB RNA levels using quantitative PCR (qPCR). The fljB transcript was increased over 100-fold in S. Typhimurium ΔqseG compared to the WT, and a corresponding, but more modest, decrease in the fliC transcript was also observed. Importantly, complementation of qseG in trans restored fljB and fliC transcripts to WT levels (Fig. 4C). As fljB is controlled by an invertible promoter, we tested whether the observed increase in fljB transcription was due to an increase in the proportion of fljB promoters that were in the on position. A qPCR-based assay was designed in which one primer was located within the invertible region and one outside, so that the molar ratio of the orientations could be calculated. In the WT strain, the off orientation was favored by a ratio of over 100:1. In the absence of qseG, the ratio was altered to closer to 1:1, with the on position being slightly favored (Fig. 4D). qseG complementation restored the on/off ratio to WT levels. These observations reveal that QseG modulates the orientation of the fljB promoter to repress fljB expression.
Absence of qseG does not affect S. Typhimurium motility, cell invasion, or intracellular replication.
Because QseG was involved in regulation of flagellin transcription, we hypothesized that the mutant might have an altered motility phenotype. Motility was assessed using a soft-agar swimming assay, but no significant alteration in motility was observed (Fig. 5A). This was not entirely unexpected, as we have not demonstrated that QseG represses overall flagellar expression, but rather, it modulates which flagellin allele is favored. In a HeLa cell-based invasion assay, the ΔqseG mutant did not display any defects in cell invasion in vitro (Fig. 5B). Similarly, no defects in intracellular replication were observed in the ΔqseG mutant in either human (Fig. 5C) or mouse (Fig. 5C) macrophages. S. Typhimurium produces two T3SSs, designated T3SS1 and T3SS2, that are deployed at distinct parts of the pathogen’s life cycle. Defects in cell invasion and intracellular replication are typically phenotypes associated with defects in T3SS1 or T3SS2, respectively, and since we did not observe any differences in the amounts of secreted proteins associated with either of these systems, a lack of phenotype was not unexpected.
FIG 5.

QseG does not contribute to motility, cell invasion, or intracellular replication in S. Typhimurium. (A) The indicated S. Typhimurium strains were grown to an OD600 of 1.0 in LB broth plus 0.2% arabinose (to induce the complementation plasmid) and then stabbed into 0.3% agar LB plates. At 6, 8, and 24 h, the diameters of the motility halos were measured. Averages and standard deviations from three replicates are reported, and representative plates at 24 h are shown. (B) HeLa cells were infected with the indicated S. Typhimurium strains to assess cell invasion efficiency. The ΔinvA ΔspiB mutant is an invasion-deficient strain. Averages and standard deviations from three replicates are shown. p.i., postinfection. (C) J774 (mouse) and THP1 (human) macrophages were infected with the indicated S. Typhimurium strains to assess intracellular replication ability. The ΔinvA ΔspiB mutant is an intracellular replication-deficient strain. The averages and standard deviations of three replicates are shown.
QseG is required for full S. Typhimurium virulence in mice.
To assess the importance of QseG in vivo, we utilized a murine model of systemic disease, in which mice were infected intraperitoneally with S. Typhimurium. At days 2 and 3 postinfection, groups of five mice were sacrificed, and spleens and livers were collected to enumerate bacterial loads. The data shown are combined from two independent experiments. Mice infected with the ΔqseG mutant survived significantly longer than those infected with WT S. Typhimurium (Fig. 6A), demonstrating that qseG is required for full virulence in this model of S. Typhimurium infection. The ΔqseG mutant-infected mice also appeared to lose slightly less weight than WT-infected mice (Fig. 6B), although this was not statistically significant. Interestingly, no significant differences in bacterial loads were observed at either day 2 or 3 in the liver or spleen in infected animals (Fig. 6C). Since QseG contributes to S. Typhimurium virulence but is not required for efficient colonization or proliferation in this model, it is possible that it augments virulence by modulating the host response to infection.
FIG 6.

QseG impacts S. Typhimurium murine infection. (A and B) BALB/c mice were infected intraperitoneally with 1 × 104 CFU of the indicated S. Typhimurium strains or sterile PBS. Survival (A) and weight (B) were monitored over time. Data from two independent experiments, each using 10 mice per group, are displayed. Significant differences between survival curves were determined using the Mantel-Cox method. (C) On day 2 or 3, 5 mice were randomly selected and sacrificed, and serial dilutions of organ homogenates were plated for CFU counts.
QseG limits inflammation during S. Typhimurium infection.
QseG modulates expression of the flagellin proteins, which are pathogen-associated molecular patterns and potent activators of the immune system. Flagellins are recognized by Toll-like receptor 5 (34), leading to cytokine and nitric oxide production (35), and by the NOD-like receptors NLRC4 and NAIP5, leading to activation of the inflammasome (36). To test whether QseG has a role in modulating the inflammatory response during S. Typhimurium infection, we exposed T84 colonic epithelial cells to supernatants from different strains of S. Typhimurium and monitored the induction of interleukin 8 (IL-8) transcription by qPCR (Fig. 7A). Cells exposed to supernatants from WT S. Typhimurium showed approximately 50-fold induction of IL-8 transcription compared to uninfected cells. Supernatants from S. Typhimurium ΔfliC ΔfljB caused no IL-8 induction, suggesting that any IL-8 induction observed in the assay can be attributed to the flagellin response. Supernatants from S. Typhimurium ΔqseG significantly increased IL-8 transcription compared to WT supernatants, resulting in nearly 100-fold induction over uninfected cells. Supernatants from a ΔqseG ΔfljB double mutant resulted in a slightly dampened IL-8 response compared to S. Typhimurium ΔqseG; however, this was not statistically significant, and IL-8 levels were not reduced to those measured in WT S. Typhimurium supernatants. We hypothesize that because there is significant cross talk between fljB and fliC expression, deletion of fljB could lead to increased fliC expression, compensating for the loss of FljB. Additionally, it is possible that S. Typhimurium ΔqseG increases inflammatory cytokine production through another mechanism other than modulating fljB expression.
FIG 7.

Altered inflammation during murine infection by the S. Typhimurium ΔqseG mutant. (A) Concentrated supernatants from the indicated S. Typhimurium strains were applied to T84 intestinal epithelial cells. Induction of IL-8 transcription was assessed via qPCR. IL-8 levels were normalized to GAPDH levels, and the values are reported as a fold change over uninfected cells. The averages and standard deviations from three replicates are displayed; statistical significance was determined using Student's t test. (B) In the murine infection experiment shown in Fig. 6, total DNA was extracted from infected spleens and livers on day 3 postinfection. The orientation of the fljB promoter was assayed via the qPCR-based method described in the legend to Fig. 3C (n = 5). Significant differences were determined using Student's t test. (C) In the infection experiment shown in Fig. 6, total RNA was extracted from infected spleens and livers on day 3 postinfection. Host NOS2 expression levels were assayed via qPCR, normalized to GAPDH levels, and reported as a fold change over levels from PBS-treated mice (n = 5). Significant differences were determined using Student's t test.
