

Abstract
The oligomeric state of membrane proteins has recently emerged in many cases as having an effect on their function. However, the intrinsic dynamics of their spatial organization in cells and model systems makes it challenging to characterize. Here we use molecular dynamics (MD) simulations at multiple resolutions to determine the dimer conformation of the human serotonin transporter (hSERT). From self-assembly simulations we predict dimer candidates and subsequently quantify their relative strength. We use umbrella sampling (US) replica exchange MD simulations for which we present extensive analysis of their efficiency and improved sampling compared to regular US MD simulations. The data shows that the most stable hSERT dimer interface is symmetrical and involves transmembrane helix 12 (TM12), similar to the crystal structure of the bacterial homologue LeuT, but with a slightly different orientation. We also describe the supramolecular organization of hSERT from a 250 μs self-assembly simulation. Finally, the effects of the presence of phosphatidylinositol bisphosphate or cholesterol in the membrane model has been quantified for the TM12-TM12 predicted interface. Collectively, the presented data bring new insight to the area of protein and lipid interplay in biological membranes.
Introduction
Oligomerization of integral proteins in crowded cell membranes has recently emerged as a dynamic process that could be used in cells as a regulatory mechanism. Such mechanism could act through the cell’s ability to translocate (traffic) proteins between organelles and compartments of the cell membrane1, thereby changing the environment of the protein, which, in turn, would affect its oligomerization propensity and thereby affect their function2. Alternatively, a direct modification of the confined lipidic environment of the protein could be used to affect protein oligomerization and function.
Neurons and the space through which they communicate, the synapse, are the stage of a multitude of such processes, contributing to the synaptic plasticity2; eg. GABA2, NMDA3 and AMPA receptor trafficking4,5, DAT function and trafficking6, SERT partitioning and function7, trafficking and oligomeric state8, and generally neurotransmitter:sodium transporters9,10. The functional role of protein oligomerization in the synapses is however still unclear in most cases.
In this study we focus on monoamine transporters (MATs). MATs are proteins located in the pre-synaptic plasma membrane and assure the re-uptake of neurotransmitters serotonin (5-hydroxytryptamine, 5HT), dopamine (DA) and norepinephrine (NE) from the synaptic cleft into the pre-synaptic neuron. Neurotransmitter re-uptake is a key step for proper neurotransmission and MATs dysfunction has been associated with multiple neuronal disorders such as depression11, ADHD12, Parkinson12 and addiction13, making MATs the primary targets for treatment of mental disorders14.
Notably, MATs have been shown experimentally to form oligomers in the synaptic plasma membrane15, but it is unclear which regulatory or functional role these may have. Many lines of evidence suggest that DA and 5-HT transporters (DAT and SERT, respectively) can form a homotetramer presumably consisting of two dimers16–19, but recent experiments have also shown that SERT exists in many different oligomeric states including monomers8,15. As for today we know that DAT oligomer formation is a prerequisite for ER export and integration into the plasma membrane20. DAT also has a cooperative effect on inhibitor binding and substrate transport in the individual protomers21. Supporting this idea, recent simulations indicate that DAT collective motions resemble more the conformational changes associated with transport when it is analyzed as a dimer than as a monomer, suggesting that DAT would be a more efficient transporter in a dimer conformation22. Access to the structures of MAT dimers and higher order oligomers at the atomistic resolution appear therefore timely to guide and inspire the design of new experiments.
A few studies have provided important clues to MATs dimer conformation but yet no consensus has emerged. Cysteine crosslinking experiments suggest that transmembrane helix (TM) 4 (Cys243) and TM6 (Cys306) are involved in the formation of a symmetrical dimer in DAT16,23. The overall organization of MAT TMs is given in Fig. 1A using hSERT-numbering. A DAT dimer interface involving TM6 was also more recently suggested by a combination of crosslinking, mutagenesis and computational experiments24. In addition, four tandem repeats of a leucine heptad (LX6LX6LX6L), a structural motif which underlies oligomerization in phospholamban25, is present but incomplete in MATs’ TM2. A mutational study of this repeat in DAT suggested that TM2 should be involved in a symmetrical dimer interface26. However, in the crystal structures of hSERT27 and dDAT28, this repeat is buried within the core of the proteins preventing it from participating directly in an interface29. Furthermore, in the case of SERT, FRET experiments have shown that the fragment pairs TM1/TM2 and TM11/TM12 might represent symmetrical intermolecular interaction sites30. A GXXXG motif in SERT’s TM12, but not conserved across MATs, was proposed to participate in the interaction site in the TM11/TM12 pair9. But again, visual inspection of the recent hSERT crystal structure revealed that this repeat is not exposed to the membrane. Nevertheless, an interface involving TM11/TM12 would be in agreement with the crystal structure of the MAT’s bacterial homologue, LeuT, which when resolved as a symmetrical dimer involves TM9, TM12 and extra-cellular loop 2 (ECL2) at the interface31. LeuT has also been resolved as a monomer. Overall, current data do not support a unique dimer model for all MATs, but instead suggests that the different transporters may have different interfaces.
Figure 1.

Systems simulated. hSERT was embedded into a series of lipid bilayers (POPC, grey tails, and tan and blue headgroups) and solvated in water (W, blue). (A) One copy of hSERT embedded in 380 POPC molecules; some transmembrane (TM) helices are highlighted for convenience, and lipid and water molecules are not shown to feature the transporter. (B) 16 copies of hSERT inserted in a POPC lipid membrane. The final configuration of ten self-assembly simulations of 30 μs each are depicted. (C) Configuration of 64 copies of hSERT after a 250-μs self-assembly simulation. In (B and C), the systems are viewed from the top of the membrane, a 1:99 protein:lipid ratio was used, TM11 (black) and TM12 (red) are highlighted, the solvent and the lipid molecules are omitted for clarity, hSERT’s backbone trace is shown in pink, the green areas represent the periodic images of the central cell (white). In (B and C), a few illustrative aggregates are highlighted in black.
Furthermore, mounting evidence has stressed the importance of the lipid composition in biological membranes for the formation and stabilization of protein oligomers32–36, a factor that could be used to regulate oligomerization in cells and thereby the function of proteins. In the case of MATs, the oligomeric state of hSERT has been shown to evolve from a dynamic exchange of the transporters between the multiple oligomer types in the ER to a more static distribution in the cell membrane8. Although the presence of phosphatidylinositol bisphosphate (PIP2) in the cell membrane is key to this transition, hSERT assembly also occurs in the absence of PIP28. The role of PIP2 remains to be determined but it was suggested to interact with the N-terminus of hDAT37. More recently, DAT nanodomains where found sensitive to cholesterol (CHOL) depletion and potentially implicated in modulating dopaminergic neurotransmission6. Also recently, mass spectroscopy studies underline the importance of lipids in the stabilization of LeuT36 dimers. In this study, cardiolipin stabilizes the LeuT dimer in addition to six phospholipids most likely bound at the interface36. Alternatively, lipids can affect protein’s function through a direct interaction as we have recently shown recently experimentally for hSERT38 and computationally for hDAT39.
Here we determine the structure of the hSERT dimer and characterize its supramolecular organization in a membrane environment. We apply our approach developed previously to determine the relative strength of membrane protein interfaces40, which consists in the use of coarse grain molecular dynamics and relies on the determination of potentials of mean force (PMF) of dimer candidates extracted from self-assembly simulations. The method is herein improved by replacing regular umbrella sampling (US) simulations by US replica exchange molecular dynamics (REMD) simulations41, which we show considerably improved convergence for. The dimer structure of hSERT extracted with this enhanced method shows an interface centered on TM12 but with a different orientation of the protein core than seen for LeuT31. We also estimated the effect of PIP2 and CHOL on the strength of this dimer interface and infer on the mechanism by which these lipids might affect the interface.
Results
Four main interfaces are formed during the self-assembly simulations
Ten self-assembly simulations of 16 hSERT molecules and one with 64 molecules embedded in a pure POPC lipid membrane collectively show that hSERT forms dimers and higher order oligomers with multiple interfaces on a μs time scale (Fig. 1B,C). Each hSERT monomer is on average in contact with two other monomers (Figs 1B,C and S1) resulting in a supramolecular organization with different sizes of aggregates. Some align almost linearly, others form small aggregates and a few combine both to form small stretches of rows-of-dimers (Example are encircled in Fig. 1B,C).
