

Abstract
Mass spectrometers equipped with ion trap analyzers have been significantly improved due to their high performance and wide application area accompanying the low costs of purchase. Despite several advantages, such as reasonable resolution at low cost, high sensitivity, and capability for multistage analysis, ion traps have an important drawback: low mass cutoff during tandem mass spectrometry analysis MSn. Although the low mass cutoff associated with the ion trap does not seriously obstruct peptide identification, it may cause a serious problem in identification of small molecules (posttranslational modifications, e.g., glycan structures) and quantification of peptides with multiplexed isobaric tag reagents. The presented approach offers the possibility to use isobaric tags for relative and absolute quantification labeling (iTRAQ) for quantitative, proteomic analysis using typical, widely available ion trap devices and manufacturer's software. We have performed series of analyses of standard protein labeled with isobaric tags in various concentration ratios to prove quantitative capabilities of this approach.
Keywords: iTRAQ, ion trap, quantitative proteomics
Graphical abstract
Introduction
The isobaric tags for relative and absolute quantification (iTRAQ) are a popular quantitation method based on the labeling of peptides with a compound that easily produces fragments, so-called reporter ions, under MS/MS conditions (Figure 1). iTRAQ technology utilizes an ester derivative to modify primary amino group by linking an isobaric tag to peptides via amide bond.1 During fragmentation of tagged peptides, the mass balancing carbonyl group is released as a neutral fragment, thus releasing the reporter ions that provide relative quantitative information on proteins.
Figure 1.
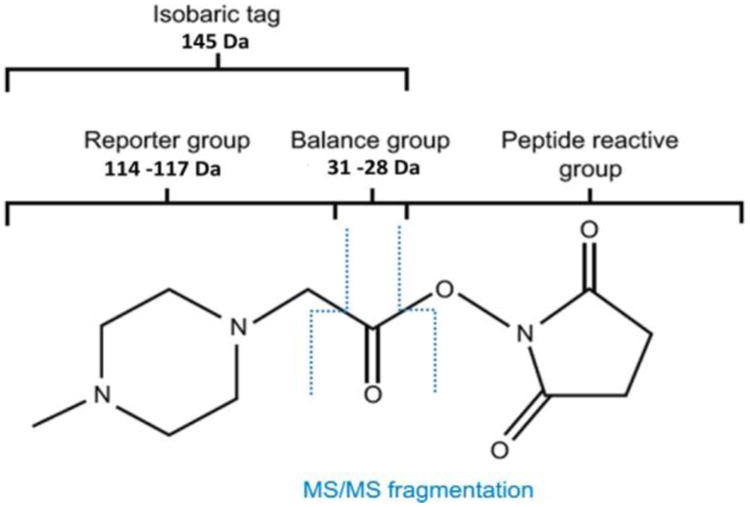
Isobaric tags for relative and absolute quantification with iTRAQ.
In the case of a 4-plex, the reporter tags possess masses between 114 and 117 Da depending on various isotopic combinations 12C/13C, 14N/15N, and 16O/18O in each individual reagent. The balance groups also differ in mass between 28 and 31 Da to ensure that isobaric tag mass remains constant and equal to 145 Da. This warrants the indistinguishably labeled peptides that elute at the same retention times during chromatographic separation and are simultaneously present on the same fragmentation spectrum. Originally, the iTRAQ approach was designed for analysis of two distinct biological samples using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF/TOF) instruments; however, it is now equally used by quadrupole-based analyzers for up to eight conditions in a single experiment, so-called 8plexes.2
The proteomic workflow for quantitation by iTRAQ usually is done in a few simple steps. The most important is that peptides from different experimental groups are labeled and pooled together prior to enrichment or fractionation, which ensures that every step of the sample preparation procedure is equal in all combined samples. To date, the major obstacle of iTRAQ method is the inability to perform experiments using ion trap instruments that are popular MS devices not only in proteomic laboratories.
