

Abstract
Primary familial and congenital polycythemia is characterized by erythropoietin hypersensitivity of erythroid progenitors due to germline nonsense or frameshift mutations in the erythropoietin receptor gene. All mutations so far described lead to the truncation of the C-terminal receptor sequence that contains negative regulatory domains. Their removal is presented as sufficient to cause the erythropoietin hypersensitivity phenotype. Here we provide evidence for a new mechanism whereby the presence of novel sequences generated by frameshift mutations is required for the phenotype rather than just extensive truncation resulting from nonsense mutations. We show that the erythropoietin hypersensitivity induced by a new erythropoietin receptor mutant, p.Gln434Profs*11, could not be explained by the loss of negative signaling and of the internalization domains, but rather by the appearance of a new C-terminal tail. The latter, by increasing erythropoietin receptor dimerization, stability and cell-surface localization, causes pre-activation of erythropoietin receptor and JAK2, constitutive signaling and hypersensitivity to erythropoietin. Similar results were obtained with another mutant, p.Pro438Metfs*6, which shares the same last five amino acid residues (MDTVP) with erythropoietin receptor p.Gln434Profs*11, confirming the involvement of the new peptide sequence in the erythropoietin hypersensitivity phenotype. These results suggest a new mechanism that might be common to erythropoietin receptor frameshift mutations. In summary, we show that primary familial and congenital polycythemia is more complex than expected since distinct mechanisms are involved in the erythropoietin hypersensitivity phenotype, according to the type of erythropoietin receptor mutation.
Introduction
Primary erythrocytosis (also known as primary familial and congenital polycythemia, PFCP) is a pathology of erythroid progenitors, which display hypersensitivity to erythropoietin.1–14 This rare inherited entity is usually passed down with an autosomal dominant pattern with complete penetrance.2–6,8–14 PFCP is characterized by an isolated primary polycythemia in which an increased red cell mass is associated with subnormal serum erythropoietin levels. It can therefore mimic the clinical presentation of polycythemia vera. However, the hematopoiesis is polyclonal and PFCP patients do not show any risk of their disease transforming into myelofibrosis or leukemia. Moreover, somatic JAK2V617F15 and JAK2 exon1216 mutations, hallmarks of polycythemia vera, are not found in PFCP,12,13 which is, however, characterized by the presence of germline mutations in the erythropoietin receptor gene (EPOR).
Erythropoietin receptor (EPOR) is a type I cytokine receptor,17 essentially expressed by erythroid progenitors in hematopoietic tissue. The binding of erythropoietin to its receptor at the cell surface leads to the transient activation of preformed EPOR-JAK2 complexes18,19 and downstream signaling pathways, including signal transducer and activators of transcription (STAT),20 phosphatidylinositol 3 kinase/Akt21 and mitogen-activated protein kinase pathways. Erythropoietin-induced signaling is crucial for the proliferation and the survival of erythroid progenitors as well as for terminal erythroid differentiation.22
Around 20 germline heterozygous nonsense and frameshift mutations located in exon 8 of EPOR have been described so far in PFCP, all leading to the truncation of the C-terminal part of the receptor.3–11,13,14,23–28 Interestingly, similar EPOR truncations have been described in BCR-ABL1-like acute lymphoblastic leukemia, due to acquired rearrangements of EPOR with immunoglobulin chain loci.29 The C-terminal part of the receptor includes several conserved tyrosine residues that are docking sites for positive and negative regulators of EPOR signaling. The erythropoietin hypersensitivity of PFCP progenitors is, therefore, usually explained by the disappearance of negative regulatory domains located in the truncated part of the receptor.30,31 A few missense EPOR mutations have also been described, but their involvement in the PFCP phenotype is not yet clear.4,7,13,26–28,32 Previous studies have suggested that truncated EPOR mutations might not be equivalent in term of underlying mechanisms leading to EPOR activation,14,27 but relatively few functional studies have been carried out. We, therefore, investigated the mechanism of some EPOR mutations in PFCP.
We identified and extensively studied a new germline frameshift EPOR mutation, c.1300dup (p.Gln434Profs*11), responsible for marked erythropoietin hypersensitivity as in JAK2V617F-positive polycythemia vera. We modeled EPOR p.Gln434Profs*11 and several other EPOR mutants already described in the Ba/F3 and UT-7 cell line and demonstrated that different mechanisms are involved in the erythropoietin hypersensitivity phenotype, according to the type of EPOR mutation, highlighting that PFCP is a more complex pathology than usually considered.
Methods
Materials
Human recombinant erythropoietin and interleukin-3 were generous gifts from Amgen (Neuilly, France). Stem cell factor was purchased from Biovitrum AB (Stockholm, Sweden).
Patients, cell purification and erythroblast cultures
Peripheral blood samples from the patient or healthy donors were collected by leukapheresis. Written informed consent was obtained from the patient in accordance with the Declaration of Helsinki and the study was approved by the ethics committee of La Pitié-Salpétrière Hospital. Mononuclear cells were separated over a Ficoll density gradient and CD34+ cells were purified by a double-positive magnetic cell sorting system (AutoMACS; Miltenyi Biotec, Paris, France). CD34+ cells were amplified in erythroid conditions for 7-10 days in Iscove modified Dulbecco medium with penicillin/streptomycin/glutamine, alpha-thioglycerol, bovine serum albumin, a mixture of sonicated lipids and insulin-transferrin in the presence of recombinant human cytokines (25 ng/mL stem cell factor, 100 U/mL interleukin-3, 1 U/mL erythropoietin).
EPOR sequencing
Genomic DNA was extracted from blood, nails and hair using standard procedures. Exon 8 of EPOR was amplified and sequenced in both directions. Mutations are numbered as recommended by the Human Genome Variation Society (http://www.hgvs.org/) using the reference sequence NM_000121.3. Participants gave written informed consent to the genetic study.
Quantification of clonogenic progenitors in semi-solid cultures
Colony assays were performed with 500 purified CD34+ progenitors per culture dish in duplicate in H4100 Methocult media (StemCell Technologies, Grenoble, France) supplemented with 12% bovine serum albumin, 30% or no fetal bovine serum, 2-β-mercaptoethanol (1 mM), 1% L-glutamine, stem cell factor (25 ng/mL), interleukin-3 (100 U/mL) and in the absence or presence of increasing concentrations of erythropoietin (0.001; 0.01; 0.1 and 1 U/mL). Burst-forming units-erythroid (BFU-E)-derived colonies were counted on day 14.