A cluster analysis of hSERT dimers formed during the ten repeats of self-assembly simulations identified four main hSERT dimer interfaces (Fig. 2A). These conformations were found within the first 10 clusters and had more than 3% of the clusters analyzed (Figure S2). The two predominant clusters involve TM12 and account for 40% of the interfaces analyzed. In the first cluster hSERT forms a symmetric interface (TM12-TM12) and in the second cluster an asymmetric one is formed, where TM12 engages in a contact with TM7 in the second monomer. The presence of TM12 at the hSERT dimer interface is in line with experimental studies pointing to its contribution to the dimer interface30,31,43. The third cluster is an asymmetric dimer, which involves TM4/9 and TM2/11. The forth cluster is symmetric and the interface is primarily formed in the aqueous phase, involving the extracellular ends of TM3 and TM4 and the connecting loop, ECL2 (Fig. 2A).
Figure 2.

hSERT interfaces. (A) Representative structures of the four most populated interfaces formed in the self-assembly CG simulations of hSERT in a POPC membrane (See text for details). From top to bottom: TM12-TM12, TM12-TM7, TM4/9-TM2/11 and TM3/4-TM3/4. For each interface the values of the ϕ1 and ϕ3 angles used to define their relative orientation in the VBA (See Figure S26 for details) and their contributions to the first 10 clusters are indicated. Atomistic structures of hSERT were fitted onto the CG configurations. (B) Top: Interface of LeuT (bacterial homologue of hSERT) as found in a crystal structure. Bottom: hSERT fitted on LeuT. TM12 (red) was omitted during the fitting procedure. One can appreciate the resulting different locations of TM12. The orange bananas illustrate the complemented crescent moons of the interface. The color code is as in Fig. 1A.
This organization may be compared to the case of other proteins. Rhodopsin, a prototypical G protein-coupled receptor (GPCR), also forms on average two contacts per monomer but linear arrays predominate their supramolecular organization in self-assembly simulations40. Similar differences of protein organization in lipid membranes were reported for model proteins in response of the protein class and shape, hydrophobic mismatch and membrane curvature42.
Notable is the similarity of the symmetric interface using TM12 (cluster 1) with the interface formed in crystals by the bacterial homologue of SERT, LeuT31. In both cases TM12 is the main contribution to the interface (Fig. 2B). However, the two interfaces differ. First, the contacts between TM12 change. While in hSERT the two TM12 seem to wrap around each other taking advantage of a kink in the middle of the helix, in LeuT TM12 is straight and the interface involves another helix, TM9, in contact with TM12 (Fig. 2B). Second, the position of TM12 relative to the rest of the helical bundle of the transporter (TM1–11) differs in LeuT and in hSERT. This difference leads to a distinct orientation of the proteins helical bundle relative to the two TM12 interfaces. This difference is apparent when the hSERT helical bundle (TM1–11) is superimposed on LeuT (Fig. 2B). This operation is equivalent to a rotation of hSERT around the membrane normal, opening a large space between the monomers, which then lack direct contacts. This operation also rotates TM12 away from the interface then mainly facing TM9 (Fig. 2B). The relaxation of this interface is described later in this manuscript.
PMF: efficiency and convergence of US-MD and US-REMD
To determine the relative strength of hSERT interfaces found in the self-assembly simulations we have calculated the potential of mean force (PMF) using the umbrella sampling (US) method44 coupled with replica exchange molecular dynamics (REMD) simulations, also called window or umbrella exchange MD simulations41. In this section we present an analysis on the efficiency and convergence of such simulations and we compare them to conventional US-MD simulations.
In the determination of a PMF using the US technique, the starting conformation of the system used at each umbrella is key because the conformational sampling might be affected in cases where the free energy surface is complex and contains multiple wells. Convergence is difficult to attain even in simple cases40,45–48. In principle, by allowing the system to walk the reaction coordinate (umbrella) space US-REMD should allow for a better sampling and therefore a faster convergence than US-MD41.
We compared US-MD simulations starting with the proteins bound or unbound in all umbrella windows. Their progression over a 1.1 μs simulation shows that both have difficulties to converge (Fig. 3A,B). While the simulation started from bound proteins has difficulties to sample the umbrella windows where the proteins are at the boundary between bound and unbound states (shoulder forming at ~5.8/6.0 nm as the simulation time increases in Fig. 3A), the simulation started from unbound proteins fails to sample the bound state (minimum of the PMF at 5.4 nm). These limitations are respectively due to the energy barrier needed to separate the proteins leading to a sudden separation of the proteins, and the trapping of lipids at the protein interface preventing them from exploring the minimum. Both simulations lead to major errors in the PMF as the system may adopt very unrealistic conformations and remain trapped for extended periods of time. Interestingly, the blind combination of both simulations leads to a reasonable PMF (Fig. 3C). On a technical note, it is actually difficult to understand how WHAM is able to converge since for many umbrella windows the histograms are significantly different in the two simulations (Figure S3).
Figure 3.

Convergence of umbrella sampling (US) MD simulations. The convergence is illustrated by following the evolution of the potentials of mean force (PMF) for one of the most populated interfaces: TM12-TM12. PMFs were obtained with (A–E) a conventional US-MD approach for membrane proteins or (F,G) with an US-replica exchange molecular dynamics (REMD). The different curves in each panel represent the PMF obtained using different time window as indicted in the color legends. The US-MD were either started from a bound (A) or unbound state of the proteins. In (C) both (A) and (B) were combined using all the umbrella windows, while in (D) subsets of umbrellas were used (see Figure S4 for details of the windows used). In (E) the convergence of the mix 2 is shown. In (F and G) the proteins were initially bound.
Similar issues were observed in our previous study of rhodopsin in which case up to 20 μs per umbrella simulation were not sufficient to overcome the energy barriers trapping the system40. We therefore used a strategy consisting of mixing umbrella simulations started from bound and unbound conformations. While, as discussed above, one can blindly combine all the umbrellas (see Fig. 3C for the example of hSERT). However, this approach typically requires the determination of an appropriate mixture of bound and unbound umbrella simulations to calculate an accurate PMF. PMFs are indeed extremely sensitive to the set of windows given to WHAM (Figs 3D,E, S3). Although, when carried out with extreme care this approach can provide reliable results40, it is a tedious task and will always be sensitive to the user’s understanding of the system and therefore undoubtedly subject to bias. We illustrate the practice of that sensitive approach on one interface of hSERT (Figs 3D,E, S4).
US-REMD simulations seem to offer an ideal practical solution to this issue. The systems or replicas are allowed to walk through the umbrella space (protein interfacial separation) and thereby explore different conformations. The proteins should bind and unbind and the lipids should get trapped and un-trapped as the distance between the proteins varies. We first tested the efficiency of US-REMD simulation as a function of the frequency of exchange trials between umbrella windows. Values of 20, 200 and 2000 ps were compared by monitoring the evolution of the PMF calculated using 0.1 μs time windows over a period of 0.9 μs (Figure S5). Although theses simulations are not converged themselves, one can appreciate that a 20 ps interval between exchange trials is the most efficient. It allows the system to overcome barriers faster. In the cases of 200 and 2000 ps between attempts of exchange of replicas the PMF seems to get trapped for longer periods of time than when attempts are tried every 20 ps (Figure S5). An interval of 20 ps between exchange trials is used in all the following simulations.
We then tested whether starting from a bound or an unbound conformation of the protein would also impact US-REMD as described above for US-MD and reported previously for US-REMD applied to more simple systems48. The comparison of two US-REMD simulations in which all replicas are started from either a bound or an unbound state shows that it does matter in a similar manner. Over a 2.6 μs period, the US-REMD simulation started from an unbound configuration fails to sample the minimum or the bound state (Figure S6), due to lipid molecules being trapped at the interface. This limitation leads to a very rough and apparently weak binding similar to the US-MD results. In contrast, the US-REMD simulation started with bound proteins produces an apparently more realistic profile (Figure S6). The distances at the minimum are well sampled but also the distances corresponding to protein separation, removing the shoulder observed in the US-MD PMF (Fig. 3A). It is unfortunately not currently possible to extend these two simulations so that both curves would converge, similar to what Domanski et al. were able to do on a more simple system48. We expect the computational cost to be prohibitive.
In line with the work of Domanski et al.48 our results, however, suggest that starting the simulation from bound proteins is more beneficial. Lipids trapped at the dimer interface when the unbound proteins come closer seems to be a severely limiting issue also in US-REMD. Following the walk of the systems in the umbrella space indicates that typically only a few of them actually explore a large range of umbrellas and therefore a large range of protein separations even with the extended simulation time used (Figure S7). Most replicas remain within a restricted range of umbrellas or distances between proteins. The benefit of REMD then relates more to its ability to sort conformations in the umbrella space than to its ability to generate different conformations. This dual aspect of REMD simulation has been described previously for temperature replica exchange simulations49. An alternative solution would be to use a mix of bound and unbound configurations of the protein that are more representative of the umbrella potential they start at. But this situation is rapidly reached (a few hundred nanoseconds) in the US-REMD simulation started from bound proteins.