To confirm the role of QseG in modulating the fljB promoter in vivo, DNA from infected livers and spleens was collected, and the orientation of the fljB promoter was assessed via the qPCR-based assay described above. A higher proportion of the fljB promoter was in the on position in livers and spleens from S. Typhimurium ΔqseG-infected mice than in those from WT-infected mice (Fig. 7B), confirming that the modulation of the fljB promoter by QseG occurs in vivo during infection. To test the effect of QseG on the inflammatory response, we collected RNA from infected livers and spleens and assayed for host expression of the inducible nitric oxide synthase NOS2. A significant increase in NOS2 expression was observed in livers infected with S. Typhimurium ΔqseG compared to those infected with the WT (Fig. 7C), suggesting that QseG does indeed dampen the inflammatory response to S. Typhimurium, perhaps through its modulation of fljB expression.
DISCUSSION
To establish a productive infection, intestinal pathogens must tightly regulate their virulence systems, deploying them at the appropriate sites and times during infection. Likewise, pathogens need to dampen expression of genes that provoke inflammatory responses when they are not needed to escape detection by the host immune system.
The signaling networks that control pathogen gene expression during infection are extremely complex and often integrate many different environmental signals. Here, we investigated the function of QseG, a protein conserved in many pathogenic Enterobacteriaceae but which lacks significant homologs in the database. qseG is cotranscribed with qseEF, a two-component system known to control critical virulence genes in both EHEC and S. Typhimurium. Notably, we found that QseG is required for full virulence in EHEC, C. rodentium, and S. Typhimurium. However, we found that the sequence similarity of qseG between EHEC and S. Typhimurium was lower than for the surrounding genes, suggesting that its functions may have diverged in the two species. Our data indeed support the notion that, although qseG is required for full virulence by both EHEC and S. Typhimurium, its mechanisms of action differ between the two pathogens.
In EHEC, we found that QseG interacts with a component of the T3SS, SepL, but the nature of this interaction and its significance remain undefined. Interestingly, SepL and its S. Typhimurium homolog, SsaL, share only 35% similarity (37), and sepL cannot fully complement an ssaL mutant (38). Therefore, we do not think that SepL and SsaL play equivalent roles in regard to their interactions with QseG.
Previous studies have shown that QseG impacts the translocation of Tir but not its secretion. This, along with its interaction with SepL, suggests that QseG is involved in regulation of T3SS function. QseG's location in the inner leaflet of the outer membrane and its lack of any predicted signal transduction motifs suggest that its regulatory role may be carried out through protein-protein interactions with SepL and potentially other components of the T3SS apparatus. The QseEF two-component system controls transcription of the T3SS effector gene, espFu; thus, the qseEGF operon may control the activity of EHEC's T3SS at several levels.
In S. Typhimurium, we found that QseG modulates the invertible promoter controlling expression of the flagellin gene fljB, leading to repression of this flagellin allele. However, QseG does not alter motility in S. Typhimurium. This flagellin switch does not exist in EHEC, and in EHEC, QseG also does not play any role in motility or flagellar expression (data not shown). QseG also has a role in controlling the inflammatory response to S. Typhimurium that we hypothesize is mediated through its role in controlling fljB expression. Previous studies have shown that strains that are locked into expressing fljB are attenuated in mouse models of infection compared to both the WT and a “locked off” strain that expresses fliC, suggesting a disadvantage to expressing fljB in a mammalian host (39). Interestingly, purified recombinant FliC and FljB activate NF-κB to comparable levels in vitro (40), suggesting that FljB is not inherently more inflammatory than FliC.
We propose that while flagella are important during the intestinal phase of infection, expression of flagellin, and particularly FljB, is detrimental to S. Typhimurium at systemic sites (Fig. 8). During the systemic phase of infection, QseG is important for modulating the fljB promoter to favor the off position, leading to diminished FljB secretion. This leads to decreased NOS2 and cytokine induction and decreased overall inflammation at systemic sites.
FIG 8.
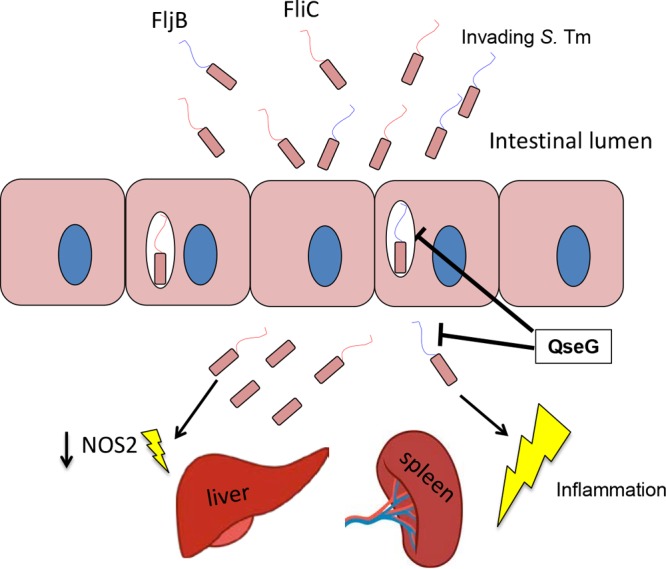
Model of the role of QseG in S. Typhimurium (S. Tm). Flagellin expression is advantageous during the intestinal portion of the life cycle, and the intestinal S. Typhimurium population is mixed, with some expressing FliC and others FljB. Once the bacteria cross the epithelial barrier and become systemic, flagellin expression is no longer advantageous. Flagellin secretion at systemic sites, particularly FljB, can lead to an increased inflammatory response and an attenuated infection. QseG is involved in limiting the inflammatory response to flagellin and modulates the fljB promoter so that the off orientation is favored. This leads to less FljB secretion, lowered NOS2 induction by the host, and a more virulent phenotype in a mouse model of systemic infection.
Several studies have demonstrated extensive cross talk between regulation of SPI-1 and flagella in S. Typhimurium (41–49). We have previously shown that QseEF activates expression of SPI-1, and now we have demonstrated that QseG regulates the expression of flagellin, suggesting that the qseEGF operon is another player in the cross talk between these two critical systems. However, further studies are needed to define whether there are interactions between QseEF and QseG and their respective regulons.