The majority of difficulties associated with ion trap analyzers, such as resolution and low mass range, have been solved along with the rapid technological development in the mass spectrometry industry. Only one problem remains an obstacle: the simultaneous analysis of complex peptide mixtures with their isobaric quantitation tags. This is because of the low mass cutoff, which usually limits the MS/MS spectra range up to 30% of its precursor m/z. This phenomenon is a physical limitation of the ion traps. Isobaric tags used for peptide quantitation are usually absent on the MS/MS spectra. During collision-induced dissociation (CID), iTRAQ labels absorb most of kinetic energy, hence increasing the probability for incomplete fragmentation and resulting in poor MS/MS spectra and a subsequent low confidence in peptide assignment.
Several approaches were used to address this issue as different fragmentation methods have been developed, including Pulsed-Q dissociation (PQD),3 high-energy C-trap dissociation (HCD),4 electron transfer dissociation (ETD),5 and MS3 procedure6 to facilitate the quantification of low mass reporter ions.
Here we present a novel solution to eliminate low mass cutoff in the Paul's ion traps as a straightforward and highly effective alternative to the possibilities listed above. This approach provides an easy solution by modifying settings for fragmentation, acquisition, and deconvolution parameters specifically used for iTRAQ labeled peptides.
Experimental Section
To establish optimal instrumental conditions, we used bovine serum albumin (BSA) as a reference protein, which was enzymatically cleaved, and finally treated with iTRAQ reagents, giving reporter ions of 1141+ and 1171+ m/z (AB Sciex, USA), according to the manufacturer's protocol. Two samples of 100 μg BSA were pretreated with dissolution buffer in the presence of sodium dodecyl sulfate (SDS), followed by reduction and cysteine residues blocking by interaction with tris-(2-carboxyethyl)phosphine (TCEP) and methyl methanethiosul-fonate (MMTS). All reagents were provided in the manufacturer's kit. Samples were submitted to tryptic digestion (Trypsin-Gold, Mass Spectrometry grade, Promega, USA) and labeled with iTRAQ reagents in the presence of 75% ethanol. Strong cation exchange spin cartridges (SCX, Thermo Fisher Scientific, USA) were used for samples desalting according to the standard protocol utilizing loading/washing buffer: 25% acetonitrile (ACN), 0.1% formic acid (FA) in water, and eluting buffer: 25% ACN, 0.1% FA, 400 mM NH4HCO3 in water. Purified peptides were freeze-dried (CentriVap, Labconco, USA).
After dissolution in 2% ACN supplemented with 0.1% FA, the samples were mixed at various ratios 1:1, 1:1.5, 1:5, 5:1, and 9:1 (reporter ions 114:117), and 50 fmol of each were separated using the Proxeon nanoLC system (Thermo Fisher Scientific, Vienna, Austria) and directly identified on line using the AmaZon ETD (Bruker-Daltonics, Bremen, Germany) mass spectrometer. For the separation purposes, the RP C18 column was used (homemade 75 μm I.D./10 cm capillary column filled with C18 SupelcoSil 5 μm beads). A shorter guard column was used (homemade 100 μm I.D./1 cm capillary column filled with C18 SupelcoSil 5 μm beads). Other separation parameters were as follows; solvent A: water with 0.1% FA; solvent B: 100% ACN with 0.1% FA. Flow rate was set to 300 nL/min. Gradient conditions: 0−20% B in 50 min; 20−70% B for 1 min; plateau 70% B for 4 min; 70−0% B for 3 min; and re-equilibration of the column with 100% A for 2 min. All remaining parameters (precolumn and analytical column washing, sample injection, etc.) were set according to standard Proxeon manufacturer's protocols to prevent any carryover.