Finally, the convergence of a US-REMD simulation started from bound hSERTs was followed over a 10 μs period (Fig. 3F,G). It is clear that a few μs are needed to remove the memory from the bound state and start collecting relevant information. Similar progression is observed for all the interfaces probed in this study (Figs 3F,G, S8–11). Overall our analysis indicates that the US-REMD simulations are able to reach a reasonable level of convergence within 10 μs. We note that in the present approach we sample only one interface at the time so the proteins do not need to rotate to probe other interfaces. This particularity certainly accelerates the convergence of the simulations.
The symmetric TM12 dimer is the most stable
The small number of binding and unbinding events in the self-assembly simulations (Figures S12, S13) precludes the interpretation of the population of the different interfaces as thermodynamics data40. These simulations are typically out of equilibrium. Instead, the relative stability of the four most populated interfaces formed in the self-assembly simulations were assessed by US-REMD simulations that allow the calculation of the PMF of the system as a function of protein separation (Fig. 4). Weak harmonic potentials are used to control the relative orientation of the hSERT monomers and thereby define specific interfaces (Fig. 4A), as done previously40.
Figure 4.
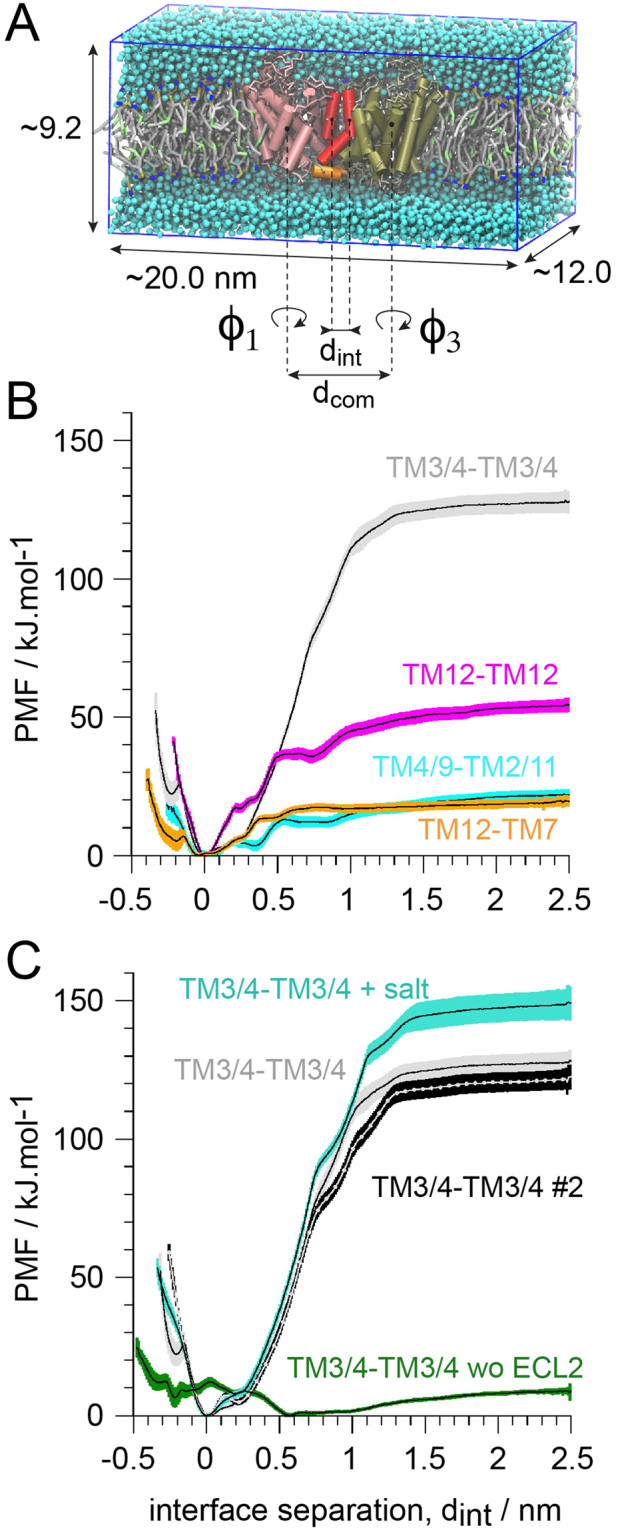
Comparative strength of hSERT dimer interfaces. (A) Simulation setup. The transporters transmembrane helices (TM) are shown in tan and pink tubes, but TM11 and TM12 are black and red, respectively, and the C-terminus helix in orange. (B) Potentials of mean forces (PMF) of the different hSERT interfaces formed in the self-assembly simulations. (C) PMFs of the TM3/4 interface variant: the original TM3/4 and a repeat (#2), in presence of 0.2 M of NaCl (+salt) and with the interaction of the ECL2 between the two monomers removed (wo ECL2).
The depths of the well in the PMF curves suggest that the symmetric interface based on TM3/4 is by far the most stable. Then comes the symmetric TM12 interface, followed by the two asymmetric interfaces, TM4/9-TM2/11 and TM7-12 (Fig. 4). The TM3/4 conformation is stabilized by ~130 kJ/mol, ca. 70 and 110 kJ/mol more than the other interfaces. This represents an unusually high value for protein-protein interactions2,40,45,48,50–53. A visual inspection of the umbrella trajectories of this interface, revealed an interaction of the ECL2 (between TM3 and TM4) in the aqueous phase that proved to be difficult to “break” in the simulations and led to an unrealistic conformation where the proteins tilt considerably (Figure S14). The section of the protein interface that spans the membrane hydrophobic interior is solubilized by lipids even in the equilibrated dimer conformation (Figure S14). These observations suggest that the strength of this interface might be the result of the so-called “stickiness” of the MARTINI force field for proteins in the aqueous phase; a known issue only sparsely documented in the literature but a few attempts of improvement have been reported54 (D de Yong, X Periole and SJ Marrink, unpublished results).
The involvement of ECL2 in the strength of the TM3/4 interface was further evaluated by determining an additional set of PMFs (Fig. 4C). We first repeated the US-REMD simulation of the TM3/4 interface. The two profiles are within the statistical error of each other. Addition of 0.2 M NaCl to the aqueous phase increased the apparent strength of the interface but the position of the minimum is not affected. Finally, we probed the effect of removing the ECL2-ECL2 interactions between the two hSERTs. The impact is drastic. The stabilization of the interface upon contact simply disappears. This observation was confirmed by the results from a simulation where the proteins were allowed to evolve freely from the dimer conformation without the ECL2-ECL2 interactions being accounted for. The two hSERTs separated within a few microseconds (Figure S15).
Furthermore, we performed simulations of the ECL2-ECL2 starting from an unusual conformation with highly tilted proteins where ECL2 interactions seemed solely responsible for holding the complex together. US-MD and unbiased simulations performed at both atomistic (AT) and coarse grain (CG) resolutions confirmed that the interface is more stable in the CG resolution than in the AT one. More details can be found in S14–17. One aspect that was not explored here is the contribution of applying an elastic network (EN) to preserve the loop structure. An increased rigidity of the loop in the CG resolution is likely to stabilize their interaction. This idea was hinted by the rapid loss of structure in the ECL2 in the AT resolution simulations (Figures S16–19). For the sake of comparison we also ran the TM12-TM12 interface at AT resolution. The interface was found to be stable starting from the dimer conformation derived from the CG simulations. A period of relaxation of the interface lasted for about 100 ns followed by 400 ns of a steady interface (Figure S20).
The strength of the other three interfaces is within a more regular range for membrane protein interactions53 (Fig. 4) and do not involve extra-membraneous sections (Fig. 2). The symmetric interface involving TM12 is more stable than the other two with an interaction energy of~55 kJ/mol against ~20 kJ/mol for the other two. The involvement of TM12 at the stronger interface, suggests that it might be the most biologically relevant one, which is fully in line with experimental studies30,31,43.
