

Abstract
Recombinant adeno-associated viruses (rAAVs) serve as vectors for in vivo gene delivery in both mice and humans, and have broad applicability for the treatment of genetic diseases. Clinical trials with AAV vectors have demonstrated promise and safety in several human diseases. However, the in vivo validation of novel AAV constructs expressing products that act specifically on human cells and tissues is limited by a paucity of effective translatable models. Humanized mice that are engrafted with human cells, tissues, and immune systems offer strong potential to test the biological effectiveness of AAV vectors on human cells and tissues. Using the BLT (bone marrow, liver, thymus) humanized NOD-scid Il2rgnull (NSG) mouse model, which enables efficient development of HLA-restricted effector and regulatory T cells (Tregs), we have evaluated the delivery and function of human interleukin (IL)-2 by an AAV vector. Humanized mice treated with an AAV vector expressing human IL-2 showed a significant and sustained increase in the number of functional human FOXP3+CD4+ Tregs. The expression of human IL-2 did not significantly change the levels or activation status of conventional T-cell subsets. Numbers of activated human natural killer cells were also increased significantly in humanized mice treated with the IL-2 vector. These data recapitulate observations in clinical trials of IL-2 therapy and collectively show that humanized mouse models offer a translational platform for testing the efficacy of AAV vectors targeting human immune cells.
Keywords: : AAV, humanized, NSG, Treg, cytokine
Introduction
The use of recombinant adeno-associated virus (rAAV) for in vivo gene delivery is a promising approach for the treatment of numerous genetic disorders and provides a basic research tool.1 AAV is a nonpathogenic, nonenveloped parvovirus that infects both dividing and nondividing cells in vivo and supports stable expression of transgenes by infected cells.2 An additional advantage for the use of AAV as a gene therapy vector is the high level of diversity in AAV serotypes that enables the targeting of a broad range of cell types in vivo.3 AAV-based vectors have been used in mice for the efficient delivery and long-term expression of transgenes with positive therapeutic results.4 Moreover, AAV vectors have shown promise and safety in clinical trials for a number of diseases including hemophilia, rheumatoid arthritis, cystic fibrosis, lipoprotein lipase deficiency, Leber's congenital amaurosis, choroideremia, Canavan's disease, muscular dystrophy, α1-antitrypsin deficiency, Parkinson's disease, macular degeneration, and heart disease.5 However, the efficient preclinical testing of novel AAV vectors expressing transgenes that act specifically on human cells and tissues is hampered by the paucity of appropriate models that recapitulate human biology.6 Previous studies using humanized mice have demonstrated the success of AAV vectors for the delivery of anticancer therapies,7,8 to express PDX1 in human liver cells,9 to target transgene expression to human CD4+ T cells,7 and to augment the development and function of human immune systems after CD34+ hematopoietic stem cell (HSC) engraftment.10–13 The success of these previous experimental strategies with AAV vectors in humanized mice supports the use of humanized mouse models to test AAV vectors for gene delivery targeting specific human immune cell populations.
FOXP3+CD4+ regulatory T cells (Tregs) are an essential component of T cell tolerance and physiological immune homeostasis,14–16 and interleukin (IL)-2 is critical to maintain normal FOXP3+CD4+ Treg homeostasis.17 Low-dose IL-2 therapies have been tested in both mice and in clinical trials as a strategy to increase functional FOXP3+CD4+ Treg levels and limit autoimmunity and suppress transplant rejection.18–21 Low-dose IL-2 therapies preferentially act through the high-affinity IL-2 receptor that is expressed by FOXP3+CD4+ Tregs.22 Studies in nonobese diabetic (NOD) mice have demonstrated that AAV delivery of IL-2 has a protective effect on the development of type 1 diabetes (T1D).18,19,23,24 Injection of NOD mice with an AAV8 vector expressing murine IL-2 under the control of the murine insulin promoter enabled long-term expression of IL-2 by pancreatic beta cells, increased the number of functional FOXP3+CD4+ Tregs, and delayed progression of T1D. Similar effects on FOXP3+CD4+ Treg levels and delay of diabetes progression were also observed in NOD mice injected with recombinant IL-2.25 Delivery of low-dose human IL-2 in clinical trials has been accomplished by repeated injections of recombinant IL-2, Proleukin (aldesleukin).21 Clinical trials have shown that injection of patients with recombinant human IL-2 at low doses increases human FOXP3+CD4+ Treg levels and reduces symptoms in uncontrolled chronic graft-versus-host disease (GVHD),26 hepatitis C virus (HCV)-induced vasculitis,27 alopecia areata,28 and systemic lupus erythematosus (SLE).29,30 However, a clinical trial of a rapamycin/IL-2 combination therapy in patients with T1D resulted in beta cell dysfunction.31 Patients with T1D treated with combination rapamycin and low-dose IL-2 showed a significant increase in levels of human FOXP3+CD4+ Tregs, but this was accompanied by a decrease in C-peptide levels and increases in numbers of natural killer (NK) cells and eosinophils.31 Studies treating patients with T1D with low-dose IL-2 have shown efficacy only for increasing the FOXP3+CD4+ Treg number, but these studies also demonstrated expansion of human NK cells and eosinophils in a dose-specific manner.20,32–34 These findings highlight the need for models to test the efficacy of strategies targeting human FOXP3+CD4+ Tregs and the development of novel approaches to deliver low-dose IL-2.
Here we use a humanized mouse model that enables efficient development of functional human T cells, including CD4+CD25+FOXP3+CD125dim Tregs, to study the effectiveness of an AAV vector expressing human IL-2. These studies use NOD-scid Il2rgnull (NSG) mice engrafted with human fetal thymus and liver tissues, abbreviated as the BLT (bone marrow/liver/thymus) model. The BLT model supports robust development of HLA-restricted conventional and Treg subsets.35 Our results show that use of an AAV8 vector expressing human IL-2 under the control of the murine insulin promoter increases the levels of FOXP3+CD4+ Tregs in the blood and spleen of NSG-BLT mice. Moreover, the IL-2-expanded human FOXP3+CD4+ Tregs maintain the ability to suppress immune responses both in vivo and in vitro. Coinciding with the FOXP3+CD4+ Treg expansion, a significant increase in the number and activation status of human NK cells is observed, recapitulating the clinical observations. These findings validate that humanized mouse models are effective for the in vivo study of AAV-based therapies that specifically target human immune cell subsets.
Materials and Methods
Mice
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NOD-scid Il2rgnull; NSG) mice were obtained from colonies maintained by L.D.S. at the Jackson Laboratory (Bar Harbor, ME). All animals were housed in a specific pathogen-free facility in microisolator cages, given autoclaved food, and maintained on acidified autoclaved water or sulfamethoxazole–trimethoprim-medicated water (Goldline Laboratories, Ft. Lauderdale, FL) provided on alternate weeks. All animal procedures were done in accordance with the guidelines of the Animal Care and Use Committee of the University of Massachusetts Medical School and the Jackson Laboratory and conformed to the recommendations in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, National Academy of Sciences, 8th edition, 2011).