While we have demonstrated that QseG modulates the fljB promoter in S. Typhimurium, we have also shown previously that it is localized to the outer membrane in EHEC (4). The localization of QseG poses an interesting question as to how it exerts its effect on the orientation of the fljB DNA promoter. One possibility is that it interacts with other proteins that relay the message from the periplasm to the cytoplasm and exert effects on the promoter. We have found that QseG has at least one direct binding partner in EHEC, SepL, but we have yet to identify any proteins that interact with QseG in S. Typhimurium. The focus of future studies will be to determine proteins that QseG may interact with to carry out its function of modulating the fljB promoter. Another potential, albeit less likely, mechanism is that QseG itself interacts with DNA or cytoplasmic DNA binding proteins before it is lipidated and secreted to the outer membrane. As there are no putative DNA binding domains, we do not expect QseG to bind DNA itself, but future studies will seek to identify protein binding partners.
QseG modulates flagellar phase variation in S. Typhimurium, but paradoxically, QseG is present in EHEC and other Enterobacteriaceae whose genomes include only a single flagellin gene, fliC. Clearly, there has been some functional divergence of QseG across species, which is reflected by the 62% sequence identify between the S. Typhimurium and EHEC proteins. This specialization is also seen in the QseEF two-component system that is cotranscribed with qseG. In both EHEC and S. Typhimurium, QseEF control the expression of critical virulence factors, but the particular genes they control are species specific. However, as the presence of the qseEGF operon has been conserved across many pathogenic Enterobacteriaceae, there may still be a common, undefined function.
The work presented here highlights the idea that systems conserved across different species can evolve and be tuned to perform species-specific functions. Thus, while QseG is required for full virulence in both EHEC and S. Typhimurium, this outer membrane protein performs distinct functions in the two species. Furthermore, this work highlights the complexity of the regulatory networks that bacterial pathogens use to modulate the expression of their virulence genes. Many systems incorporate several layers of regulation, as well as cross talk between different virulence-associated systems, such as a T3SS and flagellins, which we still do not fully understand. Uncovering the mechanistic basis for virulence gene regulation will allow us to more fully understand how pathogens cause disease and may help in developing more effective therapeutics.
MATERIALS AND METHODS
Bacterial strains and plasmids.
All the bacterial strains and plasmids used in this study are listed in Table 2. EHEC and S. Typhimurium deletion mutants were created using λ red mutagenesis (50). An S. Typhimurium QseG complementation plasmid was created via restriction digestion and ligation into the pBAD/Myc-His A backbone. The complementation plasmid was induced with 0.2% arabinose. The primers used in this study are listed in Table 3.
TABLE 2.
Strains and plasmids
| Strain or plasmid | Genotype | Reference |
|---|---|---|
| Strains | ||
| 86-24 | Stx+ EHEC strain, serotype O157:H7 | 61 |
| NRO2 | 86-24 ΔqseG | 4 |
| CG04 | ΔsepL | This study |
| CG05 | ΔsepL ΔqseG | This study |
| 86-24 ΔsepL sepL::pTOPO-2HA | This study | |
| 86-24 ΔsepL ΔqseG sepL::pTOPO-2HA | This study | |
| Yeast L40 | Yeast reporter strain; MATa trp1 leu2 his3 LYS2::lexA-HIS3 URA3::lexA-lacZ | 52 |
| B2HPC | L40 transformed with pLX-YopJ + pVP-MEKK “yeast two-hybrid positive control” | 62 |
| B2HNC | L40 transformed with pLX-Laminin + pVP16 “yeast two-hybrid negative control” | 62 |
| SL1344 | Streptomycinr S. Typhimurium | 63 |
| SL1344 ΔqseG | This study | |
| SL1344 ΔinvA ΔspiB | 59 | |
| SL1344 ΔqseG qseG::pBAD/Myc-His | This study | |
| SW762 (ΔfliC ΔfljB) | SL1344 ΔfliC fljB5001::MudJ | 64 |
| SL1344 ΔqseG ΔfljB | This study | |
| Plasmids | Reference | |
| pKD3 | λ red template plasmid | 50 |
| pKD46 | λ red helper plasmid | 50 |
| pCP20 | λ red helper plasmid | 50 |
| SepL-HA | sepL::pTOPO-2HA | 14 |
| QseGpBAD | S. Typhimurium qseG under araBAD promoter in pBAD/Myc-His A | This study |
| pLex-ADB | Yeast two hybrid | |
| QseG-Lex | Yeast two hybrid | This study |
| SepL-Lex | Yeast two hybrid | This study |
| pVP16 | Yeast two hybrid | This study |
| EspD-VP16 | Yeast two hybrid | This study |
| SepD-VP16 | Yeast two hybrid | This study |
| EspA-VP16 | Yeast two hybrid | This study |
| EscC-VP16 | Yeast two hybrid | This study |
| SepL-VP16 | Yeast two hybrid | This study |
| Tir-VP16 | Yeast two hybrid | This study |
TABLE 3.