The interface similar to the LeuT dimer is not accessible to hSERT in a membrane environment
We have also probed the strength of the hSERT dimer interface that would result from an orientation similar to the one found in the crystal structure of LeuT (Fig. 2)31. A notable difference in LeuT and hSERT is the location of TM12 relative to the rest of the helical core of hSERT (TM1–11), which results in TM12 being rotated away from the interface when the helical core is positioned as found in the LeuT dimer structure compared to the interface found in the simulations (Fig. 2B). Starting from the separation found in the LeuT crystal structure the two monomers quickly fill the empty gap (~0.6 nm) to form direct contacts between TM9 and TM12 (Figure S21). Strikingly, the prediction of hSERT interface using the web server Cluspro55 (with standard weights and using the C2 symmetry option) gave an interface very similar to the LeuT one, with a positional root-man-square distance of 0.31 nm of the backbone Cα. An overlap of the two structures is shown in Figure S21D.
The calculation of the PMF for this interface proved to be impossible with our current methodology due to the shape of the protein interface. The interface resembles two complementary crescent moons (Figs 2, S22). When the distance between the two monomers is increased, a gap forms between the proteins but it is inaccessible to the lipids due to the shape complementarity of the interface and the contacts between TM9 and TM12. Before lipid molecules may enter, the space is field by water molecules (Figure S22A). Once the channel was formed it was stable for distances between the proteins of over 2 nm from the bound conformation. In contrast, in the simulations started from unbound proteins, lipids get and remain trapped at the interface (Figure S22B). Although these observations could argue for a strong interface it was not detected in the self-assembly simulations. The reason for this absence is likely that the proteins need to operate a 20 degrees tilt one relative to the other compared to their equilibrium orientation free in the membrane bilayer (Figure S23). Overall our results suggest that the interface of hSERT corresponding to the one found in the crystal structure of LeuT31 or predicted by Cluspro is not accessible in a membrane environment. One can easily imagine that the lack of such environment in the preparation of the crystals and in Cluspro workflow may favor the formation of this interface.
Effect of phosphatidylinositol bisphosphate (PIP2) on the TM12 interface
We have quantified the perturbation of the symmetric TM12-TM12 dimer interface by the lipid 1-palmitoyl-2-oleoyl-PIP2, POPIP2 (Fig. 5). The PMF determined in a POPC:POPIP2 mixture with a 9:1 molecular ratio indicates that the presence of POPIP2 lipids in the membrane decreases the strength of the TM12 hSERT dimer interface in comparison to a pure POPC bilayer. The PMF further reveals that the presence of POPIP2 affects hSERT association in two ways. First, a conformation at an interfacial protein separation, dint, of 0.3 nm is stabilized. In the profile from the pure POPC membrane a small bump can be seen at the same dint suggesting that this conformation is also present in pure POPC but only marginally noticeable in the PMF curve. This observation suggests that the presence of POPIP2 alters the balance between the main dimer in POPC (dint = 0 nm) and this alternative conformation at dint = 0.3 nm, so that the minimum at dint = 0 is not as deep in POPIP2 as in pure POPC. Second, an energy barrier forms at dint = 0.55 nm resulting in the stabilization of an additional conformation at dint = 0.75 nm.
Figure 5.
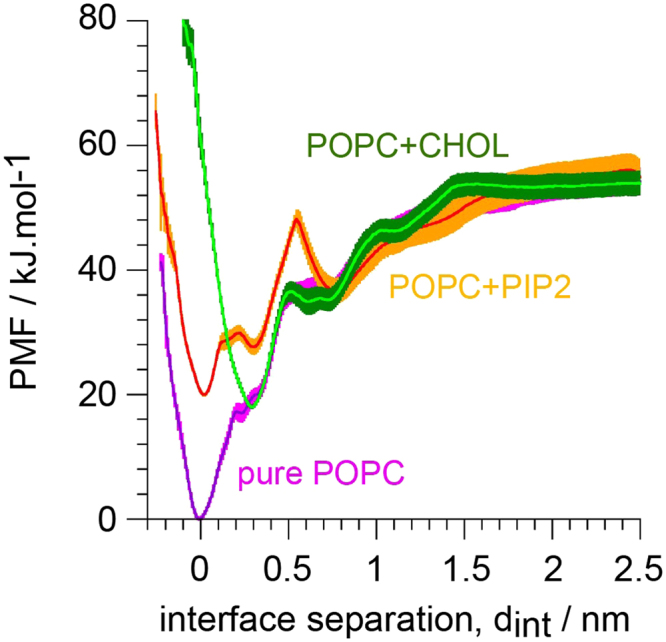
Effect of POPIP2 (C16:0/C18:1-PIP2 lipid) and of cholesterol (CHOL) on the predicted hSERT dimer interface based on the symmetric TM12-TM12 contact. A potential of mean force (PMF) of this interface is shown as determined in a pure POPC membrane (POPC), a mixture POPC:POPIP2 (ratio 9:1, 10%) and a mixture POPC:CHOL (ratio 4:1, 20%).
To rationalize the modifications of the PMF triggered by the presence of POPIP2 we calculated the occupational density of POPIP2 in the US-REMD simulations as a function of the distance between the proteins (Fig. 6A). The density maps determined for the monomeric system (Fig. 6B) are similar to the ones measured at different distances between hSERT molecules in the dimer system. There is therefore no straightforward explanation for the effect described above at short distances. However, a slight increase in density close to the interface is observed (indicated by an arrow in Fig. 6A) and could therefore alter the main minimum. Also, a small density observed at dint = 0.58 and 0.78 nm (indicated by an arrow in Fig. 6A) and not observed in the monomer, places partial POPIP2 molecules at the interface between the hSERT monomers and could therefore be responsible for the energy barrier observed at dint = 0.55 nm. A molecule of POPIP2 at the interface could indeed prevent both the association and the separation of the transporters.
Figure 6.

Lipid rearrangement at the dimer interface of the TM12-TM12 hSERT predicted dimer. We show the 3D maps of lipid densities around the hSERT dimers (A) and monomer (B). The proteins are depicted in tan and pink tubes with TM12 highlighted in red and hSERT C-term helix in orange. Lipid densities are shown in green for cholesterol and orange for POPIP2 (C16:0/C18:1-PIP2 lipid). In (A) the densities are shown for five interfacial distances, dint, and zoom in of the interface viewed from the extracellular side of the membrane are given for three representative distances. The densities were obtained using the ensemble of conformations found in the umbrella simulation with the reference dint indicated in the figure. Arrows point to relevant densities mentioned in the text. More views are given in Figures S24, 25.
Effect of cholesterol on the TM12 interface
Similarly, we have quantified the effect of CHOL on the symmetric TM12-TM12 dimer interface. The PMF determined in a POPC:CHOL mixture at a 4:1 molecular ratio indicates that, as for POPIP2, the presence of CHOL in the membrane decreases the strength of interaction between hSERTs. In the case of CHOL the perturbation of the PMF occurs mainly at the minimum (dint = 0.0 nm). CHOL does not allow this conformation to form but instead stabilizes an alternative conformation found in POPIP2 at dint = 0.3 nm. The remaining of the PMF curve is similar to the one in pure POPC although CHOL slightly modifies the plateau observed at 0.5–0.7 in pure POPC to form a small energy barrier (Fig. 5).
The 3D maps of the occupational density of CHOL as a function of the distance in our US-REMD simulation were determined to elucidate the effect of CHOL on hSERT interaction as interpreted from the PMF (Fig. 6A). As in the case of POPIP2, the densities of CHOL around each hSERT when the two monomers are far apart are identical to the ones obtained from the monomer simulations. But, in contrast with the case of POPIP2, the densities of CHOL at the interface between the hSERT are different from the monomeric system (Fig. 6B). The change of density is already visible at dint = 0.78 nm (Fig. 6A) and until dint = 0.0 nm. The change in the density maps shows the presence of CHOL right at the interface of the transporters between TM12s, joining CHOL densities from each monomers and on both side of TM12 (Fig. 6A,B). This additional density might relate to the more pronounced shoulder observed at dint = 0.55 nm. However, from the size of the density one could have expected a larger effect on the PMF. At shorter distance, the CHOL density between TM12s disappears to leave only the density on the side of the dimer interface (indicated by an arrow in Fig. 6A). The presence of CHOL at that location is most likely responsible for the lack of a dimer at dint = 0.0 nm.
Discussion
In the present study we have employed US-REMD simulations with the MARTINI CG model to probe the relative strength of hSERT dimer interfaces predominantly formed during a set of self-assembly simulations. Four interfaces were mainly observed during these simulations (Figs 1,2) and we quantified their relative strength by determining the PMF as a function of the protein separation while their relative orientations are restrained. TM12 abundantly participates to these interfaces and a symmetric arrangement of hSERT primarily involving TM12 is predicted to be the most stable dimer interface probed (Fig. 3). This result is in line with experimental evidence suggesting the involvement of TM12 in the interface of SERT oligomers30,31,43.