Fetal tissue transplantation
Human fetal thymus and fetal liver specimens (gestational age, between 16 and 20 weeks) were obtained from Advanced Bioscience Resources (Alameda, CA). On receipt, tissues were washed with RPMI supplemented with penicillin G (100 U/ml), streptomycin (100 mg/ml), Fungizone (0.25 μg/ml), and gentamicin (5 μg/ml). The thymus tissue and a portion of the liver were sectioned into 1-mm3 fragments for transplantation, as previously described.36 The remaining fetal liver was processed to recover human CD34+ hematopoietic stem cells (HSCs) as described below. Recipient NSG mice were irradiated with 200 cGy and coimplanted with fetal thymus and fetal liver fragments in the renal subcapsular space, as described previously.37
Enrichment of CD34+ HSCs from fetal liver tissue
To recover human CD34+ HSCs, fetal liver was processed as previously described.36 Briefly, fetal liver was minced and digested at 37°C for 20 min with a collagenase–Dispase buffer (Gibco liver digest medium; Thermo Fisher Scientific, Waltham, MA). Recovered cells were washed with RPMI supplemented with 10% fetal bovine serum (FBS) and filtered through a metal sieve. Red blood cells were removed by Ficoll-Hypaque density centrifugation. The fetal liver cells were then depleted of CD3+ cells, using a magnetic bead negative-selection separation technique (Miltenyi Biotec, Auburn, CA), and the percentage of CD34+ cells was determined by flow cytometry (CD34-specific antibody, clone 581; BD Biosciences, San Jose, CA). At a minimum of 4 hr after irradiation, recipient NSG mice were injected intravenously with CD3-depleted fetal liver cells to achieve 1 × 105 CD34+ HSCs per mouse.
Double-stranded AAV8 vectors
The double-stranded AAV (dsAAV) vectors (or self-complementary AAV) were engineered and packaged as previously described.38 Briefly, full-length cDNA encoding human IL-2 or enhanced green fluorescent protein (EGFP) were subcloned into a dsAAV plasmid39 containing the murine preproinsulin II promoter (mIP), which controls expression of the cloned genes19 and restricts expression to cells synthesizing murine insulin. dsAAV vector packaging was carried out as previously described19,40 or produced by the Viral Vector Core at the University of Massachusetts Medical School Horae Gene Therapy Center (Worcester, MA). Recipient mice were injected intraperitoneally with 2.5 × 1011 vector particles of the purified AAV8-huIL-2 or AAV8-EGFP.
ELISA
Blood was collected from mice into 1000 U of heparin sodium injection, USP (Pfizer Injectables, New York, NY) and thoroughly mixed to prevent coagulation. Blood was then centrifuged for 30 min at 1000 × g, and the plasma layer was collected. Plasma samples were then diluted 1:3 in 1% bovine serum albumin (BSA)–phosphate-buffered saline (PBS) and frozen until levels of IL-2 were measured with a human IL-2-specific ELISA kit (BioLegend, San Diego, CA), in accordance with the manufacturer's protocol.
Antibodies and flow cytometry
Human immune cell populations were monitored in NSG-BLT mice, using monoclonal antibodies (mAbs) specific for the following human antigens; CD45 (clone HI30), CD3 (clone UCHT1), CD4 (clone RPA-T4), CD8 (clone RPA-T8), CD16 (clone 3G8), CD20 (clone 2H7), CD25 (clones 2A3 and MA-251), CD45RA (clone HI100), CD56 (clone NCAM16.2), CD62L (clone DREG-56), CD94 (clone HP-3D9), CD127 (clone A019D5), Ki67 (clone B56), FOXP3 (clone 236A/E7), HLA-DR (clone G46-6), NKp46 (clone 9e2/Nkp46), and granzyme B (clone GB11) purchased from eBioscience (San Diego, CA), BD Biosciences, or BioLegend. Mouse cells were identified and excluded from analysis by staining with an mAb specific for murine CD45 (clone 30-F11; BD Biosciences). Single-cell suspensions of bone marrow and spleen were prepared from engrafted mice, and whole blood was collected in heparin. Single-cell suspensions of 1 × 106 cells of bone marrow or spleen or 100 μl of whole blood was washed with fluorescence-activated cell sorting (FACS) buffer (PBS supplemented with 2% FBS and 0.02% sodium azide) and then preincubated with rat anti-mouse FcR11b mAb (clone 2.4G2; BD Biosciences) to block binding to mouse Fc receptors. Specific mAbs were then added to the samples and incubated for 30 min at 4°C. Stained samples were washed and fixed with 2% paraformaldehyde for cell suspensions or treated with BD FACS lysing solution for whole blood. At least 50,000 events were acquired on an LSRII or FACSCalibur instrument (BD Biosciences). Data analysis was performed with FlowJo software (Tree Star, Ashland, OR).
Histological analyses
For histological examination, samples of liver and pancreas were recovered from NSG-BLT mice, immersed overnight in 10% neutral buffered formalin, and embedded in paraffin. Sections (5 μm) were cut and stained with hematoxylin and eosin. Immunohistochemical staining was performed with mAbs specific for human CD45 (clone 2B11 + PD7/26; Dako, Glostrup, Denmark), using a DakoCytomation EnVision Dual Link system implemented on a Dako Autostainer universal staining system (Dako).
Phospho-STAT5 staining
Phospho-STAT5 (pSTAT5) was stained with a Phosflow staining kit obtained from BD Biosciences and in accordance with the manufacturer's protocol. Briefly, splenocytes were suspended at 5 × 106 cells/ml in RPMI supplemented with 10% FBS, and 1 × 106 cells were then added to 5-ml FACS tubes (BD Biosciences). Splenocytes were then cultured at 37°C/5% CO2 for 2 hr and either treated with human IL-2 (50 IU) for 20 min at 37°C or left untreated. After the incubation splenocytes were fixed with Cytofix buffer, permeabilized with Perm Buffer III, and washed twice in staining buffer. Cells were stained at room temperature with an antibody specific for pSTAT5 (clone 47/Stat5 [pY694]; BD Biosciences) for 60 min and then washed with staining buffer. Splenocytes were then stained with mAbs specific for murine CD45 and human CD4 and CD25. Samples were analyzed immediately after staining.
In vitro FOXP3+CD4+ Treg suppression assay
Human FOXP3+CD4+ Tregs were tested for the ability to suppress in vitro proliferation of human CD4+ T cells. FOXP3+CD4+ Tregs were enriched from AAV8-huIL-2-treated and PBS-treated NSG-BLT mice, and effector CD4+ T cells (CD25–) were enriched from a cohort of NSG-BLT mice that were generated with tissues not HLA-matched to the AAV-treated NSG-BLT mice. CD4+ T cells were enriched from the spleens of NSG-BLT mice by magnetic bead negative selection, using a human CD4+ T cell isolation kit (Miltenyi Biotec). Enriched CD4+ cells were then stained with mAbs to CD25 and CD127, and FOXP3+CD4+ Tregs (CD25+CD127dim) and effector T cells (CD25–) were purified by cell sorting, using a BD FACSAria (BD Biosciences). Effector T cells were labeled with 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (CFSE; Sigma-Aldrich, St. Louis, MO) and incubated for 5 days with anti-CD3/anti-CD28-coated beads (Miltenyi Biotec) in the presence or absence of human FOXP3+CD4+ Tregs at graded ratios. After 5 days, cells were stained with fluorochrome-conjugated mAbs and LIVE/DEAD viability dye (Thermo Fisher Scientific) and analyzed via FACS for proliferation (CSFE dye dilution). The percentage of proliferation inhibition was calculated as previously described by determining the proportion of dividing effector T cells from each coculture as compared with the proportion of dividing effectors in the absence of FOXP3+CD4+ Tregs when stimulated with anti-CD3/anti-CD28-coated beads.41
In vivo killing assay
NSG-BLT mice and NSG mice not engrafted with human cells were injected intravenously with PBMCs (20 × 106 cells) that were allogeneic to the transplanted tissues in NSG-BLT mice. HLA allelic differences between the injected allogeneic PBMCs and the BLT-generated immune system were used to monitor levels of the injected PBMCs. Specifically, PBMCs from HLA-A2+ or HLA-A3+ donors were injected into BLT mice that were A2– or A3–, respectively. Three days after PBMC injection, splenocytes were recovered from the injected mice and levels of allogeneic PBMCs were determined by staining with mAbs specific for either HLA-A2 (clone BB7.2; BD Biosciences) or HLA-A3 (clone GAP.A3; BD Biosciences). For deletion of CD4+ T cells, mice were injected intraperitoneally with 200 mg of OKT-4 (anti-CD4) mAb (Bio X Cell, Lebanon, NH) on days −8, −7, and −6 before injection with allogeneic PBMCs. Depletion of CD4+ T cells was confirmed by flow cytometry, using the RPA-T4 clone.