Primers
| Primer | Sequence (5′–3′) | Use |
|---|---|---|
| SL_qseG_Redf | GGAAGTTTGTTTTCGCATCTCGCTGCCATTGCCTGCATCTGACAAACACTAATGTGTAGGCTGGAGCTGCTTC | S. Typhimurium qseG KOa |
| SL_qseG_Redr | GTAATCCGGGATCGTCATCGACCAGCAGCAGGTGCGCCGGTTTACGGCTTACATATGAATATCCTCCTTAG | S. Typhimurium qseG KO |
| SL_qseG_REDcheckF | GGAAGTTTGTTTTCGCATCTCGCTGCC | Confirming S. Typhimurium qseG KO |
| SL_qseG_REDcheckR | GTAATCCGGGATCGTCATCGACCAGC | Confirming S. Typhimurium qseG KO |
| STMqseG_pBADmychisfwd xhoI | CCGCTCGAGGAATTTAAGCCTGGTGCGTATGTCAC | S. Typhimurium qseG complementation |
| STMqseG_pBADmychisrev hindIII | GCCAAGCTTTGGCGTTACCTCATCTTGTGACG | S. Typhimurium qseG complementation |
| Stm_rpoA_RTfwd | CGCGGTCGTGGTTATGTG | S. Typhimurium qPCR |
| Stm_rpoA_RTrev | GCGCTCATCTTCTTCCGAAT | S. Typhimurium qPCR |
| Stm_fljB_RTfwd | GGTACTACACTGGATGTATCG | S. Typhimurium qPCR |
| Stm_fljB_Rtrev | CGTAGCCGCTTTAATAGC | S. Typhimurium qPCR |
| Stm_fliC_RTfwd | GGTGGTAAAACTTACGCTG | S. Typhimurium qPCR |
| Stm_fliC_RTrev | GATCAGGCTGTGCTTTAAAG | S. Typhimurium qPCR |
| Stm_Hinversion_Univ | GCGCATTACGCTGTAAATCG | fljB promoter orientation |
| Stm_Hinversion_ON | GCGGGGCAATTTAATGGTGTCCGG | fljB promoter orientation |
| Stm_Hinversion_OFF | CGCGCTGTTGATACGCAGACC | fljB promoter orientation |
| fljB_RED_fwd | GCACAAGTAATCAACACTAACAGTCTGTCGCTGCTGACCCAGAATAACCGTGTAGGCTGGAGCTGCTTC | fljB KO |
| fljB_RED_rev | CAGAGACAGCACGTTCTGCGGGACCTGGTTAGCCTGCGCCAGAACGGCATATGAATATCCTCCTTAG | fljB KO |
| fljB_up | GCACAAGTAATCAACACTAAC | Screen fljB KO |
| fljA_down | GTAGTCCGAAGACGTGATCC | Screen fljB KO |
| hIL8_RTfwd | ATGACTTCCAAGCTGGCCG | Human IL-8 qPCR |
| hIL8_RTrev | CTCAGCCCTCTTCAAAAACTT | Human IL-8 qPCR |
| hGAPDH_RTfwd | AATCCCATCACCATCTTCCA | Human GAPDH qPCR |
| hGAPDH_RTrev | TGGACTCCACGACGTACTCA | Human GAPDH qPCR |
| mNOS2_RT_fwd | CCAGCCTTGCATCCTCATTGG | Mouse NOS2 qPCR |
| mNOS2_RT_rev | CCAAACACCAAGCTCATGCGG | Mouse NOS2 qPCR |
| mGAPDH_RT_fwd | AGGTCGGTGTGAACGGATTTG | Mouse GAPDH qPCR |
| mGAPDH_RT_rev | TGTAGACCATGTAGTTGAGGTCA | Mouse GAPDH qPCR |
| TirVP16F | CAAGGATCCACAGCATGCCTATTGGTAATCTTG | Yeast two hybrid |
| TirVP16R | CAATGCGGCCGCTTAGACGAAACGATGGGATC | Yeast two hybrid |
| SepLVP16F | CCAGGATCCACAGCATGGCTAATGGTATTG | Yeast two hybrid |
| SepLVP16R | CAATGCGGCCGCTCAAATAATTTCCTCCTTATAG | Yeast two hybrid |
| SepDVP16F | CACGGATCCCCGGCATGAACAATAATAATGGC | Yeast two hybrid |
| SepDVP16R | CAATGCGGCCGCTTACACAATTCGTCCTATATC | Yeast two hybrid |
| EspAVP16F | CAAGGATCCACAGCATGGATACATCAAATGCAACATCC | Yeast two hybrid |
| EspAVP16R | CAATGCGGCCGCTTATTTACCAAGGGATATTGCTG | Yeast two hybrid |
| EscCVP16F | CAAGGATCCCCGGCCGATATAGGACGAATTGTGTAATG | Yeast two hybrid |
| EscCVP16R | CATTGCGGCCGCTTATTCGCTAGATGCAGATTTTATC | Yeast two hybrid |
| QseGVP16F | CAAGGATCCACAGCATGCGACACATTTTTCAACG | Yeast two hybrid |
| QseGVP16R | CAATGCGGCCGCTTATGGCTCATCAGGAGTGACC | Yeast two hybrid |
| SepLLexF | CACGAATTCCCCATGGCTAATGGTATTG | Yeast two hybrid |
| SepLLexR | CAACTGCAGTCAAATAATTTCCTCCTTATAG | Yeast two hybrid |
| QseGLexF | CACGAATTCCCCATGCGACACATTTTTCAACG | Yeast two hybrid |
| QseGLesR | CAACTGCAGTTATGGCTCATCAGGAGTGACC | Yeast two hybrid |
| SepLREDF | TGGCTAATGGTATTGAATTTAATCAAAACCCCGCATCTGTTTTTAATTCTAATTCATTAGGTGTAGGCTGGAGCTGCTTC | EHEC KO |
| SepLREDR | TAATTTCCTCCTTATAGTCGATAACTTTACCAATCATTAATAATGCATTCTCTCTCTGCTCATATGAATATCCTCCTTAG | EHEC KO |
KO, knockout.
Proteinase K protection assays.
Proteinase K protection assays were performed as previously described (51).
Yeast two-hybrid screen.
A yeast two-hybrid screen was used as previously described (52) to test for possible protein-protein interactions between QseG and QseG, EscC, EspA, SepD, SepL, and Tir. Briefly, the Saccharomyces cerevisiae reporter strain L40 was transformed (53) with all combinations of the proteins in both the bait (pLEX-ADE) and the library (pVP16) vectors. Dual transformants were selected on yeast minimal medium lacking leucine and tryptophan and then assayed for protein-protein interactions using the integrated lacZ and HIS3 reporters. Association of the two-protein fusions was determined by growth on yeast minimal medium lacking histidine.
Coimmunoprecipitation.
Coimmunoprecipitation of SepL and QseG was performed using SepL-HA (14), anti-HA antiserum, and an anti-QseG antiserum (4) from EHEC expressing SepL-HA on a plasmid, as previously described (54).
Live-cell imaging.
Live-cell imaging was performed as previously described (55). The Lifeact::GFP (green fluorescent protein)-expressing cell line was created using the Flip-In System (Invitrogen). HeLa cells were transfected with pLacZ::Zeocin using Fugene 6 (Promega) to create FLP recombination target (FRT) sites in the genome. Cells were then selected with 100 μg/ml zeocin. Resistant foci were grown and assayed by Southern blotting against lacZ for single insertions, and then β-galactosidase assays were performed to measure expression of the inserted locus. High-expressing single insertions were then transfected with the flippase helper plasmid pOD44 and Lifeact::GFP cloned into the pFRT plasmid. These transfected cells were then selected in 100 μg/ml hygromycin, and resistant foci were visualized under fluorescence microscopy to measure levels of Lifeact::GFP expression. These cells were then maintained in 50 μg/ml hygromycin. When they were split before infection, hygromycin was not added. EHEC bacteria were transformed with the mCherry-expressing plasmid pDP151 and grown overnight statically in LB. The Lifeact::GFP cells' medium was replaced with low-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and infected with a 1:100 dilution of the overnight culture. The infection was allowed to continue for 2 h at 37°C and 5% CO2, and then the cells were washed 3 times with DMEM and visualized by live-cell imaging with a Zeiss microscope. Images were taken every 2 min for 2 h.