DNA manipulation and retrovirus production
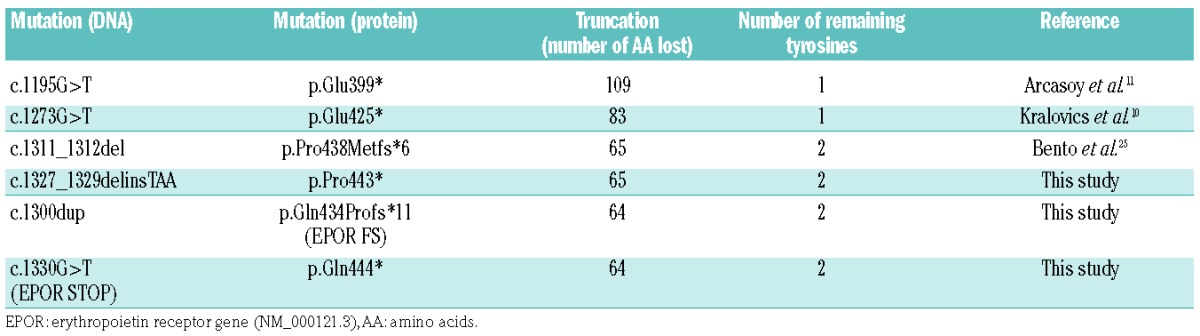
EPOR mutations were introduced into the pMX-HA-human EPOR WT-IRES-GFP plasmid by the QuikChange site-directed mutagenesis method using the PfuUltra high-fidelity DNA polymerase (Agilent Technologies, Stratagene, Les Ulis, France): c.1300dup (p.Gln434Profs*11, EPOR FS); c.1330G>T (p.Gln444*, EPOR STOP); c.1303_1304delinsGC (p.Leu435Ala, EPOR WTdiL or EPOR STOPdiL); c.1195G>T (p.Glu399*); c.1273G>T (p.Glu425*); c.1311_1312del (p.Pro438Metfs*6); c.1327_1329delinsTAA (p.Pro443*); c.[1300dup; 1311G>C] (p.[Gln434Profs*11; Ala437Arg]) (Table 1). Full-length EPOR mutant cDNA was verified by sequencing.
Table 1.
EPOR mutations investigated in this study and their functional consequences.
Cell lines
The murine Ba/F3 and human UT-7 cells were grown in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (StemCell Technologies, Grenoble, France) and 5% WEHI-conditioned medium as a source of murine interleukin-3 or granulocyte-macrophage colony-stimulating factor (10 ng/mL). Cells were transduced with pMX-IRES-GFP retrovirus to stably express the human wild-type EPOR or EPOR mutants, carrying a N-terminal HA-tag. GFP+ cells were subsequently sorted by flow cytometry (Influx, Beckton-Dickinson, Le Pont-de-claix, France).
Proliferation assays
The premixed WST-1 cell proliferation assay was carried out according to the manufacturer’s instructions (Takara Bio Europe, Clontech, Saint-Germain-en-Laye, France). Experiments were performed in triplicate. Dose-response curves to erythropoietin were expressed as percentages of viability of the maximal response.
Western blot analysis
For signaling studies, cells were starved for 5 h and then incubated with increasing concentrations of erythropoietin for 15 min or stimulated with 1 U/mL erythropoietin for 15 min and washed three times in phosphate-buffered saline 1X to remove the cytokine.
Signaling studies were performed using polyclonal antibodies against the phosphorylated forms of JAK2 (Tyr 1007/1008), STAT5 (Tyr 694), ERK1/2 (Thr 202/Tyr 204) and Akt (Ser 473) and against the pan proteins (Cell Signaling Technology, Ozyme, Montigny -le-Bretonneux, France). β-actin (Sigma, Saint Quentin-Fallaviers) was used as a loading control.
For glycosidase digestion, cell lysates were incubated with either endoglycosidase H (Endo H) or peptide:N-glycosidase F (PNGase F) (1,000 U) at 37°C for 16 h according to the recommendations of the supplier (New England Biolabs, Evry, France).
Dimerization of human erythropoietin receptor monomers assessed by split Gaussia luciferase assay
A split Gaussia subunit, Gluc1 or Gluc2, was fused to the C-terminus of each EPOR construct.33 When an EPOR monomer with a Gluc1 fusion subunit dimerizes with another EPOR monomer with a Gluc2 fusion subunit, the two Gluc subunit proteins recombine into a catalytically active luciferase that is able to degrade coelenterazine, thus emitting light. Both EPOR constructs fused to a C-terminal Gluc1 and a Gluc2 subunit were transiently transfected at a 50:50 ratio into HEK cells using Transit-LT1 (Mirus, Euromedex, Souffelweyersheim, France) as a transfecting agent. The pGL3-control vector (Promega, Charbonnières-les-Bains, France) that constitutively expresses the firefly luciferase was co-transfected in each condition as a transfection control reporter. After 48 h, luciferase signals were read by a GloMax discovery system (Promega) after addition of coelenterazine and firefly luciferin to each well. A 530LP filter was used to discriminate the luminescence of the firefly luciferase from that of the Gaussia luciferase.
Erythropoietin receptor stability assay
Ba/F3 cells were incubated for different periods of time with 50 ng/mL cycloheximide (Sigma). After cell lysis, western blotting was performed with anti-HA antibody. HA-tagged EPOR was quantified with Image J software, using β-actin as a loading control. The half-life of the receptor was calculated using GraphPad PRISM software.
Cell-surface localization of HA-tagged erythropoietin receptor by flow cytometry
Cells were labeled with monoclonal mouse anti-HA antibody conjugated to phycoerythrin (Miltenyi Biotecs) and processed by flow cytometry (FACSCanto, Beckton-Dickinson, Le Pont-de-claix, France).
Erythropoietin labeling and binding and erythropoietin receptor internalization studies
Erythropoietin labeling using IODO-GEN (Pierce, Rockford, IL, USA), erythropoietin binding and EPOR internalization studies were performed as previously described.34–36 Nonspecific binding was determined using a 250-fold excess of unlabeled erythropoietin and was less than 5% in each case. All reported data represent specific binding. The number of cell-surface receptors was determined by comparing the radioactivity of Ba/F3-EPOR cells to the radioactivity of the reference UT-7 cell line.34 For internalization experiments, after incubation with 125I-erythropoietin, 5 × 106 cells per condition were washed twice at 4°C to remove unbound ligand. An acidic wash was then performed to separate cell surface–bound from internalized erythropoietin. Cells were incubated in 0.5 mL acidic buffer (150 mM NaCl, 50 mM sodium acetate, pH 3.5) for 3 min at 4°C. The pH was then adjusted to 7.4 using 1 M Tris-HCl, pH 9 and the cell suspension was centrifuged. The radioactivity of the supernatant (cell surface–bound erythropoietin) and of the cell pellet (internalized erythropoietin) was determined. When 125I-erythropoietin was bound to the cells at 4°C to inhibit erythropoietin internalization, more than 95% of cell-bound 125I-erythropoietin was recovered in the acidic wash supernatant using this method. Each experiment was performed three times with similar results.