Statistical analyses
To compare individual pair-wise groupings, we used one-way analysis of variance (ANOVA) with Bonferroni post-tests and the Kruskal–Wallis test with Dunn's post-test for parametric and nonparametric data, respectively. Results of proliferation assays were analyzed by t test. For both analyses, significant differences were assumed for p values less than 0.05. Statistical analyses were performed with GraphPad Prism software (version 4.0c; GraphPad, San Diego, CA).
Results
Human immune system homeostasis is maintained in NSG-BLT mice treated with AAV8-huIL-2
NSG-BLT mice provide an effective humanized model to study the effects of human-specific therapies on immune system homeostasis and T cell biology. The NSG-BLT model supports the growth of human thymus tissue and enables the selection of HLA-restricted T cells on autologous human thymic epithelial cells. Representative flow cytometric data showing human T cell populations detected in the peripheral blood of NSG-BLT mice at 12 weeks postimplantation are shown in Fig. 1A and B. Human CD8+ and CD4+ conventional T cells and FOXP3+CD4+ Tregs develop efficiently and populate the periphery of NSG-BLT mice. Human FOXP3+CD4+ Tregs represented an average of 3.6 ± 0.7% of total circulating human hematolymphoid cells (human CD45+). The peripheral development of T cell subsets suggests that NSG-BLT mice are potential candidates for preclinical evaluation of immune therapies targeting human T cells.
Figure 1.

Levels of human CD4+ and CD8+ T cells are maintained in NSG-BLT mice treated with AAV8-huIL-2. NSG-BLT mice were generated as described in Materials and Methods. Human immune system development was examined in the blood of NSG-BLT mice 12 weeks after implantation of tissues and HSCs (A and B). The gating strategy is shown in (A), with identification of CD3+ T cells by gating on human CD45+ cells and then gating on CD4+ and CD8+ T cells. Within the CD4+ population, FOXP3+CD4+ Tregs were identified as CD127lowCD25+FOXP3+. Representative human cell engraftment levels are shown in (B). The data shown in (A) and (B) are representative of 40 experiments. (C) NSG-BLT mice were injected intraperitoneally with either AAV8-EGFP or AAV8-huIL-2 (2.5 × 1011 vector particles), and human IL-2 levels were monitored in the serum by ELISA. Human immune system engraftment, including overall engraftment of human CD45+ cells, was monitored in the blood of NSG-BLT mice injected with either AAV8-EGFP or AAV8-huIL-2 (D), CD3+ T cells (E), CD4+ T cells (F), and CD8+ T cells (G). These results are representative of four experiments.
Previous studies have shown that delivery of IL-2 by AAV vector is an effective approach to expand functional FOXP3+CD4+ Tregs in NOD mice and prevent the development of T1D.19 To test the effect of AAV8-delivered human IL-2 on human FOXP3+CD4+ Tregs and conventional T cell homeostasis, we used the NSG-BLT model. NSG-BLT mice were injected intraperitoneally with 2.5 × 1011 vector particles of either a human IL-2-expressing vector (AAV8-huIL-2) or a control vector (AAV8-EGFP). Human IL-2 was detected in the plasma of NSG-BLT mice injected with AAV8-huIL-2 but not in AAV8-EGFP-treated mice beginning at 2 weeks postinjection of AAV (Fig. 1C). Human cell chimerism (CD45+) and T cell (CD3+CD4+ and CD3+CD8+) levels were monitored in the peripheral blood every 2 weeks over a 10-week time frame. No significant differences were observed in the percentages of CD45+ human cells (Fig. 1D) and of CD3+ T cells (Fig. 1E) (both CD4+ and CD8+) in the peripheral blood of NSG-BLT mice injected with AAV8-huIL-2 as compared with control NSG-BLT mice injected with AAV-EGFP (Fig. 1F and G). Representative staining for human cell infiltrates in mouse pancreas and liver recovered from NSG-BLT mice that were injected with AAV8-EGFP or AAV-huIL-2 is shown in Supplementary Fig. S1 (supplementary data are available online at www.liebertpub.com/hum). No significant differences in human cell infiltration were observed in tissues recovered from either AVV8-EGFP-treated or AAV-huIL-2-treated NSG-BLT mice. These findings suggest that treatment with this AAV vector at this dose establishes constitutive low-level expression of human IL-2, which does not significantly alter human immune system homeostasis in NSG-BLT mice.
AAV8-huIL-2 treatment increases the level of human FOXP3+CD4+ Tregs in NSG-BLT mice
We next assessed the effect of AAV-huIL-2 on human FOXP3+CD4+ Tregs in NSG-BLT mice (Fig. 2). NSG-BLT mice were injected with AAV8-EGFP or AAV-huIL-2, and the levels of human FOXP3+CD4+ Tregs (human CD3+CD4+CD127–CD25+FOXP3+) were monitored in the peripheral blood every 2 weeks. Over the course of 10 weeks, the proportion of FOXP3+CD4+ Tregs increased significantly in NSG-BLT mice injected with AAV8-huIL-2 (Fig. 2). Representative staining of FOXP3+CD4+ Tregs from the blood of AAV8-EGFP-treated (Fig. 2A) and AAV8-huIL-2-treated (Fig. 2B) NSG-BLT mice shows the changes in the percentage of human FOXP3+CD4+ Tregs over time. A significant increase in human CD127–CD25+CD4+ T cells was detectable at week 4 in NSG-BLT mice after injection with AAV8-huIL-2, and this increase was maintained through week 10 (Fig. 2C), as compared with AAV8-EGFP-injected NSG-BLT mice. The majority of CD127–CD25+ T cells in both treatment groups of NSG-BLT mice were FOXP3+ at all time points, as shown by the percentage of CD4+ T cells that were CD25+FOXP3+ (Fig. 2D) or CD127–CD25+FOXP3+ (Fig. 2E).
Figure 2.

Treatment with AAV8-huIL-2 increases the percentage of human FOXP3+CD4+ Tregs in the blood of NSG-BLT mice. NSG-BLT mice were injected intraperitoneally with either AAV8-EGFP (n = 5) or AAV8-huIL-2 (n = 9) (2.5 × 1011 particles) and bled every 2 weeks to monitor human FOXP3+CD4+ Treg levels. Representative staining (CD127 and CD25) is shown for human FOXP3+CD4+ Tregs over time in NSG-BLT mice injected with AAV8-EGFP (A) and AAV8-huIL-2 (B). Percentage of CD4+ Treg subsets over time is shown: CD127lowCD25+ (C), CD25+FOXP3+ (D), and CD25+CD127lowFOXP3+ (E). These results are representative of four experiments. **p < 0.01; ***p < 0.001.
Levels of FOXP3+CD4+ Tregs were also evaluated in the spleen and thymus of NSG-BLT mice treated with AAV8-EGFP (Fig. 3A) and AAV8-huIL-2 (Fig. 3B). Significant increases in both the percentage and number of human FOXP3+CD4+ Tregs in the spleen at the 10-week time point were observed (Fig. 3C and D). To determine whether ectopic expression of IL-2 enhanced thymic development, we analyzed the FOXP3+CD4+ Treg subset within the human thymus graft of NSG-BLT mice injected with AAV8-huIL-2 (Fig. 3E). The implanted thymus grafts were excised from mice 10 weeks after treatment with either AAV8-EGFP or AAV8-huIL-2 and thymocytes were analyzed by flow cytometry. No significant increase in human FOXP3+CD4+ Treg levels was detected within the thymic tissue of NSG-BLT mice treated with AAV8-huIL-2. Together these data suggest that delivery of human IL-2 by AAV vectors increased the frequency of peripheral but not thymic human FOXP3+CD4+ Tregs in NSG-BLT mice.