Infant rabbit model of EHEC infection.
To prepare the inocula, bacteria were grown overnight in LB broth at 37°C with appropriate antibiotics, harvested by centrifugation, and resuspended in sterile phosphate-buffered saline (PBS) (pH 7.2) and adjusted to a cell density of ∼109 CFU ml−1. Infant rabbit experiments were carried out as described previously (56). Briefly, 3-day-old New Zealand White rabbits were intragastrically inoculated with ∼5 × 108 CFU of WT EHEC or the qseG mutant using a size 5 French catheter. The rabbits were monitored twice daily for signs of illness or diarrhea. Diarrhea was described as follows: (i) none, i.e., normal pellets were dark green, hard, and formed; (ii) mild, i.e., diarrhea consisting of a mixture of soft yellow-green unformed and formed pellets, resulting in light staining of the hind legs; or (iii) severe, i.e., diarrhea consisting of unformed or liquid feces, resulting in significant staining of the hind legs. The rabbits were euthanized at 2 and 5 days postinfection. At necropsy, the intestinal tract from the duodenum to the anus was removed, and samples were obtained for microbiological analyses. To limit any litter-specific effects, at least two different litters were used to test each bacterial strain.
C. rodentium murine infections.
For mouse survival experiments, 10 3.5-week-old female C3H/HeJ or 129x1/SvJ mice per group were infected by oral gavage with multiple infectious doses of the wild-type or C. rodentium ΔqseG strain by oral gavage with 100 μl of PBS. Mouse survival in each group (10 animals per group) was accessed over the course of 14 to 26 days. A Kaplan-Meyer test was used to determine statistical significance. For colon weight measurement, 3.5-week-old female C3H/HeJ mice (5 animals per group) were infected with 1 × 109 cells of the wild type or C. rodentium ΔqseG. The infected mice were sacrificed on day 6 postinfection, and their colons were removed, washed, and weighed.
Salmonella Typhimurium.
Nine- to 12-week-old female BALB/c mice were infected intraperitoneally with 1 × 104 CFU of strain SL1344 in PBS or with sterile PBS as a control. Two independent experiments were performed, each with 15 mice per group (10 for survival and 5 for organ collection). Survival was monitored and weight was measured daily. On day 2 (1st experiment) or day 3 (2nd experiment), 5 randomly selected mice were sacrificed, and their livers and spleens were homogenized and plated on LB agar plus streptomycin to enumerate CFU. Weight and survival data from the two independent experiments were combined. A Student t test was used to compare weight loss in the different groups on each day, and the survival curves were analyzed using the Mantel-Cox test.
To measure fljB promoter orientation and NOS2 expression in infected tissues, portions of livers and spleens were collected in TRIzol, and total DNA and RNA were extracted according to the manufacturer's instructions. The fljB promoter orientation was measured in the extracted DNA using the qPCR standard curve method outlined below. NOS2 levels were assayed from the extracted RNA via qPCR as described below. NOS2 readings were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) levels and reported as a fold change over levels from uninfected (PBS-treated) mice. Averages and standard deviations across the five mice were reported, and statistical significance was calculated using Student's unpaired t test.
Secreted proteome analysis.
S. Typhimurium strain SL1344 cells were grown to late log phase in LB. The cells were pelleted, and the supernatants were filtered through a 0.2-μm filter and concentrated using Amicon 10-kDa MWCO concentrators (Millipore). Samples of the concentrated supernatants were run under denaturing conditions on SDS-12% PAGE gels and stained using Bio-safe Coomassie (Bio-Rad). The unknown band that appeared in the ΔqseG sample was extracted from the gel and submitted for identification by liquid chromatography-tandem MS (LC–MS-MS) by the University of Texas (UT)-Southwestern proteomics core facility. For total protein identification and quantification, concentrated supernatants from the WT and S. Typhimurium ΔqseG were loaded onto an SDS-PAGE gel and run only until the samples entered the resolving gel. The sample was extracted from the resolving gel and submitted for LC–MS-MS analysis to the UT-Southwestern proteomics core.
Quantitative real-time PCR.
For in vitro transcription analysis, S. Typhimurium was grown to an optical density at 600 nm (OD600) of 1.0 in LB. RNA was extracted and DNase treated as described previously (57). Quantification of RNA transcript levels was performed as described previously (58). Primer pairs were validated for amplification efficiency and specificity using a standard curve of diluted RNA. Sample RNA was mixed with SYBR master mix, validated primers, RNase inhibitor, and reverse transcriptase. A one-step reaction was run using the ABI 7500 system, and data were analyzed using ABI sequence detection v. 1.2 software. Values were normalized to endogenous rpoA and analyzed by the comparative critical threshold method. Values are presented as fold change over WT levels, and averages and standard deviations from three independent replicates are represented.
fljB promoter orientation assay.
DNA was extracted from strains grown as described above using a Sigma bacterial genomic DNA (gDNA) extraction kit. A universal primer was designed within the invertible element, and on and off primers were designed outside the invertible element so that a product would be amplified only when the promoter was in the designated position. Amplicons for each orientation were synthesized via conventional PCR, purified, and diluted to make a standard curve of known quantities. The standard curves were used to determine the quantity of each orientation in unknown samples, which were converted to moles, and the molar ratio of on to off was calculated.
Swimming motility assay.
Motility assays were performed as described previously (59). Briefly, strains were grown in LB broth plus 0.2% arabinose to induce complementation plasmid shaking at 37°C until they reached an OD600 of 1.0. Cells were stabbed into LB plates containing 0.3% agar and incubated at 37°C. Motility halos were measured at the indicated time points. Averages and standard deviations of three replicates are displayed.
Cell invasion assay.
HeLa epithelial cells were routinely cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum in 5% CO2. An invasion assay was performed as described previously (60). Briefly, HeLa cells were seeded at 1 × 105 per well in 24-well plates 24 h prior to infection. Overnight S. Typhimurium cultures were diluted in fresh LB and grown to an OD600 of 1.0, washed in sterile PBS, and resuspended to 1× in prewarmed DMEM. The cells were washed 3 times with prewarmed PBS and infected at a multiplicity of infection (MOI) of 10. The plate was spun at 1,000 × g for 5 min and incubated at 37°C in 5% CO2 for 90 min. The medium was removed, the cells were washed, and DMEM plus gentamicin (30 μg/ml) was added to kill extracellular bacteria. The cells were incubated for an additional 60 min before removing the medium and washing with PBS 3 times. The cells were lysed by incubation in 1 ml 1% Triton X-100 in PBS at room temperature for 5 min. The cell lysates and the bacterial inoculum were diluted in PBS and plated on LB agar with streptomycin to enumerate CFU.