Results
Identification of a new germline EPOR mutation responsible for marked erythropoietin hypersensitivity
Primary polycythemia was diagnosed in a 28-year old woman without a history of thrombosis. She had high hemoglobin concentration (21 g/dL) and hematocrit (60%) and an increased red cell mass (65%) with a low erythropoietin level (1.2 mU/mL; laboratory standard: 5-25 mU/mL). She had no splenomegaly at physical examination. Leukocyte and platelet counts in the peripheral blood as well as bone marrow aspiration and biopsy were strictly normal. Later no JAK2V617F or JAK2 exon12 mutation was found, rendering the diagnosis of polycythemia vera unlikely. The search for abnormal hemoglobin affinity and for VHL, PHD1/2, SH2B3 (LNK) pathological mutations was negative. EPOR sequencing identified a new germline heterozygous frameshift mutation, c.1300dup (p.Gln434Profs*11), which generates a new ten-amino acid C-terminal tail and a stop codon at position 444, leading to the truncation of 64 amino acids of the wild-type receptor and the loss of the six last conserved tyrosine residues (Table 1, Figure 1A,B). The mutation was found in neutrophils and CD3+ T lymphocytes as well as in non-hematopoietic tissues such as nails and hair allowing the diagnosis of PFCP to be made. There was no familial history of polycythemia. The blood counts of the mother and two children were strictly normal. The father’s blood count was not available.
Figure 1.

Study of the EPOR c.1300dup (p.Gln434Profs*11) in primary cells. (A) Electropherograms showing the EPOR c.1300dup mutation in hematopoietic cells (CD3+ T-lymphocytes and neutrophils) and in non-hematopoietic tissues (nails, hair and buccal swab). (B) Scheme of EPOR wild-type and of the new frameshift EPOR mutants (EPOR FS). Negative signaling regulators (on the left) and internalization/degradation sites (on the right) are indicated. The tyrosine (Y) number of the mature EPOR is also indicated in parentheses. (C, D) Effect of erythropoietin (EPO) concentration on erythroid colony formation in the presence (C) or absence (D) of fetal bovine serum. BFU-E colonies were counted at day 14. Each experiment was performed twice in duplicate. The results are expressed in percentages of the number of colonies at 1 U/mL of EPO. (E) Effect of EPO concentration on EPOR signaling in erythroblasts. After 7 to 10 days in culture with stem cell factor (SCF), interleukin-3 (IL3) and EPO, CD34+ cells from the patient or healthy donors were cytokine-starved for 5 h then stimulated for 15 min with increasing concentrations of EPO. JAK2 and STAT5 phosphorylations were examined by western blotting. One of three independent experiments is presented and fold activation is indicated below. (F) Persistence of JAK2 and STAT5 phosphorylation in erythroblasts. After 7 to 10 days in culture with SCF, IL3 and EPO, CD34+ cells from the patient or healthy donors were cytokine-starved for 5 h prior to 15 min of stimulation with EPO 1 U/mL. Cells were then washed to remove the EPO and cultured in the absence of cytokine or serum. JAK2 and STAT5 phosphorylations were examined by western blotting in a time-dependent manner: after 5 h of starvation (−), after the EPO stimulation (+EPO) and at different times after EPO removal (10 min, 30 min, 1 h and 4 h). One out of two independent experiments is presented and fold activation is indicated below.
Erythroid progenitors displayed autonomous growth when cultured in the presence of serum (Figure 1C), an effect probably explained by residual erythropoietin in the serum, as in the absence of serum, no spontaneous growth was observed, but there was greater erythropoietin hypersensitivity (a nearly 5-fold increase) compared to that of control cells from a healthy donor (Figure 1D).
We performed signaling experiments with primary CD34+ cells from the PFCP patient which were cultured in vitro for 7-10 days in the presence of interleukin-3, stem cell factor and erythropoietin. Unlike control cells, PFCP erythroid progenitors exhibited constitutive and persistent phosphorylation of JAK2 as well as constitutive activation of STAT5 with a hypersensitive erythropoietin dose-response (Figure 1E,F). This erythropoietin-induced JAK2 and STAT5 activation also persisted for 4 h after removal of erythropoietin from the PFCP cells, whereas it was much more transient in control cells (Figure 1F).
Here we show that EPOR c.1300dup (p.Gln434Profs*11) is a strong gain-of-function mutation, which induces major erythropoietin hypersensitivity in primary erythroid progenitors, similar to that observed in patients with polycythemia vera, as well as constitutive and persistent activation of JAK2 and STAT5.
Functional analysis of EPOR c.1300dup (p.Gln434Profs*11) in the Ba/F3 cell line
In order to study the functional impact of the new frameshift mutant, Ba/F3 cells were transduced to express different HA-tagged human EPOR: the wild-type receptor (EPOR WT), EPOR p.Gln434Profs*11 (EPOR FS), identical to the patient’s mutation or EPOR p.Gln444* (EPOR STOP) generating a truncated receptor at position 444 (Table 1, Figure 2A). EPOR FS and EPOR STOP differed by the nature of the C-terminal ten amino acid residues, namely the new sequence PALASMDTVP in EPOR FS and the natural sequence QLLRPWTLCP in EPOR STOP. These cells expressed quite similar levels of exogenous EPOR, as detected with anti-HA antibody (Figure 2C). Interestingly, EPOR FS migrated slightly above EPOR STOP, suggesting differences in post-translational modification. However, the glycosylation states of EPOR WT, EPOR STOP and EPOR FS were similar after using Endo H and PGNase F (Online Supplementary Figure S1).
Figure 2.