Figure 3.

Treatment with AAV8-huIL-2 increases the percentage and number of human FOXP3+CD4+ Tregs in the spleen of NSG-BLT mice but does not change levels in the thymus. NSG-BLT mice were injected intraperitoneally with either AAV8-EGFP (n = 5) or AAV8-huIL-2 (n = 9) (2.5 × 1011 vector particles). At 10 weeks posttreatment FOXP3+CD4+ Treg levels were determined in the spleen and thymus. The gating strategy used for analyzing splenocytes is shown for NSG-BLT mice treated with AAV8-EGFP (A) and AAV8-huIL-2 (B). The proportion (C) and number (D) of CD127lowCD25+FOXP3+ Tregs are shown. The human thymic tissue was recovered from NSG-BLT mice injected with either AAV8-huIL-2 or AAV8-EGFP, and the proportion of FOXP3+CD4+ Tregs was determined (E). These results are representative of four experiments. ***p < 0.001. NS, not significant.
AAV8-huIL-2 treatment alters the phenotype and homeostasis of peripheral human FOXP3+CD4+ Tregs in NSG-BLT mice
Expression of human IL-2 in NSG-BLT mice stimulated a significant increase in the levels of peripheral human FOXP3+CD4+ Tregs. To assess phenotype, we first examined FOXP3+CD4+ Treg expression of CD45RA and CD62L in AAV8-treated NSG-BLT mice. CD45RA+CD62L+ human FOXP3+CD4+ Tregs are capable of robust expansion and efficiently suppress immune responses.42 The levels of CD45RA+CD62L+ human FOXP3+CD4+ Tregs were significantly higher in blood (Fig. 4A) and spleen (Fig. 4B) of AAV8-huIL-2-treated NSG-BLT mice as compared with FOXP3+CD4+ Tregs from AAV8-EGFP-treated NSG-BLT mice. AAV8-huIL-2-treated NSG-BLT mice also had a higher proportion of Ki67+ human FOXP3+CD4+ Tregs in the spleen when compared with the AAV8-EGFP-treated NSG-BLT mice (Fig. 4C). A higher proportion of Ki67+ human FOXP3+CD4+ Tregs was also detected in the blood of AAV8-huIL-2-treated mice, but this difference did not reach statistical significance (data not shown). Expression of CD25 (IL-2 receptor α chain) was increased on human FOXP3+CD4+ Tregs in the blood of NSG-BLT mice 2 weeks after treatment with AAV8-huIL-2, and CD25 expression was maintained through the 10-week experiment (Fig. 4D).
Figure 4.

AAV8-huIL-2 treatment alters the phenotype and homeostasis of human FOXP3+CD4+ Tregs in NSG-BLT mice. NSG-BLT mice were injected intraperitoneally with 2.5 × 1011 particles of either AAV8-huIL-2 (n = 9) or control AAV8-EGFP (n = 5). Expression of phenotypic and proliferation markers by the recovered human FOXP3+CD4+ Tregs was determined at 10 weeks posttreatment. The frequency of human FOXP3+CD4+ Tregs coexpressing CD45RA and CD62L was determined in blood (A) and spleen (B). To determine the proportion of human FOXP3+CD4+ Tregs undergoing cell division, expression of Ki67 by splenic FOXP3+CD4+ Tregs was evaluated (C). Expression of CD25 by FOXP3+CD4+ Tregs recovered from the blood of NSG-BLT mice was evaluated every 2 weeks after AAV vector treatment (D). The data shown in (A–D) are representative of four experiments. pSTAT5 was evaluated in FOXP3+CD4+ Tregs recovered from the spleen of NSG-BLT mice 10 weeks after AAV vector treatment (E and F). Splenocytes were stimulated with 50 IU of recombinant human IL-2 for 20 min or unstimulated (medium only), and pSTAT5 levels were evaluated by flow cytometry. The gating strategy (E) and the pSTAT5 levels detected in CD3+CD4+CD25+ cells (F) are shown. The data shown in (E and F) are representative of two experiments. **p < 0.01; ***p < 0.001; ****p < 0.0001. MFI, mean fluorescence intensity.
To test responsiveness of human FOXP3+CD4+ Tregs to IL-2 stimulation we assessed levels of phosphorylated STAT5 (pSTAT5) after exposure to IL-2 in vitro. Splenocytes from NSG-BLT mice injected 10 weeks previously with either AAV8-huIL-2 or AAV8-EGFP were stimulated in vitro with recombinant human IL-2 for 20 min and evaluated for pSTAT5 expression by flow cytometry. FOXP3+CD4+ Tregs were identified as CD3+CD4+CD25+ cells (Fig. 4E). Within this population the mean fluorescence intensity (MFI) of pSTAT5 staining was quantified. FOXP3+CD4+ Tregs from NSG-BLT mice injected with AAV8-huIL-2 or AAV8-EGFP showed a similar level of STAT5 phosphorylation in response to IL-2 stimulation (Fig. 4F). These results demonstrated that human FOXP3+CD4+ Tregs exposed to chronic IL-2 stimulation maintain responsiveness to IL-2.
Human FOXP3+CD4+ Tregs from AAV8-huIL-2-treated NSG-BLT mice maintain functionality
The results presented above show that expression of human IL-2 by an AAV vector increases the levels of FOXP3+CD4+ Tregs in NSG-BLT mice but does not indicate whether the Treg population maintains overall functionality in vitro and in vivo. For in vitro suppression assays, human FOXP3+CD4+ Tregs (CD4+CD127lowCD25+) were sorted from the spleens of AAV8-EGFP- or AAV8-huIL-2-injected NSG-BLT mice 5 weeks after treatment. Splenic effector human T cells (CD4+CD25–) were enriched from a separate cohort of otherwise untreated NSG-BLT mice (allogeneic donor). The two recovered populations were then used in a CFSE-based proliferation suppression assay.41 CFSE-labeled, effector CD4+ T cells were stimulated with CD3 and CD28 in the presence or absence of graded numbers of purified human FOXP3+CD4+ Tregs. After 5 days of in vitro culture, proliferation of the CD4+ conventional T cells was assessed by dilution of CFSE. Human FOXP3+CD4+ Tregs recovered from NSG-BLT mice injected with AAV8-huIL-2 or AAV8-EGFP were able to suppress CD4 proliferation, and this effect decreased with lower Treg:T effector ratios (Fig. 5A). No statistically significant differences in suppression were observed between FOXP3+CD4+ Tregs recovered from AAV8-huIL-2- or AAV8-EGFP-treated mice. These results indicate that human FOXP3+CD4+ Tregs responding to IL-2 in NSG-BLT mice maintain in vitro functionality.
Figure 5.