MS scan range in the CID positive ion mode was adjusted to 230−800 m/z and 90−610 m/z in the case of MS/MS spectra. Target mass optimization for MS analysis was set to 500 m/z. Peak threshold intensity selected for data-dependent fragmentation procedure was set to 2 500 000. Additionally, there was also a limit of the m/z values of the ions fragmented, which was set to the value up to 610 m/z to allow fragmentation generating iTRAQ reporter ions. The number of precursor ions simultaneously selected for fragmentation was limited to four. Critical for the experimental design, 20 MS(n) averages were collected in the ultrascan mode. After this procedure, the next ion was taken into fragmentation. The isotope distribution of each ion was excluded due to the SmartScan procedure, which eliminates signals belonging to the same precursor ion. Such option saves time and eliminates space on the disc; however, after 30 s this option was reset to allow other ions of same m/z (if any) to be fragmented. Strict active exclusion was turned on, with the group of three following isotopic peaks belonging to the peak undergoing fragmentation. Additionally, the preferred charge state for isolation was set to “double”. Importantly, MS/MS fragmentation amplitude was optimized as 1.15 V with 17% CID cutoff in the so-called panorama fragmentation mode (PAN). Fragmentation procedure was supported by the smart fragmentation mode with the CID energy amplitude in a range from 60 to 180% of the fragmentation energy set. Tickle level was tuned to 120%.
Although “panorama fragmentation” mode, which expands ion trap capabilities to trap the fragment ions down to ca. 17% of the parent ion m/z, is a property of the Bruker's ion traps, we believe that methodology proposed by us can be easily adopted for other ion traps offered on the market. For the Thermo LTQ instruments, the method of iTRAQ analysis with peptide identification based on MS/MS scan and quantification of the tag in a subsequent MS3 scan has been utilized using PQD.7
The new method of deconvolution of mass spectra was necessary to be developed to achieve the accurate results for further database investigations. We have increased the parameters representing the number of the most abundant, nondeconvoluted ions exported to mgf file up to 200 and altered the mass range of ions submitted to deconvolution above the 150 m/z to prevent the assignment of charge state to the reporter ions. For the standard deconvolution method, the information about iTRAQ reporter ions would be lost and made the quantification impossible.
Furthermore, the quantitation method in the Mascot search engine needed adjustment because there is only 4-plex and 8-plex iTRAQ approach available. We have established a 2-plex iTRAQ quantitation method using weighted standard deviation statistical method. In this method, the intensities of the set of peptides for each identified protein are added, and the protein ratio is calculated from the summed values. Additionally, the variable modifications of methionine dioxidation and S-methylthiocysteine were applied. Data were searched against the Swiss-Prot database (ver. 57.15; 515 203 sequences, 181 334 896 residues), and the peptide tolerance was set to 1.2 Da and MS/MS tolerance was set to 0.6 Da. Peptide charge was selected as +1; +2, and +3. Taxonomy was limited to mammals. The instrument was described as an ion trap.
Results and Discussion
Using the described approach, we were able to monitor low m/z fragments and obtain quantitative data for iTRAQ reporter ions 114 and 117 (Figure 2). Because reporter ions absorb significant amount of the collision energy, for more informative spectra, we adjusted collision energy to 1.15 V and prolonged the time to collect MS/MS spectra to 20 averages (instead of usually used 2−4) to provide more stable ratios between reporter ions.
Figure 2.