The predicted conformation of the preferred hSERT dimer resembles the dimer found in the crystal structures of LeuT31, a bacterial homologue of MATs. Both the predicted hSERT dimer and the crystal LeuT dimers involve TM12, but in LeuT TM9 equally participates to the interface (Fig. 2). This apparent disparity can be explained by the different position of TM12 relative to the rest of the helical bundle (TM1–11) in hSERT and in LeuT, as well as the kink in TM12 in hSERT. In hSERT, TM12 is distant from TM9 compared to in LeuT (Fig. 2). These findings suggest that TM12 might be an important interaction anchor not only for hSERT and other MATs (DAT and NET) but also for MAT homologues such as LeuT.
The predicted hSERT dimer interface involving TM12 was studied further by quantifying the effects resulting from modifying the bilayer lipid composition that the proteins are embedded in. The PMF were thus determined in POPC:CHOL and POPC:POPIP2 bilayers with 4:1 and 9:1 lipid molecular ratios, respectively, and compared with the case of a pure POPC bilayer (Fig. 5). The effects are strong. Both mixed membranes remove about 20 kJ/mol from the stabilization of the interface upon protein binding in pure POPC. Moreover, in the presence of POPIP2 an energy barrier forms at the distance where the proteins start interacting. However, only a tiny density of POPIP2 was found at the interface between the proteins at that separation. In the case of CHOL, it appeared that the dimer could not reach the same interface as in pure POPC and with POPIP2. In POPC:CHOL, an alternative interface is stabilized that is at ~0.3 nm from the minimum found in pure POPC and in POPC:POPIP2. Extensive densities of CHOL were found at the interface between the proteins that rationalize this effect.
A similar behavior of CHOL was previously reported for G protein-coupled receptors (GPCR). CHOL was suggested to change the relative stability of GPCR interfaces by destabilizing one through a direct interaction to the interface preventing the binding of the proteins56,57. Our data is also in line with CHOL affecting integral protein oligomerization by a direct interaction at the protein interface. Note, however, that our data does not exclude a contribution to protein oligomerization by CHOL from affecting bulk mechanical properties of the membrane.
In the case of PIP2, the mechanism of destabilization of the interface is less clear as only residual POPIP2 density is found at the protein interface. Nevertheless, our data suggests that anionic lipids might act on a non-binding surface of proteins by destabilizing that particular interface. This behavior contrasts with earlier reports of anionic lipids having a gluing effect on proteins32,33. Cardiolipin (CL), an anionic lipid as POPIP2 but with much bulkier alkyl chains, has been shown to stabilize interactions between complexes of the respiratory chain32. We have shown that CL operates by using specific binding sites on both protein partners33. This ability of anionic lipid to bind proteins might relate to the observation that PIP2 decreases the exchanges of hSERT molecules in oligomer aggregates8. Indeed, we found significant densities of PIP2 at the surface of hSERT (Fig. 6).
Although it is difficult to relate the effect described above in mixed membranes to experimental data, collectively, our results reveal the strong impact of the membrane composition on the strength of integral protein-protein interactions. This is not a new concept. The membrane composition was previously shown to affect the propensity of proteins to oligomerize35,58,59 and affect the interface involved35,56,57. Here, we present a first quantitative measure of such effects. These observations emphasize the importance of using a native-like membrane environment for such studies and argue in favor of the development of more reliable complex models60,61 and new experiments allowing working directly in cells6.
The determination of the PMF for the association/dissociation of two full size proteins is a great challenge that we have undertaken earlier using US-MD40. Here we have extended our methodology by using US-REMD, a method recently developed41 and used on simple systems48,62. We found that the benefits of US-REMD simulations are manyfold. First, US-REMD removes the earlier need to combine umbrellas by hand, thereby removing a potential bias. Second, US-REMD increases drastically the sampling at distances where the proteins associate and dissociate; from the distance of contact to ~1 nm. Nevertheless, the US-REMD approach is not the ultimate solution to sampling issues. It could be further improved by coupling it with methods such as Hamiltonian replica exchange, where protein-protein and protein-lipid interactions could be scaled. Third, these two improvements allowed generating reasonably converged PMFs within 10 μs per replica, thus about 300 μs cumulative simulation time. The same level of convergence could in principle be achieved with simple US-MD but with much longer simulation times. Alternatively, one could imagine a manner to combine conventional US simulations in a more automated manner so that the rapid convergence of individual US simulations could be exploited. Filizola and coworkers have combined US-MD with metadynamics to improve the sampling52. It is not clear how their approach compares with US-REMD but it allows studying systems in great detail63.
In summary, we have benchmarked US-REMD to study full size protein interactions and predicted that the most likely interface of hSERT involves TM12 in a symmetric manner. Furthermore, we have shown that the composition of the embedding membrane strongly affects the strength of the interface. We expect these findings will stimulate new modeling and experimental studies to unravel the precise mechanism behind the effects of lipids reported here. On the modeling side, we showed that US-REMD simulations allow studying protein-protein interactions with a close to atomistic resolution without requiring excessive computer resources. This approach should be easily automated for general use, opening great promise for the prediction of protein-protein interactions in a more native-like environment.
Methods
Models and resolutions
An atomistic model for hSERT (residues 74 to 617) was built from its crystal structure bound to paroxetine (PDB ID: 5I6X27) to which the following modifications were made: the ligand was removed, the missing side chains were added with Prime v. 3.9 and the protonation state of titrable residues was determined with Propka64,65, both as embedded in Maestro (the Schrödinger Suite 2015-1; Schrödinger, LLC); Glu508 was protonated; a disulfide bridge was built between Cys208 and Cys209; Ile291, Thr439 and Tyr110 were mutated back to their native side chain, Ala, Ser and Ala, respectively, using UCSF-Chimera and the Dunbrack rotamer library66. Chloride and sodium (placed at the NA1 and NA2 sites) ions and the ligand (serotonin or paroxetine) were not included in the coarse grain model because its resolution (see below) would not reflect their presence.
This atomistic model of hSERT was converted into a MARTINI-v2.2 coarse grain model using the martinize script67 and inserted into membrane bilayers using the insane script68. The MARTINI force field for lipids69, the most recent version of cholesterol70 and its extension to proteins67,71 were used in combination with the ElNeDyn approach72 to maintain the secondary and tertiary structure of the protein. This CGMD approach is well suited to study complex biological membranes53,61,73,74.
The atomistic hSERT model was also simulated at atomistic resolution. A monomer was simulated to quickly relax its structure. Dimeric conformations were extracted from the CG simulations and back-mapped to explore their stability at atomistic resolution. We used the script backwards to backmap CG structures75. The CHARMM36 force field76–78 was used to describe the protein, the lipids and the aqueous phase. Internal Cl− and Na+ ions, and water molecules found in the crystal structure (PDB ID: 5I6X27) were included in the model but not the ligand.
Simulations
All molecular dynamics (MD) simulations were performed using the GROMACS simulation package version 5.179. Conventional simulation setups associated with the MARTINI67,80,81 and CHARMM force fields76–78 were used. For the CG simulations, these include a 20 fs time step for production run, a 0.9 nm cutoff and a 500 kJ/mol/nm2 force constant for ElNeDyn. The non-bonded interactions (van der Waals and electrostatic) received a cutoff of 1.1 nm and were shifted to zero using the potential-modifier implementation in GROMACS. A relative dielectric screening constant of 15 and the Verlet neighbor search82 were used. The protein (hSERT), the membrane bilayer (POPC) and the aqueous phase (water plus ions when present) were coupled independently to an external temperature bath at 300 K using a Berendsen83 and Bussi84 thermostats (τT = 0.5 ps) for equilibration and for production, respectively. The pressure was weakly coupled83 to an external bath at 1 bar using a relaxation time of 2 ps following a semi-isotropic pressure scheme. A list of the simulations performed is given as supplementary information (Table S1). Most analysis were performed using frames saved every 1 ns.
In the atomistic simulations a 2 fs time step was used together with the LINCs algorithm85 to constrain the bonds involving hydrogens. Both temperature and pressure were weakly coupled to 310 K and 1 atm using the Berendsen approach83. The decomposition of the system for temperature and pressure control was as for the CG resolution. The non-bonded interactions received a 1.2 nm cutoff and the electrostatics were treated using PME at longer distances, while the force-switch potential modifier was used for the vdW interactions79.