Human FOXP3+CD4+ Tregs from AAV8-huIL-2-treated NSG-BLT mice function in vitro and in vivo. The ability of human FOXP3+CD4+ Tregs in NSG-BLT mice treated with AAV8-huIL-2 to suppress human effector T cell responses was evaluated with in vitro (A) and in vivo (B and C) assays. NSG-BLT mice were injected intraperitoneally with 2.5 × 1011 vector particles of AAV8-EGFP or AAV8-huIL-2. (A) Splenic human FOXP3+CD4+ Tregs (CD4+CD127lowCD25+) were sorted from AAV8-EGFP- or AAV8-IL-2-treated NSG-BLT mice 5 weeks after treatment and cocultured at the indicated ratios with CFSE-labeled splenic effector human T cells (CD4+CD25–) from a separate cohort of NSG-BLT mice (allogeneic donor). Percent inhibition after anti-CD3/CD28 antibody-coated bead stimulation was calculated as described in Material and Methods. The data shown are an average of three independent experiments. (B) Allogeneic (HLA-A2+ or HLA-A3+) human PBMCs (15 × 106) were injected into either nonengrafted NSG mice or NSG-BLT mice treated 4 weeks earlier with either AAV8-EGFP or AAV8-huIL-2 (2.5 × 1011 vector particles). Three days later, the number of A2+/A3+ donor PBMCs in blood was measured by flow cytometry. (C) To determine whether FOXP3+CD4+ Tregs suppressed the killing of allogeneic PBMCs in NSG-BLT mice treated with AAV8-huIL-2, allogeneic donor PBMCs were injected into nonengrafted NSG mice and NSG mice treated 4 weeks earlier with either AAV8-EGFP or AAV8-huIL-2 as described in (B). In addition, mice in the indicated groups were treated with an isotype control antibody or antibody specific for human CD4 to deplete CD4+ T cells (including FOXP3+CD4+ Tregs) 5 days before injection of donor PBMCs. Three days after injection of allogeneic PBMCs, the number of A2+/A3+ donor PBMCs in blood was determined. The data shown for (B and C) are representative of two independent experiments. *p < 0.05; **p < 0.01; ****p < 0.0001. NS, not significant.
To assess in vivo function, PBMCs from a donor that was HLA-mismatched with the BLT tissue donor were injected into nonengrafted NSG mice or into NSG-BLT mice treated with either AAV8-EGFP or AAV8-huIL-2. Specifically, allogeneic donors that were mismatched at HLA-A2 or -A3 were identified, as this enabled the direct monitoring of A2+ or A3+ allogeneic PBMCs in the NSG-BLT mice by flow cytometric analysis. Three days after injection of PBMCs, the levels of HLA-mismatched PBMCs recovered from the spleens of recipient mice were determined. NSG-BLT mice treated with AAV8-EGFP had significantly lower levels of mismatched PBMCs as compared with the nonengrafted NSG mice (NSG Control), indicating that the BLT immune system efficiently rejected the injected allogeneic human cells (Fig. 5B). In contrast, NSG-BLT mice treated with AAV8-huIL-2 showed a significant increase in detectable HLA-mismatched PBMCs as compared with the AAV8-EGFP group (Fig. 5B). These results indicate that treatment with AAV8-huIL-2 suppressed the ability of NSG-BLT mice to reject allogeneic human PBMCs.
To determine whether this suppressive effect was due to CD4+ T cells, we injected HLA-mismatched PBMCs into nonengrafted NSG mice (NSG Control) or AAV8-treated NSG-BLT mice that were depleted of CD4+ cells 5 days before injection of allogeneic PBMCs. Injection of AAV8-EGFP-treated NSG mice with a CD4-depleting antibody before injection of PBMCs did not reduce the number of injected PBMCs detected in the spleen of these mice (Fig. 5C). Consistent with the results shown in Fig. 5B, NSG-BLT mice treated with AAV8-huIL-2 had a significant increase in the detectable levels of HLA-mismatched PBMCs as compared with the AAV8-EGFP-treated group. However, depletion of CD4+ cells from NSG-BLT mice treated with AAV8-huIL-2 resulted in a significant reduction in detectable HLA-mismatched PBMCs as compared with the AAV8-huIL-2 group that was not CD4 depleted (Fig. 5C). The results from the CD4 depletion experiments indicate that the suppression of allospecific immune responses in NSG-BLT mice treated with AAV8-huIL-2 requires a CD4+ T cell population. Together these results demonstrate that AAV vector delivery of human IL-2 stimulates an increase in functional human FOXP3+CD4+ Tregs in NSG-BLT mice.
AAV8-huIL-2 treatment does not alter the proportion of conventional CD4+ or CD8+ T cells in NSG-BLT mice
Conventional human effector and memory CD4+ and CD8+ T cells constitutively express the IL-2 receptor β (CD122) and common γ (CD132) chains that enable intermediate IL-2 binding and signaling. Moreover, acutely activated conventional T cells will also upregulate expression of CD25 (IL-2 receptor α chain), creating a high-affinity IL-2 receptor.43 To determine whether treatment with AAV8-huIL-2 stimulates activation of human T cells in NSG-BLT mice, we first evaluated the phenotype and activation status of conventional CD4+ and CD8+ T cells (Supplementary Fig. S2). At 10 weeks posttreatment with AAV8 vectors, CD4+ and CD8+ T cells from the peripheral blood of NSG-BLT mice were evaluated for expression of HLA-DR as a marker of acute activation and for the presence of naive cells (CD45RA+CD62L+), effector/memory cells (CD45RA–CD62L–), and central memory human CD45RA–CD62L+ T cells. Consistent with the increased function of human FOXP3+CD4+ Tregs, the percentage of conventional CD4+ and CD8+ T cells expressing HLA-DR in NSG-BLT mice treated with AAV8-huIL-2 was significantly lower as compared with control NSG-BLT mice (Supplementary Fig. S2A and E). However, no significant differences were observed in the percentages of naive, effector/memory, and central memory CD4+ (Supplementary Fig. S2B–D) and CD8+ (Supplementary Fig. S2F–H) T cells. These data suggest that AAV8-huIL-2 treatment has a minimal impact on conventional T cell phenotype.
AAV8-huIL-2 treatment increases the number and activation of human NK cells in NSG-BLT mice
Human NK cells defined as NKp46+CD94high/low constitutively express the low-affinity IL-2 receptor complex and will proliferate in response to treatment with IL-2.19 Accordingly, the number and activation status of NK cells in AAV8-EGFP- and AAV8-huIL-2-treated NSG-BLT mice were evaluated. Representative flow panels of NK cells in the spleen at 10 weeks are shown in Supplementary Fig. S3A. NSG-BLT mice treated with AAV8-huIL-2 had significantly higher percentages and numbers of human NK cells in the spleen as compared with AAV8-EGFP-treated NSG-BLT mice (Supplementary Fig. S3B and C). Treatment with AAV8-huIL-2 also resulted in a significant increase in the number of NK cells that expressed granzyme B, which identifies NK cells that have cytotoxic activity (Supplementary Fig. S3D). Overall, these data demonstrate that the treatment with AAV8-huIL-2 results in a significant increase in NK cell numbers and their activation status.
Discussion
There is a paucity of validated models for efficient preclinical testing of human-specific therapies using AAV as a gene delivery vector.44 The development of immunodeficient mice that support efficient engraftment of functional human cells, tissues, and immune systems has provided the opportunity to study human-specific therapies.45–47 Here we demonstrate that the use an AAV vector to deliver human IL-2 in a humanized mouse model reproduces the effects of low-dose IL-2 therapies on human immune system homeostasis that were observed in clinical trials.48 For these studies, we used the NSG-BLT mouse model, which enables robust development of conventional and FOXP3+CD4+ Tregs and supports the generation of HLA-restricted human T cells.49–51 Our findings demonstrate that elevated levels of human IL-2 in NSG-BLT mice treated with AAV8-huIL-2 stimulate an increase in the number of functional human FOXP3+CD4+ Tregs and have minor effects on conventional T cell numbers and function. However, a concurrent increase in the numbers and activation of human NK cells was evident in NSG-BLT mice treated with AAV8-huIL-2. These results support the utilization of the BLT model to study therapies targeting human FOXP3+CD4+ Tregs and indicate that BLT mice are effective tools to study the impact of AAV vector therapies on human immune system function and homeostasis.