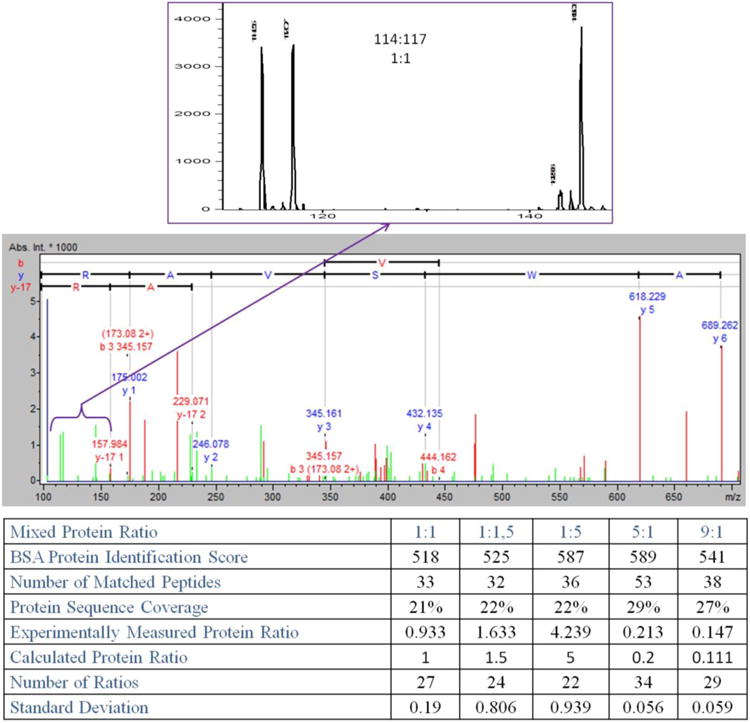
BSA tryptic digests labeled with 114 and 117 isobaric tags samples and mixed in quantity ratio 1:1. All of the remaining ratios were summarized in the table. BSA protein identification score was determined using Mascot standard scoring. The number of matched peptides included redundancy of species to enhance the quantitation statistics. Protein ratio value was calculated based on weighted average for all MS/MS spectra acquired with complete set of reporter ions. Standard deviation was considered for all measured tags ratios. Number of ratios represents the total quantity of MS/MS spectra utilized to estimate the protein ratio. Protein sequence coverage was established based on all of the identified peptides.
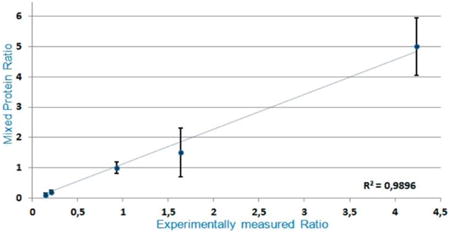
Quantitative analysis of the BSA samples was performed for all of the investigated ratios (Figure 2). The standard deviation for little differences in relative amounts between reporter ions was small and was increasing along with the ratio intervals. However, the total variation of the presented technique calculated as a linear regression coefficient achieved the value of R2 = 0.9896, providing evidence of its accuracy and precision. All of the appointed ratios analyzed by the proposed approach were statistically significant.
The results of our experiments show the capability of ion traps for quantitative analyses of the samples labeled by iTRAQ methodology. The following rules must be applied for successful analysis.
Collision energy must be optimized to achieve good-quality MS/MS spectra along with the clearly visible reporter ions. In general, this parameter should be increased as compared with standard settings, although the final value should not exceed 20% of the regular settings to maintain qualitative and quantitative information of the acquired spectra. CID energy needed for the reporter ions dissociation from the peptide ion structure was tested before analysis. We checked that reporter ions were fully dissociated from the peptide structure at much lower CID energy than that used for peptide fragmentation during LC-MS/MS analyses. This observation suggests that iTRAQ cluster formation during CID procedure is effective enough for quantitative detection.
Samples must be processed by the enzyme capable of cleaving the protein to short peptides (three to seven amino acids). Longer peptides (above seven amino acids) are not suitable for iTRAQ analysis in the ion trap instrument. They might be overlooked while isolating precursor ions when their charge is insufficient for the narrow fragmentation window. Even if their charge is high enough, their fragment ions would be multiply charged, which causes problems during deconvolution procedure. In summary, we recommend using enzymes like trypsin or pepsin. The first one generates 39 fragments between 3 and 10 amino acids, suitable for proposed analysis. Those fragments cover ca. 50% of the whole albumin sequence, which is more than satisfactory for the successful identification and quantification. Using enzymes like Arg-C or Asp-N will cause the generation of longer sequences which, in effect, may result in unsuccessful analysis.