Functional study of EPOR c.1300dup (p.Gln434Profs*11) in the Ba/F3 cell line. (A) Ba/F3 cells were transduced with pMX-HA-huEPOR-IRES-GFP retrovirus to stably express the wild-type receptor (EPOR WT), a truncated mutant at position 444 (p.Gln444*, EPOR STOP) or the frameshift mutant EPOR c.1300dup (p.Gln434Profs*11, EPOR FS). (B) Proliferation was assessed 48 h after culturing Ba/F3-EPOR cells in the absence or presence of increasing doses of erythropoietin (EPO) (0.01, 0.02, 0.03, 0.05, 0.1, 0.3, and 1 U/mL) by a WST-1 proliferation assay. Dose-response curves are means expressed in percentages of maximum growth value ± SEM (n = 3 in triplicate). Two-tailed t-test, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. (C) Effect of EPO concentration on EPOR signaling. Ba/F3 cells expressing different EPOR constructs were examined by western blotting for the presence and phosphorylation status of various signaling molecules. Cells were serum- and cytokine-starved for 5 h prior to stimulation for 15 min with increasing doses of EPO (0, 0.001, 0.01, 0.1 and 1 U/mL). Expression of β-actin was used as a loading control. One of three independent experiments is presented. (D) Phosho-STAT5/actin spontaneous phosphorylation was quantified compared to WT using Image J. Results represent the mean ± SEM (n = 3). Two-tailed t-test, *P<0.05, ***P<0.001, (E) Persistence of STAT5 phosphorylation. Ba/F3-EPOR cells were serum- and cytokine-starved for 5 h prior to 15 min of stimulation with EPO 1 U/mL. Cells were then washed to remove the EPO and cultured in the absence of cytokine or serum. STAT5 phosphorylation was examined by western blotting in a time-dependent manner: after 5 h of starvation (−), after EPO stimulation (+EPO) and at different times after EPO removal (5 min, 30 min, 1 h, 2 h and 4 h). One out of two independent experiments is presented.
MTT-like assays were performed to investigate the potential effects of EPOR STOP and EPOR FS on cell proliferation. None of these mutants was able to induce cytokine-independent cell growth. However, EPOR FS conferred a 4- to 5-fold greater erythropoietin hypersensitivity to Ba/F3 cells compared to EPOR WT. Interestingly the erythropoietin-induced growth of EPOR STOP Ba/F3 cells was similar to that of EPOR WT cells (Figure 2B). To confirm these results in a human setting, we transduced EPOR WT, STOP and FS in the human UT-7 cell line, which expresses endogenous EPOR and found similar results as in the Ba/F3 cell line (Online Supplementary Figure S2A).
We checked the signaling pathways and observed that EPOR FS induced constitutive phosphorylation of STAT5, AKT and ERK compared to EPOR WT and to a higher extent than EPOR STOP in Ba/F3 cells (Figure 2C) and UT-7 cells (Online Supplementary Figure S2B). Semi-quantitative analysis of spontaneous STAT5 phosphorylation showed a significant increase in EPOR FS compared to EPOR WT and EPOR STOP (Figure 2D). Moreover, we observed a similar persistent STAT5 activation in both EPOR FS and EPOR STOP Ba/F3 cells compared to EPOR WT cells (4 h, 2 h and 30 min, respectively, after erythropoietin removal) (Figure 2E).
These results show that EPOR FS confers a similar erythropoietin hypersensitivity to Ba/F3 and UT-7 cells as that observed in PFCP erythroid progenitors, while the truncated mutant EPOR STOP did not confer such erythropoietin hypersensitivity to these cells. The greater erythropoietin hypersensitivity induced by EPOR p.Gln434Profs*11 cannot, therefore, be explained by the receptor truncation itself and the loss of the two SHP-1 and SOCS3 binding sites which are responsible for a persistent activation, but rather by the appearance of a new C-terminal tail that confers spontaneous signaling.
Effects of the c.1300dup (p.Gln434Profs*11) mutation on erythropoietin receptor stability, cell surface expression and dimerization
To further characterize this new EPOR mutant, the cell surface expression of the wild-type receptor and both mutants was studied by flow cytometry. At similar levels of GFP (transduction of cells with IRES-GFP retroviruses), EPOR FS was significantly more abundant at the cell surface (more than 2-fold, P=0.0002) than EPOR WT and EPOR STOP in both Ba/F3 and UT-7 cell lines (Figure 3A and Online Supplementary Figure S3). These results were further confirmed in Ba/F3 cells using radiolabeled 125I-erythropoietin (2,412±494 receptors for EPOR FS, P=0.015, 1,018±284 receptors for EPOR STOP and 1,201±304 receptors for EPOR WT) (Figure 3b). We next investigated the stability of the receptors using treatment with the protein synthesis inhibitor cycloheximide. EPOR FS was more stable than both EPOR WT and EPOR STOP (half-life of 2 h and 1 h, respectively) (Figure 3C). We also studied the dimerization of human EPOR monomers by split Gaussia luciferase assay in steady-state conditions in HEK cells, in the absence of erythropoietin (Figure 3D). Close proximity between the C-terminal cytosolic domains of EPOR was increased by 4-fold with EPOR FS compared to control and EPOR STOP, as assessed by reconstitution of the split Gaussia luciferase activity (Figure 3E).
Figure 3.

Effects of c.1300dup (p.Gln434Profs*11) mutation on EPOR stability, dimerization and cell surface expression. (A) Cell-surface expression of the different EPOR was assessed by flow cytometry using PE fluorescent labeling of the extracellular HA-tag. The histogram shows the ratio of mean fluorescence intensiy (MFI) of PE-labeled cell-surface erythropoietin (EPO) on the respective MFI of GFP. Results are the mean ± SEM of seven independent experiments. (B) Cell-surface expression of the different EPOR was assessed with radiolabeled 125I-EPO. The results are expressed in cpm normalized to EPOR WT. The number of cell-surface receptors was determined by comparison between the radioactivity of transduced Ba/F3 cells and parental UT-7 cells that express 7000 receptors. (C) EPOR stability. Ba/F3-EPOR cells were incubated with cycloheximide for different times (0 min, 15 min, 30 min, 1 h, 2 h, 4 h, 6 h) and HA expression was studied by western blotting. HA-EPOR and β-actin were quantified using Image J software. The curves represent the HA/β-actin ratios. Three independent experiments were done. (D) Schematic representation of split Gaussia princeps luciferase complementation assay used to test EPOR dimerization in HEK293-derived BOSC cells. (E) Dimerization of human EPOR monomers was assessed by split Gaussia luciferase assay in steady-state conditions, in the absence of EPO in HEK cells. Close proximity between the C-terminal cytosolic domains of EPOR is significantly promoted by EPOR FS. The results represent the mean ± SEM from three independent experiments, each performed with eight biological replicates per condition. For each experiment, raw values were normalized to the average of the EPOR WT condition before pooling the data of all experiments together. Unpaired two-tailed t-test with Welch correction, ****P<0.0001.
The increased stability and cell surface localization of EPOR FS may also result in a defect in the receptor internalization pattern due to the loss of specific domains located in the C-terminal part of the wild-type receptor. However, EPOR WT, STOP and FS displayed the same internalization pattern as assessed by 125I-erythropoietin labeling experiments (Figure 4A,B). Furthermore, we noted that a dileucine motif, known to be a potential clathrin-dependent endocytosis signal,37 was lost in the new C-terminal tail of the EPOR FS. We assumed that this particular modification could be involved in the increased cell surface expression of EPOR FS. Thus, based on EPOR WT and EPOR STOP constructs, we generated two other mutants, EPOR WT/diL and EPOR STOP/diL, where the dileucine motif was removed (Figure 4C). However its abrogation did not modify either the erythropoietin sensitivity of EPOR WT or STOP Ba/F3 cells (Figure 4D) or the localization of the receptors at the cell surface (Figure 4E). Likewise, EPOR WT/diL and EPOR STOP/diL Ba/F3 cells displayed a similar signaling pattern to that of EPOR WT and EPOR STOP cells, respectively (Figure 4F).