A major breakthrough for the in vivo study of human immune systems followed the development in the mid-2000s of immunodeficient mice with a mutation in the interleukin-2 (IL-2) receptor γ-chain locus (Il2rg; also known as γc and CD132).52–55 The targeted mutation of Il2rg completely prevents murine natural killer (NK) cell development, and cripples host innate immune function. The NSG strain has become the most widely used immunodeficient mouse strain for the engraftment of human cells and tissues worldwide.56 NSG mice support engraftment with human HSCs and immune system development57–59 due, in part, to expression of a signal regulatory protein α (Sirpa) allele that has a close homology to human SIRPA.60 The NSG mouse also supports the implantation of human fetal thymic and liver tissues for the generation of BLT (Thy/Liv) mice.36,61 The robust development of human immune systems in NSG-BLT mice has enabled the study of numerous aspects of human immunity including (1) infectious diseases with pathogens such as John Cunningham virus, chlamydia, dengue virus, Epstein–Barr virus (EBV), Ebola virus, HIV, Kaposi's sarcoma-associated virus, and Mycobacterium; (2) immune responses to immunization with viral glycoproteins; and (3) immune responses to tissue allografts.46,62–69 An important advantage of the BLT model is the growth of human thymic tissues engrafted with autologous human HSCs that enables education of human conventional and FOXP3+CD4+ Tregs on human thymic epithelium and the development of HLA-restricted T cells.35 Moreover, the human FOXP3+CD4+ Tregs that develop in BLT mice are functional, making this model attractive to study the impact of immune therapies on HLA-restricted FOXP3+CD4+ Treg function and homeostasis.70,71 Our findings with IL-2 treatment of NSG-BLT mice reproduce observations from clinical trials showing that low-dose IL-2 therapies increase functional FOXP3+CD4+ Tregs and NK cells in patients20,72 and validate the use of the NSG-BLT model as a platform to test approaches targeting human FOXP3+CD4+ Tregs. There are limitations with currently available humanized mouse models, such as NSG-BLT mice, that include the absence of HLA expression on mouse cells and tissues, the limited development of functional human B cells that can undergo class switching and affinity maturation, the absence of mature germinal center formation, and reduced levels and maturation status of human innate immune cells.73 These limitations may impact the interpretation of any immunological studies done with humanized mice.
Low-dose IL-2 therapies increase levels of functional FOXP3+CD4+ Tregs in humans but also have the potential to stimulate immune cell populations that may be detrimental to the patient, including NK cells, eosinophils, and memory T cells.21 Humanized mouse models have been used to study the role of IL-2 in human immune system homeostasis. Ito and colleagues developed a NOD/Shi-scid-Il2rgnull (NOG) mouse strain expressing a human IL-2 transgene (NOG-IL-2).74 Human HSC-engrafted NOG-IL-2 mice showed a significant increase in functional human NK cells as compared with NOG mice but no effect on human T cells was reported. The NOG-IL-2 transgenic mice also supported enhanced engraftment of human CD8+ T cells purified from peripheral blood, and the engrafted CD8+ T cells mediated severe xeno-GVHD.75 A previous study also demonstrated that hydrodynamic delivery of a plasmid expressing human IL-2 into NSG mice enhanced survival of in vitro-expanded human Tregs.76 However, hydrodynamic injection of the IL-2-expressing plasmid in NSG mice engrafted with PBMCs also accelerated the development of xeno-GVHD. One study examined the impact of treating humanized NSG mice with IL-2–antibody complexes, which in standard immunocompetent mouse models alters T cell and NK cell homeostasis.77 NSG mice expressing human HLA-A2 and engrafted with A2+ HSCs from umbilical cord blood were challenged with a human melanoma cell line and then treated with human IL-2–antibody complexes (Hu-IL2c). One Hu-IL2c tested did not significantly alter the frequencies of human conventional CD4+ and CD8+ T cells but did stimulate an increase in T-bet-positive, effector T cells and an increase in human FOXP3+CD4+ Tregs. Our studies show that IL-2 delivered by AAV vector stimulates a significant increase in FOXP3+CD4+ Tregs and NK cells in NSG-BLT mice but does not dramatically alter the frequency or phenotype of conventional T cells. Moreover, the FOXP3+CD4+ Tregs in NSG-BLT mice treated with AAV8-huIL-2 maintained functionality. Tregs recovered from NSG-BLT mice treated with AAV8-huIL-2 showed an equivalent ability to suppress T cell function in vitro as compared with AAV8-EGFP-treated mice. In addition, our data suggest that Tregs in NSG-BLT mice treated with AAV8-huIL-2 are functional in vivo by preventing the rejection of allogeneic PBMCs. However, it is possible that in these experiments the detection of higher levels of allogeneic PBMCs in AAV8-huIL-2-treated NSG-BLT mice may be a result of IL-2 acting directly on the injected PBMCs to increase frequency or a result of IL-2 acting indirectly on the resident immune cells to promote engraftment of the injected PBMCs. Experiments are currently underway using AAV vector with inducible IL-2 expression that will enable IL-2 synthesis to be turned off before injection of allogeneic PBMCs. We are also performing studies to determine the functionality of NK cells recovered from NSG-BLT mice treated with AAV8-huIL-2.
The significant increase in functional human NK cells in NSG-BLT mice injected with AAV8-huIL-2 and in clinical trials with low-dose IL-2 therapies21 highlights the ability of human NK cells to respond to low levels of human IL-2. IL-2 binding and signaling is mediated by expression of three distinct receptor subunits including CD25 (IL-2Rα), CD122 (IL-2Rβ), and CD132 (IL-2Rγ) in varying combinations on the cell surface.78 The high-affinity IL-2 receptor (IL-2R), which is constitutively expressed on FOXP3+CD4+ Tregs, is a heterotrimer composed of the three subunits (CD25, CD122, and CD132).79 The majority of NK cells express the intermediate-affinity receptor, a dimeric IL-2R that consists of CD122 and CD132, which are the signaling subunits of the receptor, and are thought to respond only to high levels of IL-2.80 A small population of CD56-bright human NK cells expresses CD25 and responds efficiently to low-dose IL-2, suggesting that these may be the NK cell subset that expands in vivo with low-dose IL-2 therapies.81 One study has also shown that trans-presentation of IL-2 bound to CD25 activates CD25-negative NK cells,82 suggesting an additional mechanism for activation of NK cells by low-dose IL-2 therapies. The generation of novel IL-2 therapies that selectively target Tregs but do not activate NK cells may be possible using IL-2-specific antibody83 or by the development of modified IL-2 molecules.84 Regardless of the approach, the NSG-BLT mouse model will be an effective tool to assess the efficacy of new IL-2 strategies.
Overall, our study highlights the utility of humanized mice engrafted with human immune systems in validating AAV vectors expressing immune-modulatory therapies. NSG-BLT mice treated with AAV8-huIL-2 displayed the expected effects on human immune system homeostasis that were observed in clinical trials. These results support the use of the NSG-BLT model to study more targeted approaches to IL-2 therapy that avoid the activation of NK cell populations while still inducing a more tolerogenic environment through selectively increasing the FOXP3+CD4+ Treg population. Humanized mice present a convenient and safe preclinical system for testing such novel therapies before they are used in human clinical trials. More importantly, NSG-BLT mice are more likely to yield results that are relevant to human physiology than testing in comparable immunocompetent small-animal models given the significant differences between human and mouse immunobiology.85,86
Supplementary Material
Acknowledgments
The authors thank Darcy Reil and Pamela St. Louis for technical support.
Funding: This work was supported in part by National Institutes of Health grants 1R24 OD018259, UC4 DK104218 (M.A.B., L.D.S.), 1R01 DK103546 (R.M.T., M.A.B.), 1R01 AI132963 (M.A.B., L.D.S.), and CA034196 (L.D.S.).
Author Disclosure
Competing financial interests: M.A.B. is a consult for the Jackson Laboratory. Personal financial interests: The authors have no personal financial interests to report. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health. Employment: The authors have no employment disclosures. Other competing interests: The authors have no other competing interests to report.