Quantity of the MS/MS spectra averaging must be carefully optimized. Too low number of the MS/MS spectra averages will result in unstable signal (intensity) of the iTRAQ reporter ions. It is widely known that ion traps are not the best quantification analyzers but averaging more spectra in a single MS/MS experiment results in much better, more repeatable data. On the other side, the extended time for spectra acquisition and averaging will cause loss of data from another, closely eluted peptide emerging from the liquid chromatography (LC) column. In our experiments, we set a number of acquired and averaged MS/MS spectra from a single parent ion to 20, which allowed us to collect quantitative information. Averaging only three to five MS/MS spectra, typically used for identification of proteins during LC-MS/MS analyses, leads to the loss of quantitative information. More than 25−40 averaged MS/MS spectra did not significantly improve the results, causing unwanted delays of the next spectra acquisition and a loss of adjacent peptides. This resulted in a poor-quality identification of the protein (low Mascot scores) with quantitative information at a quality level similar to that received with smaller number of the averaged MS/MS spectra. To avoid loss of fragmentation of closely eluting peptides, we recommend the application of slightly longer LC gradients than usually applied. It minimizes peaks overlapping on the LC column and provides additional time for the mass spectrometer for successful fragmentation. Additionally, careful optimization of the fragmentation threshold will contribute to the high-quality MS/MS spectra from the high-intensity parent ions.
Deconvolution process must be carefully prepared to avoid charge assignment to reporter ions. Therefore, m/z range from 100 to 120 should definitely be excluded. Otherwise, reporter ions with assigned charges will equally contribute to Mascot identification of the protein instead of providing quantitative information.
It is widely known that resolution of the ion traps is inversely proportional to the scanning speed. This parameter must also be optimized to gain proper MS/MS scan quality. The instrument should not be set to its maximal resolution because it causes significant delays in MS/MS spectra acquisition and formation of the “pseudo” isotopic pattern for iTRAQ reporter ions. This phenomenon causes incorrect iTRAQ tags ratio quantitation due to the fact that the total intensity of the reporter ion will be divided between two neighboring “pseudo” isotopic peaks. Resolution must be sufficient to provide good-quality spectra for peptide identification. The easiest way to overcome this contradiction is to cleave protein with the aid of an enzyme releasing short peptide sequences. Such approach allows for gaining relatively low resolution, fast scanning speed, good sequence coverage, reliable deconvolution, and successful identification along with good quantification of the identified peptides.
We have also tested this approach to more complex samples like human serum. Analyzed serum samples were immunodepleted (HSA and IgG) and enriched with the use of lectin affinity chromatography in glycoproteins fraction. Proteins were treated by reducing reagent, cysteine blocking agent, and finally trypsin. Obtained peptides were analyzed by the mass spectrometer with Paul ion-trap analyzer using two quantitation approaches: iTRAQ labeling and label-free methodology. We have identified over 3200 proteins using label-free technique and 2464 proteins based on iTRAQ method; furthermore, we were able to quantify in a statistically valid manner more than 300 proteins by the label-free approach and 147 glycoproteins by means of iTRAQ approach. This phenomenon is due to the fact that iTRAQ quantitation method requires the elongated collection of MS/MS spectra, compared with the label-free approach. Therefore, during peptides elution from capillary column, part of the information is lost, especially about less abundant mixture components. The label-free method involves two subsequent analyses; consequently, the entire time of examination is two-fold extended. The number of quantified peptides and final point proteins is strongly dependent on the data processing method. Therefore, selecting a very strict threshold for ratio calculation causes the reduction of the high-quality amount of MS/MS spectra. One has to remember to use only reliable peptide matches to calculate protein ratios for the reason that if the peptide sequence is not reliable then the reporter ions cannot be properly assigned itself. Hence, the smaller number of quantified proteins is observed compared with the identified proteins.