Figure 4.

Role of diLeucine motif (diL) loss in the EPOR c.1300dup (p.Gln434Profs*11, EPOR FS) mechanism. (A, B) EPOR internalization was performed with radio -labeled 125I-erythropoietin (EPO). Cells were incubated with 125I-EPO and washed at 4°C to remove unbound ligand. An acidic wash was then performed to separate cell surface–bound from internalized EPO. The radioactivity levels of the (A) supernatant (cell surface–bound EPO) and of (B) the cell pellet (internalized EPO) were determined. Each experiment was performed three times with similar results. (C) To study the potential role of the dileucine motif loss in the EPO hypersensitivity phenotype induced by EPOR FS, Ba/F3 cells were transduced with pMX-HA-huEPOR-IRES-GFP retrovirus to stably express EPOR WT or EPOR STOP receptor with abrogation of the dileucine motif EPOR WT/diL and EPOR STOP/diL respectively. (D) Proliferation was assessed 48 h after culturing Ba/F3-EPOR cells in the absence or presence of increasing doses of EPO (0.01, 0.02, 0.03, 0.05, 0.1, 0.3, and 1 U/mL) by a WST-1 proliferation assay. Dose-response curves are means expressed in percentages of maximum growth value ± SEM (n = 3 in triplicate). Two-tailed t-test, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001. (E) Cell-surface expression of the different EPOR was assessed by flow cytometry using PE fluorescence labeling of the extracellular HA-tag. The histogram shows the ratio of mean fluorescence intensity (MFI) of PE-labeled cell-surface EPOR on the respective MFI of GFP. Results are the mean ± SEM of three independent experiments. (F) Effect of EPO concentration on EPOR signaling. Ba/F3 cells expressing different EPOR constructs were examined by western blotting for the presence and phosphorylation status of various signaling molecules. Cells were serum- and cytokine-starved for 5 h prior to stimulation for 15 min with increasing doses of EPO (0, 0.001, 0.01, 0.1 and 1 U/mL). Expression of β-actin was used as a loading control. One of three independent experiments is presented and fold activation is indicated below.
Collectively these results show that p.Gln434Profs*11 increases EPOR stability, dimerization and localization at the cell surface without modifying internalization of the receptor.
Different mechanisms are involved in the erythropoietin hypersensitivity phenotype in primary erythrocytosis according to the type of EPOR mutation
We wondered whether this model could be extended to other EPOR mutations already described in PFCP. We therefore investigated the impact of the frameshift EPOR c.1311_1312del (p.Pro438Metfs*6) mutation25 and its designed nonsense mutant counterpart, EPOR c.1327_1329delinsTAA (p.Pro443*) (Figure 5A) on the proliferation rate of Ba/F3 cells. These mutants lack the last 65 amino acids of the receptor, retaining two of the eight conserved tyrosine residues, Tyr-368 and Tyr-426 (Table 1, Figure 6A,C,D). Interestingly, EPOR p.Pro438Metfs*6 and EPOR p.Gln434Profs*11 (EPOR FS) share the same five-amino acid terminal sequence (MDTVP). MTT-like assays showed that erythropoietin hypersensitivity was induced by EPOR p.Pro438Metfs*6, but not by EPOR p.Pro443* (Figure 5B), similarly to the results obtained with EPOR p.Gln434Profs*11 (EPOR FS) and EPOR p.Gln444* (EPOR STOP), respectively. We also studied two proximal nonsense mutations, EPOR c.1195G>T (p.Glu399*)11 and c.1273G>T (p.Glu425*)10 (Figure 5A), which are responsible for more extensive truncations (109 and 83 amino acids, respectively) and the loss of seven of the eight cytoplasmic tyrosine residues, retaining only Tyr-368 (Table 1, Figure 6A,B). Interestingly, these nonsense mutants, unlike EPOR p.Pro434* and p.Gln444*, were able to confer erythropoietin hypersensitivity to Ba/F3 cells (Figure 5C). These results show that extensive truncations, unlike shorter ones, are sufficient per se to induce the erythropoietin hypersensitivity phenotype. In comparison, erythropoietin hypersensitivity induced by frameshift EPOR mutations is due to a distinct common mechanism based on the appearance of new amino acid sequences.
Figure 5.

Different mechanisms are involved in the erythropoietin hypersensitivity phenotype of primary familial and congenital polycythemia depending on the type of EPOR mutation. (A) Ba/F3 cells were transduced with pMX-HA-huEPOR-IRES-GFP retrovirus to stably express different kind of EPOR mutations already described: the frameshift EPOR c.1311_1312del mutant (p.Pro438Metfs*6) or its nonsense designed counterpart EPOR c.1327_1329delinsTAA (p.Pro443*), more proximal truncations due to the nonsense mutants EPOR c.1195G>T (p.Glu399*) and c.1273G>T (p.Glu425*). (B, C) Proliferation was assessed 48 h after culturing EPOR p.Pro438Metfs*6 and p.Pro443* Ba/F3 cells (B) or EPOR p.Glu399* and p.Glu425* Ba/F3 cells (C) in the absence or presence of increasing doses of EPO (0.01, 0.02, 0.03, 0.05, 0.1, 0.3, and 1 U/mL) and compared to EPOR WT and EPOR FS growth by a WST-1 proliferation assay. Dose-response curves are means expressed in percentages of maximum growth value ± SEM (n = 3 in triplicate). Two-tailed t-test, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Figure 6.

Regulation of EPOR signaling and positions of the EPOR truncations. (A) Negative regulators of EPOR signaling and their binding sites in the C-terminal part of the receptor: negative signaling regulators (on the left) and internalization/degradation sites (on the right). The tyrosine (Y) number of the mature EPOR is also indicated in parentheses. (B) Proximal truncations due to EPOR p.Glu399* and p.Glu425* lack all negative regulation sites of the receptor. (C and D) More distal truncations due to frameshift EPOR mutants and their designed counterparts retain the Tyr-426.