Intracellular replication assay.
J774 mouse macrophages or THP-1 human macrophages were seeded in 24-well plates and grown to confluence over 2 days. To prepare inocula, bacteria were diluted 1:10 from an overnight culture in fresh LB medium and grown to an OD600 of 1.0 at 37°C with shaking. The bacteria were washed with PBS 2 times and then resuspended in cell culture medium. The cells were infected at an MOI of 5, and the plates were spun at 1,000 × g for 5 min to maximize bacterium-cell contact. The infected cells were incubated for 30 min at 37°C, 5% CO2. The medium was removed, and the cells were washed 3 times with warm PBS. Cell culture medium plus 30 μg/ml gentamicin was added to kill extracellular bacteria. After 3 h, samples were collected by removing the medium, washing the wells 3 times with PBS, and then adding 1 ml 0.1% Triton X-100 to each well. After incubating for 5 min, the cell lysates were collected. For 24-h samples, medium was removed at 3 h and replaced with medium containing 10 μg/ml gentamicin. At 24 h postinfection, the wells were washed, and the cells were lysed as described above. Serial dilutions of the lysates were plated on LB agar plus streptomycin to enumerate CFU. Averages and standard deviations for three independent experiments are shown.
IL-8 expression by T84 cells.
The T84 (ATCC CCL-248) human colorectal carcinoma cell line was routinely cultured in DMEM-F12 medium (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (Gibco). Cells were seeded in 6-well plates at a density of 1 × 105 cells per well and grown for 48 h. Twenty-four hours prior to infection, the cell culture medium was removed and replaced with serum-free medium. Strain SL1344 cells were grown to an OD600 of 1.0 with shaking in LB medium. The cells were pelleted, and the supernatants were filtered and concentrated ∼60× with 10-kDa MWCO Amicon concentrators (Millipore). The concentrated supernatants or purified flagellin (InvivoGen) was added to T84 cells in triplicate. One hour postaddition, the medium was removed and the cells were washed 3 times with prewarmed sterile PBS. One milliliter of TRIzol was added to each well, and cells were collected into Eppendorf tubes. RNA was extracted according to the manufacturer's instructions. DNA was degraded using Turbo DNase (Ambion), and cDNA was synthesized using Superscript II reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Fifty nanograms of cDNA was used per reaction with SYBR master mix and validated primers using an ABI 7500 system. To ensure no gDNA remained, control reactions were run with RNA as the template. The data were analyzed using ABI sequence detection v. 1.2 software. Values were normalized to those of endogenous GAPDH and analyzed by the comparative critical threshold method. Values are presented as fold change over untreated cells. Averages and standard deviations were calculated across triplicate wells, and statistically significant differences were determined via Student's t test. The results of one representative experiment are shown, but three independent experiments were performed to ensure reproducibility.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grants AI053067, AI05135, AI077613, and AI114511. C.C.G. was supported through NIH Training Grant 5 T32 AI7520-14. E.A.C. was supported by NIH grant 1F32AI126722-01.
The contents are solely the responsibility of the authors and do not represent the official views of the NIH NIAID.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00936-17.
REFERENCES
- 1.Cameron EA, Sperandio V. 2015. Frenemies: signaling and nutritional integration in pathogen-microbiota-host interactions. Cell Host Microbe 18:275–284. doi: 10.1016/j.chom.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beier D, Gross R. 2006. Regulation of bacterial virulence by two-component systems. Curr Opin Microbiol 9:143–152. doi: 10.1016/j.mib.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 3.Reading NC, Sperandio V. 2006. Quorum sensing: the many languages of bacteria. FEMS Microbiol Lett 254:1–11. doi: 10.1111/j.1574-6968.2005.00001.x. [DOI] [PubMed] [Google Scholar]
- 4.Reading NC, Rasko DA, Torres AG, Sperandio V. 2009. The two-component system QseEF and the membrane protein QseG link adrenergic and stress sensing to bacterial pathogenesis. Proc Natl Acad Sci U S A 106:5889–5894. doi: 10.1073/pnas.0811409106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campellone KG, Robbins D, Leong JM. 2004. EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev Cell 7:217–228. doi: 10.1016/j.devcel.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Garmendia J, Phillips AD, Carlier MF, Chong Y, Schuller S, Marches O, Dahan S, Oswald E, Shaw RK, Knutton S, Frankel G. 2004. TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell Microbiol 6:1167–1183. doi: 10.1111/j.1462-5822.2004.00459.x. [DOI] [PubMed] [Google Scholar]
- 7.Cheng HC, Skehan BM, Campellone KG, Leong JM, Rosen MK. 2008. Structural mechanism of WASP activation by the enterohaemorrhagic E. coli effector EspF(U). Nature 454:1009–1013. doi: 10.1038/nature07160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moreira CG, Sperandio V. 2012. Interplay between the QseC and QseE bacterial adrenergic sensor kinases in Salmonella enterica serovar Typhimurium pathogenesis. Infect Immun 80:4344–4353. doi: 10.1128/IAI.00803-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lostroh CP, Lee CA. 2001. The Salmonella pathogenicity island-1 type III secretion system. Microbes Infect 3:1281–1291. doi: 10.1016/S1286-4579(01)01488-5. [DOI] [PubMed] [Google Scholar]
- 10.Tullman-Ercek D, DeLisa MP, Kawarasaki Y, Iranpour P, Ribnicky B, Palmer T, Georgiou G. 2007. Export pathway selectivity of Escherichia coli twin arginine translocation signal peptides. J Biol Chem 282:8309–8316. doi: 10.1074/jbc.M610507200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeLisa MP, Tullman D, Georgiou G. 2003. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci U S A 100:6115–6120. doi: 10.1073/pnas.0937838100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mills E, Baruch K, Charpentier X, Kobi S, Rosenshine I. 2008. Real-time analysis of effector translocation by the type III secretion system of enteropathogenic Escherichia coli. Cell Host Microbe 3:104–113. doi: 10.1016/j.chom.2007.11.007. [DOI] [PubMed] [Google Scholar]
- 13.Kenny B, DeVinney R, Stein M, Reinscheid DJ, Frey EA, Finlay BB. 1997. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91:511–520. doi: 10.1016/S0092-8674(00)80437-7. [DOI] [PubMed] [Google Scholar]