Potential of mean force
To calculate the potential of mean force (PMF) between two hSERTs we used our combination of virtual bond algorithm86 (VBA, Figure S26) with a slightly improved umbrella sampling approach (US)40. VBA is used to hold the relative orientation of the two proteins so that a particular interface is probed. In addition to the conventional US method, we have used the approach that combines US with replica exchange MD41. This straightforward implementation takes advantage of the exchange of replicas between the umbrella windows to accelerate sampling convergence over the whole range of the reaction coordinate48,62,87. The umbrella trajectories were unbiased and combined using a WHAM88,89 implementation that takes into account the use of VBA40. The equilibrium distance of each umbrella potentials were first defined every 0.1 nm. Smaller intervals were used when test simulations (500 ns) indicated a low exchange (<10%) between consecutive umbrellas. The rates of exchange varied between 0.15 and 0.40 and the production runs were 10 to 18 μs (Table S1). The final PMF and the associated statistical errors were obtained from the average of 100 PMF curves generated by a bootstrap procedure where half-sized umbrella windows were used for resampling.
Systems
Two different setups for the CG self-assembly simulations were used. A system with sixteen hSERT monomers was run in 10 repeats each lasting 30 μs and a system consisting of 64 hSERT monomers was run for 250 μs. The small setup was started with the sixteen copies of hSERT placed on a four-by-four grid using a pre-equilibrated system containing one hSERT in a membrane patch made of 99 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid molecules (Fig. 1). A 500 ns simulation with soft position restrain (100 kJ/mol/nm2 in the membrane plane) on a central residue of hSERT (Leu338) was used to randomize the orientation of the sixteen copies. The large system with 64 hSERT monomers was prepared in a similar s.
For the calculations of the potential of mean force we used systems composed of a pair of hSERT molecules inserted into a rectangular box (Fig. 2A) containing ~630 POPC molecules, or a mixture of 572 POPC and 63 1-palmitoyl-2-oleoyl-PIP2 (POPIP2) molecules (9:1 ratio) and a mixture of 507 POPC and 126 CHOL molecules (4:1 ratio). The pairs of hSERTs were dimer conformations extracted from the self-assembly simulations, which were relaxed in the corresponding lipid mixture prior to US simulations. The orientation of the hSERT dimers was chosen such that their distance (the reaction coordinate) would vary along the long side of the box. A position restrain (10 kJ/mol/nm2) was applied on each hSERT molecule (backbone bead of Leu338) such that they remain close to the center of the small side of the box.
The systems studied at the atomistic resolution contained a monomeric structure of hSERT embedded into a 380 POPC lipid bilayer and two dimer conformations back-mapped from CG structures used in the calculation of the PMFs. These systems were neutralized by adding 0.2 M NaCl in the aqueous phase.
Cluster analysis
Clustering of the hSERT dimers formed in the self-assembly simulations was performed as previously described40. Briefly, dimer conformations with a center-of-mass separation less than 5.3 nm were collected from the 10 simulations containing sixteen hSERTs and concatenated into a single file. Clusters were constructed using the GROMOS approach90 using a similarity cutoff of 0.4 nm. Modifying the cutoff value changed the relative population of the clusters but not their presence and only slightly their order.
Limitations of the methodology
Although, overall, our CGMD methodology and, in particular, the determination of protein-protein interaction strength have been very successful in bringing to light new principles of membrane proteins and their interactions42,53,61,73,74, it is important to point out some of the limitations underlying our model and methodology. First, the processes studied involve slow motion of proteins and lipids. Although we have significantly improved our methodology by using US-REMD instead of US-MD simulation as in our previous work, and increased the total simulation time used to generate a PMF by more than a factor of 10, most processes are still under-sampled in our simulations. The lack of sampling is especially true in the self-assembly simulations. Moreover, the US-REMD approach is not a final solution to interface sampling. It still shows signs of sampling limitation and could be combined with alternative enhancing methods. A second limitation of our approach might be the use of a CG representation of the interactions in the system. A known issue for protein-protein interactions modeled by the MARTINI coarse grain is the apparent over-estimation of their strength in the aqueous phase54. We have shown here using a series of CG and AT simulations that the symmetric interface TM3/4-TM3/4 is unrealistically stabilized by loop-loop interactions in the extracellular side of the membrane. There is however no evidence that interactions between full size protein are over-stabilized by the MARTINI force field in a membrane environment. Although it might seem the case from a recent study of simple transmembrane helices in a lipid bilayer91, protein/protein and protein/lipid interactions are most likely not additive. More studies will be required to reveal the delicate balances of the forces involved. The model of POPIP2 also showed unexpected collective behavior. We observed arrays of a few molecules interacting through the stacking of their rings. Considerable efforts have been and are being put into fixing these issues. The next version of the MARTINI FF should provide remedies to most of theses limitations.
Electronic supplementary material
Acknowledgements
Funding from the Danish Council for Independent Research | Natural Sciences (DFF-4002-00502 and DFF-7014-00192B) and Medical Sciences (DFF – 4004-00309), Lundbeck Foundation, and the Carlsberg Foundation made the research possible. Computations were possible through allocations of time at the Centre for Scientific Computing Aarhus, and Abacus 2.0.
Author Contributions
X.P. designed the models and simulations, built the models and conducted the simulations. X.P. and T.Z. analyzed the trajectories. X.P., T.Z. and B.S. interpreted the data and wrote the paper.
Competing Interests
The authors declare no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-22912-7.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Xavier Periole, Email: x.periole@chem.au.dk.
Birgit Schiøtt, Email: birgit@chem.au.dk.
References
- 1.Collingridge GL, Isaac JTR, Wang YT. Receptor trafficking and synaptic plasticity. Nat. Rev. Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- 2.Sarabipour S, Del Piccolo N, Hristova K. Characterization of membrane protein interactions in plasma membrane derived vesicles with quantitative imaging Förster resonance energy transfer. Acc. Chem. Res. 2015;48:2262–2269. doi: 10.1021/acs.accounts.5b00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007;8:413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 4.Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu. Rev. Cell Dev. Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, et al. Regulation of AMPA receptor surface trafficking and synaptic plasticity by a cognitive enhancer and antidepressant molecule. Mol. Psychiatry. 2013;18:471–484. doi: 10.1038/mp.2012.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rahbek-Clemmensen T, et al. Super-resolution microscopy reveals functional organization of dopamine transporters into cholesterol and neuronal activity-dependent nanodomains. Nat. Comms. 2017;8:740. doi: 10.1038/s41467-017-00790-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magnani F, Tate CG, Wynne S, Williams C, Haase J. Partitioning of the serotonin transporter into lipid microdomains modulates transport of serotonin. J. Biol. Chem. 2004;279:38770–38778. doi: 10.1074/jbc.M400831200. [DOI] [PubMed] [Google Scholar]
- 8.Anderluh A, et al. Direct PIP2 binding mediates stable oligomer formation of the serotonin transporter. Nat. Comms. 2017;8:14089. doi: 10.1038/ncomms14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sitte HH, Farhan H, Javitch JA. Sodium-dependent neurotransmitter transporters: oligomerization as a determinant of transporter function and trafficking. Mol. Interv. 2004;4:38–47. doi: 10.1124/mi.4.1.38. [DOI] [PubMed] [Google Scholar]
- 10.Borroni MV, Vallés AS, Barrantes FJ. The lipid habitats of neurotransmitter receptors in brain. Biochim. Biophys. Acta. 2016;1858:2662–2670. doi: 10.1016/j.bbamem.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 11.Andersen J, Kristensen AS, Bang-Andersen B, Strømgaard K. Recent advances in the understanding of the interaction of antidepressant drugs with serotonin and norepinephrine transporters. Chem. Commun. (Camb.) 2009;30:3677–3692. doi: 10.1039/b903035m. [DOI] [PubMed] [Google Scholar]