Discussion
In this study, we identified a new germline heterozygous EPOR mutation, c.1300dup (p.Gln434Profs*11) in a patient suffering from PFCP. Like the other mutations already described in this pathology, c.1300dup is located in exon 8 of EPOR. The frameshift generates a new cytoplasmic tail of ten amino acids and a premature stop codon at position 444 leading to the loss of 64 amino acids in the C-terminal part of the receptor. Two other mutations, c.1281dup (p.Ile428Tyrfs*17)7 and c.1288dup (p.Asp430Glyfs*15),4 leading to a similar truncation at position 444 had been previously reported but, as for most other EPOR mutants, no extensive functional study had been carried out. Here we show that EPOR p.Gln434Profs*11 is a strong gain-of-function mutant that induces major erythropoietin hypersensitivity in primary cells as well as in transduced Ba/F3 cells, without spontaneous growth, in accordance with the diagnosis of PCFP.
Previous reports assumed that the erythropoietin hypersensitivity phenotype observed in PFCP is due to the loss of negative regulatory domains located in the C-terminal part of the receptor, especially some conserved tyrosine residues.30,31 Both the magnitude and duration of EPOR signaling are indeed crucial for erythropoiesis and are therefore tightly regulated by several mechanisms induced as soon as erythropoietin binds to its cognate receptor. A classic negative feedback loop is achieved by signaling inhibitors such as the tyrosine phosphatase SHP-138 and the suppressor of cytokine signaling (SOCS) proteins SOCS3 and CIS,39 which bind the conserved Tyr-426, -454 and -456 on the cytoplasmic tail of EPOR (Figure 6A). Rapid internalization and degradation of the receptor induced by erythropoietin binding34 also contribute to the negative regulation of EPOR signaling. Several possible cooperative and partially redundant mechanisms are involved in this process, but their relative contributions remain unclear. The C-terminal part of EPOR is degraded at the cell surface by the proteasome.34,35 This step requires binding of the E3-ligase βTrcp to a conserved motif Asp461-Ser462-Gly463 of EPOR (Figure 6A).36 The erythropoietin-EPOR complex is then internalized in a process dependent on Tyr-454, -456 and -504 and the p85 subunit of phosphatidylinositol 3 kinase, and targeted to the lysosomes for degradation (Figure 6A).30,31,35 The loss of these negative signaling regulatory and degradation domains due to EPOR truncation is usually thought to explain erythropoietin hypersensitivity in PFCP.6,7,14,27,29–31,36,40 In the present study, we studied two extensive truncations, p.Glu399* and p.Glu425*, which lack seven of the eight conserved tyrosine residues (Table 1, Figure 6B) and all sequences that constitute the EPOR negative regulatory domains. These mutants conferred greater erythropoietin hypersensitivity to Ba/F3 cells compared to EPOR WT. Thus, confirming previous reports, we showed that extensive truncations lacking all EPOR negative regulatory sites are sufficient in themselves to induce the PFCP phenotype.
Our results highlight that a different mechanism underlies erythropoietin hypersensitivity due to frameshift EPOR mutations. The study of EPOR p.Gln434Profs*11 (EPOR FS) and EPOR p.Pro438Metfs*6 and their designed nonsense counterparts, EPOR p.Gln444* (EPOR STOP) and EPOR p.Pro434*, respectively, allowed us to discriminate between the effects due to the truncations themselves leading to the loss of SHP-1 and SOCS3 binding sites and those due to the appearance of a new cytoplasmic tail. In accordance with the loss of negative regulatory sites and previous reports,9,14,27,29 EPOR FS and EPOR STOP Ba/F3 cells displayed persistent phosphorylation of STAT5 after erythropoietin removal, but only EPOR FS was able to induce constitutive STAT5 phosphorylation and erythropoietin hypersensitivity. In a similar way to EPOR p.Gln434Profs*11 (EPOR FS), Ba/F3 cells expressing EPOR p.Pro438Metfs*6 displayed erythropoietin hypersensitivity, unlike EPOR p.Pro443*, suggesting a common mechanism for the frameshift EPOR mutations in PFCP due to the presence of a new, partially common cytoplasmic tail (MDTVP). Of note EPOR p.Pro443* and p.Gln444* have never been identified in PFCP patients. Furthermore, a previously described murine mutant that lacks the last 40 amino acids and has a new cytoplasmic tail, which does not include the MDTVP motif, is also responsible for erythropoietin hypersensitivity. This suggests that other sequences of amino acids could be involved in erythropoietin hypersensitivity.41 Furthermore, another mutant, EPOR p.Trp439*, was found in a large family with erythrocytosis, with a mild phenotype probably associated with incomplete penetrance of the mutation.3 This finding suggests that our in vitro cellular model may be insufficiently sensitive to unmask weak functional effects or that the wild-type WTLCP motif may act as a negative regulator of erythropoietin sensitivity.
We also demonstrated that the increased stability and the greater localization at the cell surface of EPOR FS compared to EPOR WT and EPOR STOP were not due to the loss of internalization sites located in the cytoplasmic tail of the protein as these receptors displayed similar internalization patterns. These results suggest better addressing to the cell surface, increased stability or greater recycling of EPOR FS. Moreover, we found that EPOR FS increased the basal dimerization of the receptor, correlating with the spontaneous activation of JAK2 and downstream signaling. Two recent studies have emphasized the role of EPOR dimerization and conformation in signal transduction. Indeed, while the EPO R150Q mutant was unable to mediate full signaling due to decreased EPOR dimerization and JAK2 activation,42 alteration of EPOR conformation by diabodies was able to finely tune the receptor signaling.43 The conformation of EPOR is indeed crucial for its optimal activation. In the absence of ligand, inactive EPOR dimers are pre-formed at the cell surface44 through their transmembrane domains.45 Erythropoietin binding to its receptor induces conformational changes, such as the formation of a 120° angle between the D1 domains of the two EPOR molecules46 and the reorientation of the continuous rigid alpha helix formed by the transmembrane domain and the cytosolic juxtamembrane region that contains a rigid hydrophobic motif, composed of residues Leu278, Ile282 and Trp283.47–49 It has been shown that the orientation of this conserved motif is crucial for JAK2 activation and JAK2-induced EPOR phosphorylation.47 Other active EPOR conformations have been described but this particular dimer orientation is the optimal one for signaling.50 As EPOR is not permissive for self-activation in terms of conformation, the appearance of a new cytoplasmic tail due to frameshift mutations might induce reorientation of EPOR transmembrane and/or juxtamembrane domains, leading to pre-activation of the EPOR-JAK2 complex at the cell surface.
To our knowledge this is the first extensive functional study of EPOR mutations in PFCP. We demonstrated here that different mechanisms, depending on the type and location of EPOR mutations, contribute to the erythropoietin hypersensitivity phenotype, as suggested previously. Extensive truncations lacking all negative regulatory and degradation domains are sufficient by themselves to confer erythropoietin hypersensitivity, whereas more distal truncations induced by frameshift mutants confer erythropoietin hypersensitivity that depends on the appearance of a new C-terminal tail. The latter, by increasing EPOR dimerization and stability at the cell surface, cause pre-activation of EPOR and JAK2, constitutive signaling and hypersensitivity to erythropoietin similar to that occurring in JAK2V617F-positive polycythemia vera.