- 14.Deng W, Li Y, Hardwidge PR, Frey EA, Pfuetzner RA, Lee S, Gruenheid S, Strynakda NC, Puente JL, Finlay BB. 2005. Regulation of type III secretion hierarchy of translocators and effectors in attaching and effacing bacterial pathogens. Infect Immun 73:2135–2146. doi: 10.1128/IAI.73.4.2135-2146.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Connell CB, Creasey EA, Knutton S, Elliott S, Crowther LJ, Luo W, Albert MJ, Kaper JB, Frankel G, Donnenberg MS. 2004. SepL, a protein required for enteropathogenic Escherichia coli type III translocation, interacts with secretion component SepD. Mol Microbiol 52:1613–1625. doi: 10.1111/j.1365-2958.2004.04101.x. [DOI] [PubMed] [Google Scholar]
- 16.Deng W, Puente JL, Gruenheid S, Li Y, Vallance BA, Vazquez A, Barba J, Ibarra JA, O'Donnell P, Metalnikov P, Ashman K, Lee S, Goode D, Pawson T, Finlay BB. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc Natl Acad Sci U S A 101:3597–3602. doi: 10.1073/pnas.0400326101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D, Roe AJ, McAteer S, Shipston MJ, Gally DL. 2008. Hierarchal type III secretion of translocators and effectors from Escherichia coli O157:H7 requires the carboxy terminus of SepL that binds to Tir. Mol Microbiol 69:1499–1512. doi: 10.1111/j.1365-2958.2008.06377.x. [DOI] [PubMed] [Google Scholar]
- 18.Reading NC, Rasko D, Torres AG, Sperandio V. 2010. A transcriptome study of the QseEF two-component system and the QseG membrane protein in enterohaemorrhagic Escherichia coli O157:H7. Microbiology 156:1167–1175. doi: 10.1099/mic.0.033027-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng W, Li Y, Vallance BA, Finlay BB. 2001. Locus of enterocyte effacement from Citrobacter rodentium: sequence analysis and evidence for horizontal transfer among attaching and effacing pathogens. Infect Immun 69:6323–6335. doi: 10.1128/IAI.69.10.6323-6335.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wadolkowski EA, Sung LM, Burris JA, Samuel JE, O'Brien AD. 1990. Acute renal tubular necrosis and death of mice orally infected with Escherichia coli strains that produce Shiga-like toxin type II. Infect Immun 58:3959–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luperchio SA, Schauer DB. 2001. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes Infect 3:333–340. doi: 10.1016/S1286-4579(01)01387-9. [DOI] [PubMed] [Google Scholar]
- 22.Mallick EM, McBee ME, Vanguri VK, Melton-Celsa AR, Schlieper K, Karalius BJ, O'Brien AD, Butterton JR, Leong JM, Schauer DB. 2012. A novel murine infection model for Shiga toxin-producing Escherichia coli. J Clin Invest 122:4012–4024. doi: 10.1172/JCI62746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simmons CP, Goncalves NS, Ghaem-Maghami M, Bajaj-Elliott M, Clare S, Neves B, Frankel G, Dougan G, MacDonald TT. 2002. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching-effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J Immunol 168:1804–1812. doi: 10.4049/jimmunol.168.4.1804. [DOI] [PubMed] [Google Scholar]
- 24.Ghaem-Maghami M, Simmons CP, Daniell S, Pizza M, Lewis D, Frankel G, Dougan G. 2001. Intimin-specific immune responses prevent bacterial colonization by the attaching-effacing pathogen Citrobacter rodentium. Infect Immun 69:5597–5605. doi: 10.1128/IAI.69.9.5597-5605.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reading NC, Torres AG, Kendall MM, Hughes DT, Yamamoto K, Sperandio V. 2007. A novel two-component signaling system that activates transcription of an enterohemorrhagic Escherichia coli effector involved in remodeling of host actin. J Bacteriol 189:2468–2476. doi: 10.1128/JB.01848-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moreira CG, Russell R, Mishra AA, Narayanan S, Ritchie JM, Waldor MK, Curtis MM, Winter SE, Weinshenker D, Sperandio V. 2016. Bacterial adrenergic sensors regulate virulence of enteric pathogens in the gut. mBio 7:e00826-16. doi: 10.1128/mBio.00826-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrewes FW. 1922. Studies in group-agglutination I. The Salmonella group and its antigenic structure. J Pathol 25:505–521. [Google Scholar]
- 28.Fujita H, Yamaguchi S, Iino T. 1973. Studies on H-O variants in Salmonella in relation to phase variation. J Gen Microbiol 76:127–134. doi: 10.1099/00221287-76-1-127. [DOI] [PubMed] [Google Scholar]
- 29.Kutsukake K, Iino T. 1980. Inversions of specific DNA segments in flagellar phase variation of Salmonella and inversion systems of bacteriophages P1 and Mu. Proc Natl Acad Sci U S A 77:7338–7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lederberg J, Iino T. 1956. Phase variation in Salmonella. Genetics 41:743–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearce UB, Stocker BA. 1967. Phase variation of flagellar antigens in Salmonella: abortive transduction studies. J Gen Microbiol 49:335–349. doi: 10.1099/00221287-49-2-335. [DOI] [PubMed] [Google Scholar]
- 32.Silverman M, Zieg J, Simon M. 1979. Flagellar-phase variation: isolation of the rh1 gene. J Bacteriol 137:517–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suzuki H, Iino T. 1973. In vitro synthesis of phase-specific flagellin of Salmonella. J Mol Biol 81:57–70. doi: 10.1016/0022-2836(73)90247-7. [DOI] [PubMed] [Google Scholar]
- 34.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. 2001. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature 410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 35.Mizel SB, Honko AN, Moors MA, Smith PS, West AP. 2003. Induction of macrophage nitric oxide production by Gram-negative flagellin involves signaling via heteromeric Toll-like receptor 5/Toll-like receptor 4 complexes. J Immunol 170:6217–6223. doi: 10.4049/jimmunol.170.12.6217. [DOI] [PubMed] [Google Scholar]
- 36.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. 2011. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 37.Kresse AU, Beltrametti F, Muller A, Ebel F, Guzman CA. 2000. Characterization of SepL of enterohemorrhagic Escherichia coli. J Bacteriol 182:6490–6498. doi: 10.1128/JB.182.22.6490-6498.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coombes BK, Brown NF, Valdez Y, Brumell JH, Finlay BB. 2004. Expression and secretion of Salmonella pathogenicity island-2 virulence genes in response to acidification exhibit differential requirements of a functional type III secretion apparatus and SsaL. J Biol Chem 279:49804–49815. doi: 10.1074/jbc.M404299200. [DOI] [PubMed] [Google Scholar]