- 12.Sharp SI, McQuillin A, Gurling HMD. Genetics of attention-deficit hyperactivity disorder (ADHD) Neuropharmacology. 2009;57:590–600. doi: 10.1016/j.neuropharm.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 13.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–1223. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 14.Elliott JM, Beveridge TJR. Psychostimulants and monoamine transporters: upsetting the balance. Curr. Opin. Pharm. 2005;5:94–100. doi: 10.1016/j.coph.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Anderluh A, et al. Single molecule analysis reveals coexistence of stable serotonin transporter monomers and oligomers in the live cell plasma membrane. J. Biol. Chem. 2014;289:4387–4394. doi: 10.1074/jbc.M113.531632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hastrup H, Sen N, Javitch JA. The human dopamine transporter forms a tetramer in the plasma membrane: cross-linking of a cysteine in the fourth transmembrane segment is sensitive to cocaine analogs. J. Biol. Chem. 2003;278:45045–45048. doi: 10.1074/jbc.C300349200. [DOI] [PubMed] [Google Scholar]
- 17.Jess U, Betz H, Schloss P. The membrane-bound rat serotonin transporter, SERT1, is an oligomeric protein. FEBS Letters. 1996;394:44–46. doi: 10.1016/0014-5793(96)00916-7. [DOI] [PubMed] [Google Scholar]
- 18.Kilic F, Rudnick G. Oligomerization of serotonin transporter and its functional consequences. Proc. Natl. Acad. Sci. USA. 2000;97:3106–3111. doi: 10.1073/pnas.97.7.3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milner HE, Béliveau R, Jarvis SM. The in situ size of the dopamine transporter is a tetramer as estimated by radiation inactivation. Biochim. Biophys. Acta. 1994;1190:185–187. doi: 10.1016/0005-2736(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 20.Sorkina T, Doolen S, Galperin E, Zahniser NR, Sorkin A. Oligomerization of dopamine transporters visualized in living cells by fluorescence resonance energy transfer microscopy. J. Biol. Chem. 2003;278:28274–28283. doi: 10.1074/jbc.M210652200. [DOI] [PubMed] [Google Scholar]
- 21.Zhen J, et al. Dopamine transporter oligomerization: impact of combining protomers with differential cocaine analog binding affinities. J. Neurochem. 2015;133:167–173. doi: 10.1111/jnc.13025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gur M, Cheng MH, Zomot E, Bahar I. Effect of Dimerization on the Dynamics of Neurotransmitter:Sodium Symporters. J. Phys. Chem. B. 2017;121:3657–3666. doi: 10.1021/acs.jpcb.6b09876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hastrup H, Karlin A, Javitch JA. Symmetrical dimer of the human dopamine transporter revealed by cross-linking Cys-306 at the extracellular end of the sixth transmembrane segment. Proc. Natl. Acad. Sci. USA. 2001;98:10055–10060. doi: 10.1073/pnas.181344298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng MH, Garcia-Olivares J, Wasserman S, DiPietro J, Bahar I. Allosteric modulation of human dopamine transporter activity under conditions promoting its dimerization. J. Biol. Chem. 2017;292:12471–12482. doi: 10.1074/jbc.M116.763565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simmerman HK, Kobayashi YM, Autry JM, Jones LR. A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J. Biol. Chem. 1996;271:5941–5946. doi: 10.1074/jbc.271.10.5941. [DOI] [PubMed] [Google Scholar]
- 26.Torres GE, et al. Oligomerization and trafficking of the human dopamine transporter. Mutational analysis identifies critical domains important for the functional expression of the transporter. J. Biol. Chem. 2003;278:2731–2739. doi: 10.1074/jbc.M201926200. [DOI] [PubMed] [Google Scholar]
- 27.Coleman JA, Green EM, Gouaux E. X-ray structures and mechanism of the human serotonin transporter. Nature. 2016;532:334–339. doi: 10.1038/nature17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503:85–90. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alguel Y, Cameron AD, Diallinas G, Byrne B. Transporter oligomerization: form and function. Biochem. Soc. Trans. 2016;44:1737–1744. doi: 10.1042/BST20160217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Just H, Sitte HH, Schmid JA, Freissmuth M, Kudlacek O. Identification of an additional interaction domain in transmembrane domains 11 and 12 that supports oligomer formation in the human serotonin transporter. J. Biol. Chem. 2004;279:6650–6657. doi: 10.1074/jbc.M306092200. [DOI] [PubMed] [Google Scholar]
- 31.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl–dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 32.Zhang M, Mileykovskaya E, Dowhan W. Gluing the Respiratory Chain Together. Cardiolipin is Required for Supercomplex Formation in the Inner Mitochondrial Membrane. J. Biol. Chem. 2002;277:43553–43556. doi: 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- 33.Arnarez C, Marrink SJ, Periole X. Molecular mechanism of cardiolipin-mediated assembly of respiratory chain supercomplexes. Chemical Science. 2016;7:4435–4443. doi: 10.1039/C5SC04664E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Botelho AV, Huber T, Sakmar TP, Brown MF. Curvature and Hydrophobic Forces Drive Oligomerization and Modulate Activity of Rhodopsin in Membranes. Biophys. J. 2006;91:4464–4477. doi: 10.1529/biophysj.106.082776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Periole X, Huber T, Marrink S-J, Sakmar TP. G Protein-Coupled Receptors Self-Assemble in Dynamics Simulations of Model Bilayers. J. Am. Chem. Soc. 2007;129:10126–10132. doi: 10.1021/ja0706246. [DOI] [PubMed] [Google Scholar]
- 36.Gupta K, et al. The role of interfacial lipids in stabilizing membrane protein oligomers. Nature. 2017;541:421–424. doi: 10.1038/nature20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamilton PJ, et al. PIP2 regulates psychostimulant behaviors through its interaction with a membrane protein. Nat. Chem. Biol. 2014;10:582–589. doi: 10.1038/nchembio.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laursen, L. et al. Cholesterol binding to a conserved site modulates conformation, pharmacology and transport kinetics of the human serotonin transporter. J. Biol. Chem. 10.1074/jbc.M117.809046 (2018). [DOI] [PMC free article] [PubMed]
- 39.Zeppelin T, Ladefoged LK, Sinning S, Periole X, Schiøtt B. A direct interaction of cholesterol with the dopamine transporter prevents its out-to-inward transition. PLoS Comput. Biol. 2018;14:e1005907. doi: 10.1371/journal.pcbi.1005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Periole X, Knepp AM, Sakmar TP, Marrink SJ, Huber T. Structural Determinants of the Supramolecular Organization of G Protein-Coupled Receptors in Bilayers. J. Am. Chem. Soc. 2012;134:10959–10965. doi: 10.1021/ja303286e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sugita Y, Kitao A, Okamoto Y. Multidimentional replica-exchange method for free-energy calculations. J. Comput. Phys. 2000;113:6042–6051. [Google Scholar]
- 42.Parton DL, Klingelhoefer JW, Sansom MSP. Aggregation of Model Membrane Proteins, Modulated by Hydrophobic Mismatch, Membrane Curvature, and Protein Class. Biophys. J. 2011;101:691–699. doi: 10.1016/j.bpj.2011.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neubauer HA, Hansen CG, Wiborg O. Dissection of an allosteric mechanism on the serotonin transporter: a cross-species study. Molecular Pharmacology. 2006;69:1242–1250. doi: 10.1124/mol.105.018507. [DOI] [PubMed] [Google Scholar]
- 44.Torrie GM, Valleau JP. Monte Carlo study of a phase‐separating liquid mixture by umbrella sampling. J. Chem. Phys. 1977;66:1402–1408. doi: 10.1063/1.434125. [DOI] [Google Scholar]
- 45.Sengupta D, Marrink SJ. Lipid-mediated interactions tune the association of glycophorin A helix and its disruptive mutants in membranes. Phys. Chem. Chem. Phys. 2010;12:12987–10. doi: 10.1039/c0cp00101e. [DOI] [PubMed] [Google Scholar]
- 46.Neale C, Bennett WFD, Tieleman DP, Pomès R. Statistical Convergence of Equilibrium Properties in Simulations of Molecular Solutes Embedded in Lipid Bilayers. J. Chem. Theory Comput. 2011;7:4175–4188. doi: 10.1021/ct200316w. [DOI] [PubMed] [Google Scholar]
- 47.Neale C, Madill C, Rauscher S, Pomès R. Accelerating Convergence in Molecular Dynamics Simulations of Solutes in Lipid Membranes by Conducting a Random Walk along the Bilayer Normal. J. Chem. Theory Comput. 2013;9:3686–3703. doi: 10.1021/ct301005b. [DOI] [PubMed] [Google Scholar]