Supplementary Material
Acknowledgments
We are deeply grateful to the patient involved in the study. We thank the Imaging and Cytometry Platform (PFIC) of Gustave Roussy, especially Philippe Rameau and Yann Lecluse. We also thank Gwendoline Leroy for the sequencing.
This work was supported by grants from l’Agence Nationale de la Recherche (ANR-13-JVSV1-GERMPN-01), the Laurette Fugain foundation, the GIS-Institute for rare diseases for high throughput sequencing (AO9102LS), the Association de Recherche sur la Moelle Osseuse (ARMO), the Association pour la Recherche contre le Cancer (ARC) (Fondation ARC libre 2012) and the regional PHRC AOR07014. The “Investissements d’avenir” program is funding the Labex GR-Ex (IP, WV and FV). FP was supported by Ph.D. grants from the ARC. CM was supported by ANR-Blanc 2013 GERMPN. WV is a recipient of a research fellowship from IGR INSERM (contrats d’interface). TB is a Télévie PhD fellow and SNC is supported by the Ludwig Institute for Cancer, Fondation Contre le Cancer, Fondation Salus Sanguinis, FNRS-FRSM-PDR, Project ARC10/15-027 and PAI Belgian Medical Genetics Initiative Project.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/4/575
References
- 1.Yoshimura A, Longmore G, Lodish HF. Point mutation in the exoplasmic domain of the erythropoietin receptor resulting in hormone-independent activation and tumorigenicity. Nature. 1990;348(6302): 647–649. [DOI] [PubMed] [Google Scholar]
- 2.Juvonen E, Ikkala E, Fyhrquist F, Ruutu T. Autosomal dominant erythrocytosis caused by increased sensitivity to erythropoietin. Blood. 1991;78(11):3066–3069. [PubMed] [Google Scholar]
- 3.de la Chapelle A, Traskelin AL, Juvonen E. Truncated erythropoietin receptor causes dominantly inherited benign human erythrocytosis. Proc Natl Acad Sci USA. 1993;90(10):4495–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sokol L, Luhovy M, Guan Y, Prchal JF, Semenza GL, Prchal JT. Primary familial polycythemia: a frameshift mutation in the erythropoietin receptor gene and increased sensitivity of erythroid progenitors to erythropoietin. Blood. 1995;86(1):15–22. [PubMed] [Google Scholar]
- 5.Arcasoy MO, Degar BA, Harris KW, Forget BG. Familial erythrocytosis associated with a short deletion in the erythropoietin receptor gene. Blood. 1997;89(12):4628–4635. [PubMed] [Google Scholar]
- 6.Furukawa T, Narita M, Sakaue M, et al. Primary familial polycythaemia associated with a novel point mutation in the erythropoietin receptor. Br J Haematol. 1997;99(1): 222–227. [DOI] [PubMed] [Google Scholar]
- 7.Kralovics R, Indrak K, Stopka T, Berman BW, Prchal JF, Prchal JT. Two new EPO receptor mutations: truncated EPO receptors are most frequently associated with primary familial and congenital polycythemias. Blood. 1997;90(5):2057–2061. [PubMed] [Google Scholar]
- 8.Kralovics R, Sokol L, Prchal JT. Absence of polycythemia in a child with a unique erythropoietin receptor mutation in a family with autosomal dominant primary polycythemia. J Clin Invest. 1998;102(1):124–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watowich SS, Xie X, Klingmuller U, et al. Erythropoietin receptor mutations associated with familial erythrocytosis cause hypersensitivity to erythropoietin in the heterozygous state. Blood. 1999;94(7): 2530–2532. [PubMed] [Google Scholar]
- 10.Kralovics R, Prchal JT. Genetic heterogeneity of primary familial and congenital polycythemia. Am J Hematol. 2001;68(2):115–121. [DOI] [PubMed] [Google Scholar]
- 11.Arcasoy MO, Karayal AF, Segal HM, Sinning JG, Forget BG. A novel mutation in the erythropoietin receptor gene is associated with familial erythrocytosis. Blood. 2002;99(8):3066–3069. [DOI] [PubMed] [Google Scholar]
- 12.Rives S, Pahl HL, Florensa L, et al. Molecular genetic analyses in familial and sporadic congenital primary erythrocytosis. Haematologica. 2007;92(5):674–677. [DOI] [PubMed] [Google Scholar]
- 13.Al-Sheikh M, Mazurier E, Gardie B, et al. A study of 36 unrelated cases with pure erythrocytosis revealed three new mutations in the erythropoietin receptor gene. Haematologica. 2008;93(7):1072–1075. [DOI] [PubMed] [Google Scholar]
- 14.Perrotta S, Cucciolla V, Ferraro M, et al. EPO receptor gain-of-function causes hereditary polycythemia, alters CD34 cell differentiation and increases circulating endothelial precursors. PLoS One. 2010;5(8):e12015. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.James C, Ugo V, Le Couedic JP, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037): 1144–1148. [DOI] [PubMed] [Google Scholar]
- 16.Scott LM, Tong W, Levine RL, et al. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med. 2007;356(5):459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.D’Andrea AD, Fasman GD, Lodish HF. Erythropoietin receptor and interleukin-2 receptor beta chain: a new receptor family. Cell. 1989;58(6):1023–1024. [DOI] [PubMed] [Google Scholar]
- 18.Huang LJ, Constantinescu SN, Lodish HF. The N-terminal domain of Janus kinase 2 is required for Golgi processing and cell surface expression of erythropoietin receptor. Mol Cell. 2001;8(6):1327–1338. [DOI] [PubMed] [Google Scholar]
- 19.Witthuhn BA, Quelle FW, Silvennoinen O, et al. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74(2):227–236. [DOI] [PubMed] [Google Scholar]
- 20.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98(12):3261–3273. [DOI] [PubMed] [Google Scholar]
- 21.Bouscary D, Pene F, Claessens YE, et al. Critical role for PI 3-kinase in the control of erythropoietin-induced erythroid progenitor proliferation. Blood. 2003;101(9):3436–3443. [DOI] [PubMed] [Google Scholar]
- 22.Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83(1):59–67. [DOI] [PubMed] [Google Scholar]
- 23.Petersen KB, Hokland P, Petersen GB, Nyvold CG. Erythropoietin receptor defect: a cause of primary polycythaemia. Br J Haematol. 2004;125(4):537–538. [DOI] [PubMed] [Google Scholar]
- 24.O’Rourke K, Fairbairn DJ, Jackson KA, Morris KL, Tey SK, Kennedy GA. A novel mutation of the erythropoietin receptor gene associated with primary familial and congenital polycythaemia. Int J Hematol. 2011;93(4):542–544. [DOI] [PubMed] [Google Scholar]