- 39.Ikeda JS, Schmitt CK, Darnell SC, Watson PR, Bispham J, Wallis TS, Weinstein DL, Metcalf ES, Adams P, O'Connor CD, O'Brien AD. 2001. Flagellar phase variation of Salmonella enterica serovar Typhimurium contributes to virulence in the murine typhoid infection model but does not influence Salmonella-induced enteropathogenesis. Infect Immun 69:3021–3030. doi: 10.1128/IAI.69.5.3021-3030.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simon R, Samuel CE. 2007. Activation of NF-kappaB-dependent gene expression by Salmonella flagellins FliC and FljB. Biochem Biophys Res Commun 355:280–285. doi: 10.1016/j.bbrc.2007.01.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baxter MA, Jones BD. 2005. The fimYZ genes regulate Salmonella enterica serovar Typhimurium invasion in addition to type 1 fimbrial expression and bacterial motility. Infect Immun 73:1377–1385. doi: 10.1128/IAI.73.3.1377-1385.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Clegg S, Hughes KT. 2002. FimZ is a molecular link between sticking and swimming in Salmonella enterica serovar Typhimurium. J Bacteriol 184:1209–1213. doi: 10.1128/jb.184.4.1209-1213.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ellermeier CD, Slauch JM. 2003. RtsA and RtsB coordinately regulate expression of the invasion and flagellar genes in Salmonella enterica serovar Typhimurium. J Bacteriol 185:5096–5108. doi: 10.1128/JB.185.17.5096-5108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iyoda S, Kamidoi T, Hirose K, Kutsukake K, Watanabe H. 2001. A flagellar gene fliZ regulates the expression of invasion genes and virulence phenotype in Salmonella enterica serovar Typhimurium. Microb Pathog 30:81–90. doi: 10.1006/mpat.2000.0409. [DOI] [PubMed] [Google Scholar]
- 45.Lin D, Rao CV, Slauch JM. 2008. The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J Bacteriol 190:87–97. doi: 10.1128/JB.01323-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lucas RL, Lostroh CP, DiRusso CC, Spector MP, Wanner BL, Lee CA. 2000. Multiple factors independently regulate hilA and invasion gene expression in Salmonella enterica serovar Typhimurium. J Bacteriol 182:1872–1882. doi: 10.1128/JB.182.7.1872-1882.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saini S, Pearl JA, Rao CV. 2009. Role of FimW, FimY, and FimZ in regulating the expression of type I fimbriae in Salmonella enterica serovar Typhimurium. J Bacteriol 191:3003–3010. doi: 10.1128/JB.01694-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thijs IM, De Keersmaecker SC, Fadda A, Engelen K, Zhao H, McClelland M, Marchal K, Vanderleyden J. 2007. Delineation of the Salmonella enterica serovar Typhimurium HilA regulon through genome-wide location and transcript analysis. J Bacteriol 189:4587–4596. doi: 10.1128/JB.00178-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saini S, Slauch JM, Aldridge PD, Rao CV. 2010. Role of cross talk in regulating the dynamic expression of the flagellar Salmonella pathogenicity island 1 and type 1 fimbrial genes. J Bacteriol 192:5767–5777. doi: 10.1128/JB.00624-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Norris SJ, Carter CJ, Howell JK, Barbour AG. 1992. Low-passage-associated proteins of Borrelia burgdorferi B31: characterization and molecular cloning of OspD, a surface-exposed, plasmid-encoded lipoprotein. Infect Immun 60:4662–4672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vojtek AB, Hollenberg SM. 1995. Ras-Raf interaction: two-hybrid analysis. Methods Enzymol 255:331–342. doi: 10.1016/S0076-6879(95)55036-4. [DOI] [PubMed] [Google Scholar]
- 53.Gietz D, St Jean A, Woods RA, Schiestl RH. 1992. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res 20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Golemis E. 2002. A molecular cloning manual: protein-protein interactions. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 55.Gruber CC, Sperandio V. 2014. Posttranscriptional control of microbe-induced rearrangement of host cell actin. mBio 5:e01025-13. doi: 10.1128/mBio.01025-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ritchie JM, Thorpe CM, Rogers AB, Waldor MK. 2003. Critical roles for stx2, eae, and tir in enterohemorrhagic Escherichia coli-induced diarrhea and intestinal inflammation in infant rabbits. Infect Immun 71:7129–7139. doi: 10.1128/IAI.71.12.7129-7139.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gruber CC, Sperandio V. 2015. Global analysis of posttranscriptional regulation by GlmY and GlmZ in enterohemorrhagic Escherichia coli O157:H7. Infect Immun 83:1286–1295. doi: 10.1128/IAI.02918-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hughes DT, Clarke MB, Yamamoto K, Rasko DA, Sperandio V. 2009. The QseC adrenergic signaling cascade in Enterohemorrhagic E. coli (EHEC). PLoS Pathog 5:e1000553. doi: 10.1371/journal.ppat.1000553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moreira CG, Weinshenker D, Sperandio V. 2010. QseC mediates Salmonella enterica serovar Typhimurium virulence in vitro and in vivo. Infect Immun 78:914–926. doi: 10.1128/IAI.01038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Finlay BB, Falkow S. 1988. Comparison of the invasion strategies used by Salmonella cholerae-suis, Shigella flexneri and Yersinia enterocolitica to enter cultured animal cells: endosome acidification is not required for bacterial invasion or intracellular replication. Biochimie 70:1089–1099. doi: 10.1016/0300-9084(88)90271-4. [DOI] [PubMed] [Google Scholar]
- 61.Griffin PM, Ostroff SM, Tauxe RV, Greene KD, Wells JG, Lewis JH, Blake PA. 1988. Illnesses associated with Escherichia coli O157:H7 infections. A broad clinical spectrum. Ann Intern Med 109:705–712. [DOI] [PubMed] [Google Scholar]
- 62.Orth K, Palmer LE, Bao ZQ, Stewart S, Rudolph AE, Bliska JB, Dixon JE. 1999. Inhibition of the mitogen-activated protein kinase kinase superfamily by a Yersinia effector. Science 285:1920–1923. doi: 10.1126/science.285.5435.1920. [DOI] [PubMed] [Google Scholar]
- 63.Hoiseth SK, Stocker BA. 1981. Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291:238–239. doi: 10.1038/291238a0. [DOI] [PubMed] [Google Scholar]
- 64.Winter SE, Winter MG, Poon V, Keestra AM, Sterzenbach T, Faber F, Costa LF, Cassou F, Costa EA, Alves GE, Paixao TA, Santos RL, Baumler AJ. 2014. Salmonella enterica serovar Typhi conceals the invasion-associated type three secretion system from the innate immune system by gene regulation. PLoS Pathog 10:e1004207. doi: 10.1371/journal.ppat.1004207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.