References
- 1.Mueller C, Flotte TR. Clinical gene therapy using recombinant adeno-associated virus vectors. Gene Ther 2008;15:858–863 [DOI] [PubMed] [Google Scholar]
- 2.Samulski RJ, Muzyczka N. AAV-mediated gene therapy for research and therapeutic purposes. Annu Rev Virol 2014;1:427–451 [DOI] [PubMed] [Google Scholar]
- 3.Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 2014;15:445–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duan D. Systemic delivery of adeno-associated viral vectors. Curr Opin Virol 2016;21:16–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kotterman MA, Chalberg TW, Schaffer DV. Viral vectors for gene therapy: translational and clinical outlook. Annu Rev Biomed Eng 2015;17:63–89 [DOI] [PubMed] [Google Scholar]
- 6.Kattenhorn LM, Tipper CH, Stoica L, et al. Adeno-associated virus gene therapy for liver disease. Hum Gene Ther 2016;27:947–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munch RC, Muth A, Muik A, et al. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat Commun 2015;6:6246. [DOI] [PubMed] [Google Scholar]
- 8.Pepin D, Sosulski A, Zhang L, et al. AAV9 delivering a modified human Mullerian inhibiting substance as a gene therapy in patient-derived xenografts of ovarian cancer. Proc Natl Acad Sci U S A 2015;112:E4418–E4427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hashimoto H, Mizushima T, Ogura T, et al. Study on AAV-mediated gene therapy for diabetes in humanized liver mouse to predict efficacy in humans. Biochem Biophys Res Commun 2016;478:1254–1260 [DOI] [PubMed] [Google Scholar]
- 10.Sharma A, Wu W, Sung B, et al. Respiratory syncytial virus (RSV) pulmonary infection in humanized mice induces human anti-RSV immune responses and pathology. J VIrol 2016;90:5068–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X, Huang J, Zhang M, et al. Human CD8+ T cells mediate protective immunity induced by a human malaria vaccine in human immune system mice. Vaccine 2016;34:4501–4506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, Li X, Coelho-dos-Reis JG, et al. Human immune system mice immunized with Plasmodium falciparum circumsporozoite protein induce protective human humoral immunity against malaria. J Immunol Methods 2015;427:42–50 [DOI] [PubMed] [Google Scholar]
- 13.Huang J, Li X, Coelho-dos-Reis JG, et al. An AAV vector-mediated gene delivery approach facilitates reconstitution of functional human CD8+ T cells in mice. PLoS One 2014;9:e88205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 2001;27:68–73 [DOI] [PubMed] [Google Scholar]
- 15.Caudy AA, Reddy ST, Chatila T, et al. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J Allergy Clin Immunol 2007;119:482–487 [DOI] [PubMed] [Google Scholar]
- 16.Wildin RS, Ramsdell F, Peake J, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 2001;27:18–20 [DOI] [PubMed] [Google Scholar]
- 17.Hulme MA, Wasserfall CH, Atkinson MA, et al. Central role for interleukin-2 in type 1 diabetes. Diabetes 2012;61:14–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goudy KS, Johnson MC, Garland A, et al. Inducible adeno-associated virus-mediated IL-2 gene therapy prevents autoimmune diabetes. J Immunol 2011;186:3779–3786 [DOI] [PubMed] [Google Scholar]
- 19.Johnson MC, Garland AL, Nicolson SC, et al. Beta-cell-specific IL-2 therapy increases islet Foxp3+ Treg and suppresses type 1 diabetes in NOD mice. Diabetes 2013;62:3775–3784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Todd JA, Evangelou M, Cutler AJ, et al. Regulatory T cell responses in participants with type 1 diabetes after a single dose of interleukin-2: a non-randomised, open label, adaptive dose-finding trial. PLoS Med 2016;13:e1002139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pham MN, von Herrath MG, Vela JL. Antigen-specific regulatory T cells and low dose of IL-2 in treatment of type 1 diabetes. Front Immunol 2015;6:651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, et al. Interleukin 2 signaling is required for CD4+ regulatory T cell function. J Exp Med 2002;196:851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flores RR, Zhou L, Robbins PD. Expression of IL-2 in beta cells by AAV8 gene transfer in pre-diabetic NOD mice prevents diabetes through activation of FoxP3-positive regulatory T cells. Gene Ther 2014;21:715–722 [DOI] [PubMed] [Google Scholar]
- 24.Churlaud G, Jimenez V, Ruberte J, et al. Sustained stimulation and expansion of Tregs by IL2 control autoimmunity without impairing immune responses to infection, vaccination and cancer. Clin Immunol 2014;151:114–126 [DOI] [PubMed] [Google Scholar]
- 25.Grinberg-Bleyer Y, Baeyens A, You S, et al. IL-2 reverses established type 1 diabetes in NOD mice by a local effect on pancreatic regulatory T cells. J Exp Med 2010;207:1871–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koreth J, Matsuoka K, Kim HT, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. N Engl J Med 2011;365:2055–2066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saadoun D, Rosenzwajg M, Joly F, et al. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. N Engl J Med 2011;365:2067–2077 [DOI] [PubMed] [Google Scholar]
- 28.Castela E, Le Duff F, Butori C, et al. Effects of low-dose recombinant interleukin 2 to promote T-regulatory cells in alopecia areata. JAMA Dermatol 2014;150:748–751 [DOI] [PubMed] [Google Scholar]
- 29.He J, Zhang X, Wei Y, et al. Low-dose interleukin-2 treatment selectively modulates CD4+ T cell subsets in patients with systemic lupus erythematosus. Nat Med 2016;22:991–993 [DOI] [PubMed] [Google Scholar]
- 30.Humrich JY, von Spee-Mayer C, Siegert E, et al. Rapid induction of clinical remission by low-dose interleukin-2 in a patient with refractory SLE. Ann Rheum Dis 2015;74:791–792 [DOI] [PubMed] [Google Scholar]
- 31.Long SA, Rieck M, Sanda S, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes 2012;61:2340–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartemann A, Bensimon G, Payan CA, et al. Low-dose interleukin 2 in patients with type 1 diabetes: a phase 1/2 randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol 2013;1:295–305 [DOI] [PubMed] [Google Scholar]
- 33.Yu A, Snowhite I, Vendrame F, et al. Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes 2015;64:2172–2183 [DOI] [PubMed] [Google Scholar]
- 34.Rosenzwajg M, Churlaud G, Mallone R, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun 2015;58:48–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wege AK, Melkus MW, Denton PW, et al. Functional and phenotypic characterization of the humanized BLT mouse model. Curr Top Microbiol Immunol 2008;324:149–165 [DOI] [PubMed] [Google Scholar]
- 36.Aryee KE, Shultz LD, Brehm MA. Immunodeficient mouse model for human hematopoietic stem cell engraftment and immune system development. Methods Mol Biol 2014;1185:267–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Covassin L, Jangalwe S, Jouvet N, et al. Human immune system development and survival of non-obese diabetic (NOD)-scid IL2rγnull (NSG) mice engrafted with human thymus and autologous haematopoietic stem cells. Clin Exp Immunol 2013;174:372–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.He Y, Weinberg MS, Hirsch M, et al. Kinetics of adeno-associated virus serotype 2 (AAV2) and AAV8 capsid antigen presentation in vivo are identical. Hum Gene Ther 2013;24:545–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther 2001;8:1248–1254 [DOI] [PubMed] [Google Scholar]
- 40.Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc 2006;1:1412–1428 [DOI] [PubMed] [Google Scholar]
- 41.Schneider A, Buckner JH. Assessment of suppressive capacity by human regulatory T cells using a reproducible, bi-directional CFSE-based in vitro assay. Methods Mol Biol 2011;707:233–241 [DOI] [PubMed] [Google Scholar]
- 42.Hoffmann P, Eder R, Boeld TJ, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood 2006;108:4260–4267 [DOI] [PubMed] [Google Scholar]
- 43.Brusko TM, Wasserfall CH, Hulme MA, et al. Influence of membrane CD25 stability on T lymphocyte activity: implications for immunoregulation. PLoS One 2009;4:e7980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weber T. Do we need marker gene studies in humans to improve clinical AAV gene therapy? Gene Ther 2017;24:72–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Theocharides AP, Rongvaux A, Fritsch K, et al. Humanized hemato-lymphoid system mice. Haematologica 2016;101:5–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walsh NC, Kenney LL, Jangalwe S, et al. Humanized mouse models of clinical disease. Annu Rev Pathol 2017;12:187–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shultz LD, Brehm MA, Garcia-Martinez JV, et al. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol 2012;12:786–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Long SA, Buckner JH, Greenbaum CJ. IL-2 therapy in type 1 diabetes: “Trials” and tribulations. Clin Immunol 2013;149:324–331 [DOI] [PubMed] [Google Scholar]
- 49.Covassin L, Jangalwe S, Jouvet N, et al. Human immune system development and survival of non-obese diabetic (NOD)-scid IL2rγnull (NSG) mice engrafted with human thymus and autologous haematopoietic stem cells. Clin Exp Immunol 2013;174:372–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lan P, Wang L, Diouf B, et al. Induction of human T-cell tolerance to porcine xenoantigens through mixed hematopoietic chimerism. Blood 2004;103:3964–3969 [DOI] [PubMed] [Google Scholar]
- 51.Melkus MW, Estes JD, Padgett-Thomas A, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med 2006;12:1316–1322 [DOI] [PubMed] [Google Scholar]
-
52.Ito M, Hiramatsu H, Kobayashi K, et al.