- 25.Bento C, Almeida H, Maia TM, et al. Molecular study of congenital erythrocytosis in 70 unrelated patients revealed a potential causal mutation in less than half of the cases (where is/are the missing gene(s)?). Eur J Haematol. 2013;91(4):361–368. [DOI] [PubMed] [Google Scholar]
- 26.Bento C, Percy MJ, Gardie B, et al. Genetic basis of congenital erythrocytosis: mutation update and online databases. Hum Mutat. 2014;35(1):15–26. [DOI] [PubMed] [Google Scholar]
- 27.Gross M, Ben-Califa N, McMullin MF, et al. Polycythaemia-inducing mutations in the erythropoietin receptor (EPOR): mechanism and function as elucidated by epidermal growth factor receptor-EPOR chimeras. Br J Haematol. 2014;165(4):519–528. [DOI] [PubMed] [Google Scholar]
- 28.Chauveau A, Luque Paz D, Lecucq L, et al. A new point mutation in EPOR inducing a short deletion in congenital erythrocytosis. Br J Haematol. 2016;172(3):475–477. [DOI] [PubMed] [Google Scholar]
- 29.Iacobucci I, Li Y, Roberts KG, et al. Truncating erythropoietin receptor rearrangements in acute lymphoblastic leukemia. Cancer Cell. 2016;29(2):186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sulahian R, Cleaver O, Huang LJ. Ligand-induced EpoR internalization is mediated by JAK2 and p85 and is impaired by mutations responsible for primary familial and congenital polycythemia. Blood. 2009;113(21):5287–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bulut GB, Sulahian R, Yao H, Huang LJ. Cbl ubiquitination of p85 is essential for Epo-induced EpoR endocytosis. Blood. 2013;122(24):3964–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Couedic JP, Mitjavila MT, Villeval JL, et al. Missense mutation of the erythropoietin receptor is a rare event in human erythroid malignancies. Blood. 1996;87(4):1502–1511. [PubMed] [Google Scholar]
- 33.Remy I, Michnick SW. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods. 2006;3(12):977–979. [DOI] [PubMed] [Google Scholar]
- 34.Verdier F, Walrafen P, Hubert N, et al. Proteasomes regulate the duration of erythropoietin receptor activation by controlling down-regulation of cell surface receptors. J Biol Chem. 2000;275(24):18375–18381. [DOI] [PubMed] [Google Scholar]
- 35.Walrafen P, Verdier F, Kadri Z, Chretien S, Lacombe C, Mayeux P. Both proteasomes and lysosomes degrade the activated erythropoietin receptor. Blood. 2005;105(2): 600–608. [DOI] [PubMed] [Google Scholar]
- 36.Meyer L, Deau B, Forejtnikova H, et al. beta-Trcp mediates ubiquitination and degradation of the erythropoietin receptor and controls cell proliferation. Blood. 2007;109(12):5215–5222. [DOI] [PubMed] [Google Scholar]
- 37.Letourneur F, Klausner RD. A novel dileucine motif and a tyrosine-based motif independently mediate lysosomal targeting and endocytosis of CD3 chains. Cell. 1992;69(7):1143–1157. [DOI] [PubMed] [Google Scholar]
- 38.Jiao H, Berrada K, Yang W, Tabrizi M, Platanias LC, Yi T. Direct association with and dephosphorylation of Jak2 kinase by the SH2-domain-containing protein tyrosine phosphatase SHP-1. Mol Cell Biol. 1996;16(12):6985–6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sasaki A, Yasukawa H, Shouda T, Kitamura T, Dikic I, Yoshimura A. CIS3/SOCS-3 suppresses erythropoietin (EPO) signaling by binding the EPO receptor and JAK2. J Biol Chem. 2000;275(38):29338–29347. [DOI] [PubMed] [Google Scholar]
- 40.Arcasoy MO, Harris KW, Forget BG. A human erythropoietin receptor gene mutant causing familial erythrocytosis is associated with deregulation of the rates of Jak2 and Stat5 inactivation. Exp Hematol. 1999;27(1):63–74. [DOI] [PubMed] [Google Scholar]
- 41.D’Andrea AD, Yoshimura A, Youssoufian H, Zon LI, Koo JW, Lodish HF. The cytoplasmic region of the erythropoietin receptor contains nonoverlapping positive and negative growth-regulatory domains. Mol Cell Biol. 1991;11(4):1980–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim AR, Ulirsch JC, Wilmes S, et al. Functional selectivity in cytokine signaling revealed through a pathogenic EPO mutation. Cell. 2017;168(6):1053–1064 e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moraga I, Wernig G, Wilmes S, et al. Tuning cytokine receptor signaling by reorienting dimer geometry with surrogate ligands. Cell. 2015;160(6):1196–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Livnah O, Stura EA, Middleton SA, Johnson DL, Jolliffe LK, Wilson IA. Crystallographic evidence for preformed dimers of erythropoietin receptor before ligand activation. Science. 1999;283(5404):987–990. [DOI] [PubMed] [Google Scholar]
- 45.Constantinescu SN, Keren T, Socolovsky M, Nam H, Henis YI, Lodish HF. Ligand-independent oligomerization of cell-surface erythropoietin receptor is mediated by the transmembrane domain. Proc Natl Acad Sci USA. 2001;98(8):4379–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Syed RS, Reid SW, Li C, et al. Efficiency of signalling through cytokine receptors depends critically on receptor orientation. Nature. 1998;395(6701):511–516. [DOI] [PubMed] [Google Scholar]
- 47.Constantinescu SN, Huang LJ, Nam H, Lodish HF. The erythropoietin receptor cytosolic juxtamembrane domain contains an essential, precisely oriented, hydrophobic motif. Mol Cell. 2001;7(2):377–385. [DOI] [PubMed] [Google Scholar]
- 48.Lu X, Gross AW, Lodish HF. Active conformation of the erythropoietin receptor: random and cysteine-scanning mutagenesis of the extracellular juxtamembrane and transmembrane domains. J Biol Chem. 2006;281(11):7002–7011. [DOI] [PubMed] [Google Scholar]
- 49.Li Q, Wong YL, Huang Q, Kang C. Structural insight into the transmembrane domain and the juxtamembrane region of the erythropoietin receptor in micelles. Biophys J. 2014;107(10):2325–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seubert N, Royer Y, Staerk J, et al. Active and inactive orientations of the transmembrane and cytosolic domains of the erythropoietin receptor dimer. Mol Cell. 2003;12(5):1239–1250. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.