The comparison of iTRAQ and label-free quantitation methods using Paul ion trap mass spectrometry was presented during 19th International Mass Spectrometry Conference.8
Conclusions
We demonstrated the capability of the ion trap instruments to quantitatively measure iTRAQ-labeled samples for accurate determination of protein concentration. The averaging process of collected MS/MS spectra significantly increases the accuracy of quantitation. Also, optimizing collision energy is an important factor contributing to quantitation. A compromise between the quantitative analysis of reporter ions and qualitative peptide sequence identification is mandatory. The presented experiments straddling the 1−9 fold ratio suggest that sensitivity and precision of the proposed approach are compared with the other previously described approaches for quantification based on iTRAQ labeling. Our strategy for the study of iTRAQ labeling experimental design was proven to be a valuable proteomic tool.
Acknowledgments
This project was supported by the grant EuroNanoMed “META” No. 5/EuroNanoMed/2012, and the grant from The Polish National Science Center 3744/B/H03/2011/40.
Abbrevations
- iTRAQ
isobaric tags for relative and absolute quantification
- MALDI-TOF/TOF
matrix-assisted laser desorption/ionization-time-of-flight
- CID
collision-induced dissociation
- PQD
pulsed-q dissociation
- HCD
high-energy c-trap dissociation
- ETD
electron transfer dissociation
- BSA
bovine serum albumin
- SDS
sodium dodecyl sulfate
- TCEP
tris-(2-carboxyethyl)phosphine
- MMTS
methyl methanethiosulfonate
- SCX
strong cation exchange
- ACN
acetonitrile
- FA
formic acid
- PAN
panorama fragmentation mode
- LC
liquid chromatography
Footnotes
Disclosure: The authors declare no competing financial interest.
References
- 1.Wiese S, Reidegeld KA, Meyer HE, Warscheid B. Protein labeling by iTRAQ: a new tool for quantitative mass spectrometry in proteome research. Proteomics. 2007;7:340–350. doi: 10.1002/pmic.200600422. [DOI] [PubMed] [Google Scholar]
- 2.Pottiez G, Wiederin J, Fox H, Ciborowski PJ. Comparison of 4-plex to 8-plex iTRAQ quantitative measurements of proteins in human plasma samples. Proteome Res. 2012;11:3774–3781. doi: 10.1021/pr300414z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L, Jiang J, Arellano M, Zhang L, Yan X, Wong DT, Hu S. Quantification of Serum Proteins of Metastatic Oral Cancer Patients Using LC-MS/MS and iTRAQ Labeling. Open Proteomics J. 2008;1:72–78. doi: 10.2174/1875039700801010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McAlister GC, Phanstiel DH, Brumbaugh J, Westphall MS, Coon JJ. Higher-energy collision-activated dissociation without a dedicated collision cell. Mol Cell Proteomics. 2011;10:1–6. doi: 10.1074/mcp.O111.009456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mischerikow N, van Nierop P, Li KW, Bernstein HG, Smit AB, Heck AJ, Altelaar AF. Gaining efficiency by parallel quantification and identification of iTRAQ-labeled peptides using HCD and decision tree guided CID/ETD on an LTQ Orbitrap. Analyst. 2010;135:2643–2652. doi: 10.1039/c0an00267d. [DOI] [PubMed] [Google Scholar]
- 6.Yang YH, Lee K, Jang KS, Kim YG, Park SH, Lee CS, Kim BG. Low mass cutoff evasion with q(z) value optimization in ion trap. Anal Biochem. 2009;387:133–135. doi: 10.1016/j.ab.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 7.Griffin TJ, Xie H, Bandhakavi S, Popko J, Mohan A, Carlis JV, Higgins L. iTRAQ reagent-based quantitative proteomic analysis on a linear ion trap mass spectrometer. J Proteome Res. 2007;11:4200–4209. doi: 10.1021/pr070291b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drabik A, Suder P, SierzĀ˙ga M, Pach R, Kulig J, Bodzonń-Kułakowska A, Silberring J. Ion-trap quantitative proteomics of glycoproteome from pancreatic cancer patients based on the label-free and iTRAQ approaches. IMSC2012 Kyoto 19th International Mass Spectrometry Conference; Kyoto. 15th September − 21st September 2012; abstracts. [Google Scholar]