- 48.Domański J, Hedger G, Best RB, Stansfeld PJ, Sansom MSP. Convergence and Sampling in Determining Free Energy Landscapes for Membrane Protein Association. J. Phys. Chem. B. 2017;121:3364–3375. doi: 10.1021/acs.jpcb.6b08445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Periole X, Mark AE. Convergence and sampling efficiency in replica exchange simulations of peptide folding in explicit solvent. J. Chem. Phys. 2007;126:014903. doi: 10.1063/1.2404954. [DOI] [PubMed] [Google Scholar]
- 50.Hénin J, Pohorille A, Chipot C. Insights into the Recognition and Association of Transmembrane α-Helices. The Free Energy of α-Helix Dimerization in Glycophorin A. J. Am. Chem. Soc. 2005;127:8478–8484. doi: 10.1021/ja050581y. [DOI] [PubMed] [Google Scholar]
- 51.Johnston JM, et al. Making Structural Sense of Dimerization Interfaces of Delta Opioid Receptor Homodimers. Biochemistry. 2011;50:1682–1690. doi: 10.1021/bi101474v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnston JM, Wang H, Provasi D, Filizola M. Assessing the relative stability of dimer interfaces in g protein-coupled receptors. PLoS Comput. Biol. 2012;8:e1002649. doi: 10.1371/journal.pcbi.1002649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Periole X. Interplay of G Protein-Coupled Receptors with the Membrane: Insights from Supra-Atomic Coarse Grain Molecular Dynamics Simulations. Chem. Rev. 2017;117:156–185. doi: 10.1021/acs.chemrev.6b00344. [DOI] [PubMed] [Google Scholar]
- 54.Stark AC, Andrews CT, Elcock AH. Toward optimized potential functions for protein-protein interactions in aqueous solutions: osmotic second virial coefficient calculations using the MARTINI coarse-grained force field. J. Chem. Theory Comput. 2013;9:4176–4185. doi: 10.1021/ct400008p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Comeau SR, Gatchell DW, Vajda S, Camacho CJ. ClusPro: a fully automated algorithm for protein-protein docking. Nucleic Acids Res. 2004;32:W96–9. doi: 10.1093/nar/gkh354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prasanna X, Chattopadhyay A, Sengupta D. Cholesterol Modulates the Dimer Interface of the β2-Adrenergic Receptor via Cholesterol Occupancy Sites. Biophys. J. 2014;106:1290–1300. doi: 10.1016/j.bpj.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pluhackova K, Gahbauer S, Kranz F, Wassenaar TA, Böckmann RA. Dynamic Cholesterol-Conditioned Dimerization of the G Protein Coupled Chemokine Receptor Type 4. PLoS Comput. Biol. 2016;12:e1005169. doi: 10.1371/journal.pcbi.1005169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Killian JA. Hydrophobic mismatch between proteins and lipids in membranes. Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes. 1998;1376:401–416. doi: 10.1016/S0304-4157(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 59.Jastrzebska B, et al. Functional and structural characterization of rhodopsin oligomers. J. Biol. Chem. 2006;281:11917–11922. doi: 10.1074/jbc.M600422200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ingólfsson HI, et al. Lipid organization of the plasma membrane. J. Am. Chem. Soc. 2014;136:14554–14559. doi: 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- 61.Koldsø H, Sansom MSP. Organization and Dynamics of Receptor Proteins in a Plasma Membrane. J. Am. Chem. Soc. 2015;137:14694–14704. doi: 10.1021/jacs.5b08048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park S, Kim T, Im W. Transmembrane helix assembly by window exchange umbrella sampling. Phys. Rev. Lett. 2012;108:108102. doi: 10.1103/PhysRevLett.108.108102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lelimousin M, Limongelli V, Sansom MSP. Conformational Changes in the Epidermal Growth Factor Receptor: Role of the Transmembrane Domain Investigated by Coarse-Grained MetaDynamics Free Energy Calculations. J. Am. Chem. Soc. 2016;138:10611–10622. doi: 10.1021/jacs.6b05602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olsson MHM, Søndergaard CR, Rostkowski M, Jensen JH. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011;7:525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 65.Søndergaard CR, Olsson MHM, Rostkowski M, Jensen JH. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011;7:2284–2295. doi: 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- 66.Shapovalov MV, Dunbrack RL. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure. 2011;19:844–858. doi: 10.1016/j.str.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.de Jong DH, et al. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013;9:687–697. doi: 10.1021/ct300646g. [DOI] [PubMed] [Google Scholar]
- 68.Wassenaar TA, Ingólfsson HI, Böckmann RA, Tieleman DP, Marrink SJ. Computational Lipidomics with insane: A Versatile Tool for Generating Custom Membranes for Molecular Simulations. J. Chem. Theory Comput. 2015;11:2144–2155. doi: 10.1021/acs.jctc.5b00209. [DOI] [PubMed] [Google Scholar]
- 69.Marrink SJ, de Vries AH, Mark AE. Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B. 2004;108:750–760. doi: 10.1021/jp036508g. [DOI] [Google Scholar]
- 70.Melo MN, Ingólfsson HI, Marrink SJ. Parameters for Martini sterols and hopanoids based on a virtual-site description. J. Chem. Phys. 2015;143:243152. doi: 10.1063/1.4937783. [DOI] [PubMed] [Google Scholar]
- 71.Monticelli L, et al. The MARTINI Coarse-Grained Force Field: Extension to Proteins. J. Chem. Theory Comput. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 72.Periole X, Cavalli M, Marrink S-J, Ceruso MA. Combining an Elastic Network With a Coarse-Grained Molecular Force Field: Structure, Dynamics, and Intermolecular Recognition. J. Chem. Theory Comput. 2009;5:2531–2543. doi: 10.1021/ct9002114. [DOI] [PubMed] [Google Scholar]
- 73.Marrink SJ, Tieleman DP. Perspective on the Martini model. Chem. Soc. Rev. 2013;42:6801–22. doi: 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- 74.Ingólfsson HI, Arnarez C, Periole X, Marrink SJ. Computational ‘microscopy’ of cellular membranes. J. Cell. Sci. 2016;129:257–268. doi: 10.1242/jcs.176040. [DOI] [PubMed] [Google Scholar]
- 75.Wassenaar TA, Pluhackova K, Böckmann RA, Marrink SJ, Tieleman DP. Going Backward: A Flexible Geometric Approach to Reverse Transformation from Coarse Grained to Atomistic Models. J. Chem. Theory Comput. 2014;10:676–690. doi: 10.1021/ct400617g. [DOI] [PubMed] [Google Scholar]
- 76.Best RB, et al. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone ϕ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Foloppe N, MacKerell AD., Jr. All-atom empirical force field for nucleic acids: I. parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000;21:86–104. doi: 10.1002/(SICI)1096-987X(20000130)21:2<86::AID-JCC2>3.0.CO;2-G. [DOI] [Google Scholar]
- 78.Mackerell AD, Feig M, Brooks CL. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 79.Abraham MJ, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- 80.Periole X, Marrink S-J. The Martini coarse-grained force field. Methods Mol. Biol. 2013;924:533–565. doi: 10.1007/978-1-62703-017-5_20. [DOI] [PubMed] [Google Scholar]
- 81.de Jong DH, Baoukina S, Ingólfsson HI, Marrink SJ. Martini straight: boosting performance using a shorter cutoff and GPUs. Comp. Phys. Comm. 2016;199:1–7. doi: 10.1016/j.cpc.2015.09.014. [DOI] [Google Scholar]
- 82.Verlet L. Computer ‘Experiments’ on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967;159:98–103. doi: 10.1103/PhysRev.159.98. [DOI] [Google Scholar]
- 83.Berendsen HJC, Postma JPM, Van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. doi: 10.1063/1.448118. [DOI] [Google Scholar]
- 84.Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007;126:014101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- 85.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H. [DOI] [Google Scholar]
- 86.Boresch S, Tettinger F, Leitgeb M, Karplus M. Absolute Binding Free Energies: A Quantitative Approach for Their Calculation. J. Phys. Chem. B. 2003;107:9535–9551. doi: 10.1021/jp0217839. [DOI] [Google Scholar]
- 87.Park S, Im W. Two Dimensional Window Exchange Umbrella Sampling for Transmembrane Helix Assembly. J. Chem. Theory Comput. 2013;9:13–17. doi: 10.1021/ct3008556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kumar S, Rosenberg JM, Bouzida D, Swendsen RH, Kollman PA. THE weighted histogram analysis method for free‐energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992;13:1011–1021. doi: 10.1002/jcc.540130812. [DOI] [Google Scholar]
- 89.Souaille M, Roux B. Extension to the weighted histogram analysis method: combining umbrella sampling with free energy calculations. Comp. Phys. Comm. 2001;135:40–57. doi: 10.1016/S0010-4655(00)00215-0. [DOI] [Google Scholar]
- 90.Daura X, et al. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed. 1999;38:236–240. doi: 10.1002/(SICI)1521-3773(19990115)38:1/2<236::AID-ANIE236>3.0.CO;2-M. [DOI] [Google Scholar]
- 91.Javanainen M, Martinez-Seara H, Vattulainen I. Excessive aggregation of membrane proteins in the Martini model. PLoS ONE. 2017;12:e0187936. doi: 10.1371/journal.pone.0187936. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.