NOD/SCID/
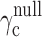 mouse: an excellent recipient mouse model for engraftment of human cells. Blood
2002;100:3175–3182 [DOI] [PubMed] [Google Scholar]
mouse: an excellent recipient mouse model for engraftment of human cells. Blood
2002;100:3175–3182 [DOI] [PubMed] [Google Scholar] - 53.Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2Rγ null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 2005;174:6477–6489 [DOI] [PubMed] [Google Scholar]
- 54.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by γc family cytokines. Nat Rev Immunol 2009;9:480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Traggiai E, Chicha L, Mazzucchelli L, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science 2004;304:104–107 [DOI] [PubMed] [Google Scholar]
- 56.Goyama S, Wunderlich M, Mulloy JC. Xenograft models for normal and malignant stem cells. Blood 2015;125:2630–2640 [DOI] [PubMed] [Google Scholar]
- 57.Brehm MA, Cuthbert A, Yang C, et al. Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the IL2rγnull mutation. Clin Immunol 2010;135:84–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lepus CM, Gibson TF, Gerber SA, et al. Comparison of human fetal liver, umbilical cord blood, and adult blood hematopoietic stem cell engraftment in NOD-scid/γc–/–, Balb/c-Rag2–/–γc–/–, and C.B-17-scid/bg immunodeficient mice. Hum Immunol 2009;70:790–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDermott SP, Eppert K, Lechman ER, et al. Comparison of human cord blood engraftment between immunocompromised mouse strains. Blood 2010;116:193–200 [DOI] [PubMed] [Google Scholar]
- 60.Takenaka K, Prasolava TK, Wang JCY, et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol 2007;8:1313–1323 [DOI] [PubMed] [Google Scholar]
- 61.Stoddart CA, Maidji E, Galkina SA, et al. Superior human leukocyte reconstitution and susceptibility to vaginal HIV transmission in humanized NOD-scid IL-2Rγ–/– (NSG) BLT mice. Virology 2011;417:154–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berges BK, Rowan MR. The utility of the new generation of humanized mice to study HIV-1 infection: transmission, prevention, pathogenesis, and treatment. Retrovirology 2011;8:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Biswas S, Chang H, Sarkis PT, et al. Humoral immune responses in humanized BLT mice immunized with West Nile virus and HIV-1 envelope proteins are largely mediated via human CD5+ B cells. Immunology 2011;134:419–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akkina R. Human immune responses and potential for vaccine assessment in humanized mice. Curr Opin Immunol 2013;25:403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brehm MA, Wiles MV, Greiner DL, et al. Generation of improved humanized mouse models for human infectious diseases. J Immunol Methods 2014;410:3–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ernst W. Humanized mice in infectious diseases. Comp Immunol Microbiol Infect Dis 2016;49:29–38 [DOI] [PubMed] [Google Scholar]
- 67.Ito R, Takahashi T, Katano I, et al. Current advances in humanized mouse models. Cell Mol Immunol 2012;9:208–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kalscheuer H, Danzl N, Onoe T, et al. A model for personalized in vivo analysis of human immune responsiveness. Sci Transl Med 2012;4:125ra130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bird BH, Spengler JR, Chakrabarti AK, et al. Humanized mouse model of Ebola virus disease mimics the immune responses in human disease. J Infect Dis 2016;213:703–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Onoe T, Kalscheuer H, Danzl N, et al. Human natural regulatory T cell development, suppressive function, and postthymic maturation in a humanized mouse model. J Immunol 2011;187:3895–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hahn SA, Bellinghausen I, Trinschek B, et al. Translating Treg therapy in humanized mice. Front Immunol 2015;6:623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Long SA, Rieck M, Sanda S, et al. Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs beta-cell function. Diabetes 2012;61:2340–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brehm MA, Shultz LD, Luban J, et al. Overcoming current limitations in humanized mouse research. J Infect Dis 2013;208(Suppl 2):S125–S130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Katano I, Takahashi T, Ito R, et al. Predominant development of mature and functional human NK cells in a novel human IL-2-producing transgenic NOG mouse. J Immunol 2015;194:3513–3525 [DOI] [PubMed] [Google Scholar]
- 75.Ito R, Katano I, Kawai K, et al. A novel xenogeneic graft-versus-host disease model for investigating the pathological role of human CD4+ or CD8+ T cells using immunodeficient NOG mice. Am J Transplant 2017;17:1216–1228 [DOI] [PubMed] [Google Scholar]
- 76.Abraham S, Pahwa R, Ye C, et al. Long-term engraftment of human natural T regulatory cells in NOD/SCID IL2rγcnull mice by expression of human IL-2. PLoS One 2012;7:e51832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Klevorn LE, Berrien-Elliott MM, Yuan J, et al. Rescue of tolerant CD8+ T cells during cancer immunotherapy with IL2:antibody complexes. Cancer Immunol Res 2016;4:1016–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Minami Y, Kono T, Miyazaki T, et al. The IL-2 receptor complex: its structure, function, and target genes. Annu Rev Immunol 1993;11:245–268 [DOI] [PubMed] [Google Scholar]
- 79.Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol 2012;12:180–190 [DOI] [PubMed] [Google Scholar]
- 80.Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 2013;38:13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hirakawa M, Matos T, Liu H, et al. Low-dose IL-2 selectively activates subsets of CD4+ Tregs and NK cells. JCI Insight 2016;1:e89278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim M, Kim TJ, Kim HM, et al. Multi-cellular natural killer (NK) cell clusters enhance NK cell activation through localizing IL-2 within the cluster. Sci Rep 2017;7:40623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boyman O, Kovar M, Rubinstein MP, et al. Selective stimulation of T cell subsets with antibody–cytokine immune complexes. Science 2006;311:1924–1927 [DOI] [PubMed] [Google Scholar]
- 84.Levin AM, Bates DL, Ring AM, et al. Exploiting a natural conformational switch to engineer an interleukin-2 “superkine.” Nature 2012;484:529–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Seok J, Warren HS, Cuenca AG, et al. ; Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 2013;110:3507–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol 2004;172:2731–2738 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.