

Abstract
Despite improvements in our understanding of the molecular basis of acute myeloid leukemia (AML), the association between genetic mutations with morphological dysplasia remains unclear. In this study, we evaluated and scored dysplasia in bone marrow (BM) specimens from 168 patients with de novo AML; none of these patients had cytogenetic abnormalities according to the 2016 World Health Organization Classification. We then performed targeted sequencing of diagnostic BM aspirates for recurrent mutations associated with myeloid malignancies. We found that cohesin pathway mutations [q (FDR-adjusted P)=0.046] were associated with a higher degree of megakaryocytic dysplasia and STAG2 mutations were marginally associated with greater myeloid lineage dysplasia (q=0.052). Frequent megakaryocytes with separated nuclear lobes were more commonly seen among cases with cohesin pathway mutations (q=0.010) and specifically in those with STAG2 mutations (q=0.010), as well as NPM1 mutations (q=0.022 when considering the presence of any vs. no megakaryocytes with separated nuclear lobes). RAS pathway mutations (q=0.006) and FLT3-ITD (q=0.006) were significantly more frequent in cases without evaluable erythroid cells. In univariate analysis of the 153 patients treated with induction chemotherapy, NPM1 mutations were associated with longer event-free survival (EFS) (P=0.042), while RUNX1 (P=0.042), NF1 (P=0.040), frequent micromegakaryocytes (P=0.018) and presence of a subclone (P=0.002) were associated with shorter EFS. In multivariable modeling, NPM1 was associated with longer EFS, while presence of a subclone and frequent micromegakaryocytes remained significantly associated with shorter EFS.
Introduction
Acute myeloid leukemia (AML) is a complex and dynamic disease, characterized by multiple somatically acquired driver mutations, co-existing competing clones, and disease evolution over time.1–3 A major challenge in the application of mutation information to clinical management of AML patients is how to integrate it into existing AML risk stratification and classification schemes. The 2016 WHO Classification incorporates cytogenetics, clinical ontogeny, dysplastic morphology in background hematopoietic cells, and the status of certain mutations (NPM1, RUNX1, and CEBPA).4 One main distinction between the WHO Classification and other AML risk stratification schemes, such as the European LeukemiaNet5 and National Comprehensive Cancer Network (NCCN),6 is its use of dysplastic morphology to categorize cases in an adverse risk group, AML with myelodysplasia-related changes (AML-MRC). Although there is general agreement as to the adverse effect of MDS-related cytogenetics on the outcome of de novo AML, the clinical and biological significance of morphological dysplasia in this setting is controversial.7 Studies have shown that, although dysplasia is frequently seen in NPM1-mutated AML, it does not confer a worse clinical outcome.8,9 Another study found that in AML patients with wild-type NPM1, those with multilineage dysplasia showed an inferior response to induction and, in younger patients, a lower 5-year survival, suggesting that the prognostic relevance of multilineage dysplasia in AML might depend on NPM1 mutation status.10
In a recent study of patients with de novo AML lacking specific cytogenetic findings, we reported that WHO-defined multilineage dysplasia had no impact on outcome, but the presence of certain specific dysplastic features (micromegakaryocytes and hypogranulated myeloid cells) was associated with adverse EFS.11 However, this study did not evaluate the impact of gene mutations other than FLT3 and NPM1. Moreover, despite the recently expanded knowledge of the mutations affecting multiple functional pathways in AML, an association of mutations with specific dysplastic features has not been closely evaluated. In myelodysplastic syndromes (MDS), Della Porta et al. found an association between granulocytic dysplasia and mutations in ASXL1, RUNX1, TP53 and SRSF2.12 Devillier et al. evaluated 94 patients with AML-MRC and found that ASXL1 mutations were associated with a higher degree of dysgranulopoiesis, but not dyserythropoiesis or dysmegakaryopoiesis.13 However, this study did not evaluate individual dysplastic features, and included patients with both MDS-related cytogenetics and a prior history of MDS, who are known to have a poor prognosis and frequent ASXL1 mutations.14,15 In AML cases without a history of MDS or MDS-associated karyotype findings, it is uncertain if the presence of background morphological dysplasia indicates a true relationship to MDS.
The goal of the current study is to analyze the association between dysplastic findings and somatic mutations in de novo AML. We investigated the associations between specific dysplastic features with individual mutations, gene pathway alterations, and clonal architecture, and explored the effects of these parameters on patients’ outcome.
Methods
Patients
Cases of newly diagnosed de novo AML were identified from the pathology archives of Brigham and Women’s Hospital/Dana-Farber Cancer Institute and Massachusetts General Hospital between 2009–2016. This study has been approved by an institutional review board (IRB) (IRB Protocol n. 2009P001369). All cases had bone marrow (BM) aspirate smear and biopsy slides available for review that were diagnosed as AML prior to any therapy being administered. Only cases with adequate karyotype and clinical follow-up information were included. Patients who had received any prior cytotoxic therapy, had a prior diagnosis of any myeloid neoplasm, or had defining cytogenetic abnormalities of AML-MRC or AML with recurrent genetic abnormalities according to the 2016 WHO Classification4 were excluded.
Morphology assessment
Bone marrow aspirate and biopsy smears from each case were viewed in a blinded fashion by 3 hematopathologists (OW, RH and OP) who scored dysplasia in each lineage in increments of 10%, as previously described;11 the median score for all 3 observers was used for all analyses. A minimum of 10 megakaryocytes (on biopsies and/or aspirate smears) and 20 erythroids and 20 myeloid elements (in aspirate smears) were required, otherwise a lineage was designated as “not evaluable”. Specific dysplastic features in each lineage were also scored on a semi-quantitative scale (<10% cells showing the dysplastic feature = 0, 10–25%=1, 26–50%=2, 51–75%=3, >75%=4). Specific dysplastic features scored in the erythroid lineage were: 1) megaloblastoid change; 2) multinucleation; 3) nuclear irregularities; and 4) pyknosis. Dysgranulopoiesis features scored were: 1) abnormal nuclear shape (including pseudo Pelger-Huet anomaly); and 2) hypogranulation. Dysmegakaryopoiesis features scored were: 1) micromegakaryocytes; 2) presence of two or multiple separated nuclear lobes; and 3) megakaryocytes with hypolobated or monolobated nuclei.
Clinical data
The complete blood count (CBC) and white blood cell (WBC) count differential results at the time of AML diagnosis were recorded. Type of treatment, including date of any allogeneic stem cell transplant (SCT), date of relapse or disease refractoriness to two induction regimens, and status at last follow up were recorded for each patient. Complete remission (CR) was determined as defined by clinical standards13 and follow-up information (relapse, death) was recorded.
Targeted sequencing
We performed targeted sequencing on BM aspirates obtained at the time of diagnosis for all 168 patients, as previously described.16–18 We enriched target regions of 87 genes (Baylor Custom SureSelect hybrid capture system, Agilent Technologies, Santa Clara, CA, USA) in 105 patients or 95 genes [Rapid Heme Panel kit, Illumina Truseq Custom Amplicon (TSCA), San Diego, CA, USA] in 66 patients, which were selected on the basis of pathogenic involvement in myeloid malignancies, on either banked DNA samples or diagnostic BM aspirate smears. We classified variants as pathogenic driver mutations based on mutation type and position, and on frequency in publicly available single nucleotide polymorphism databases. The median coverage was 1200X across all the genes. Average coverage across the entire hybrid capture run was 500X. The minimal coverage was 50X, or at least 5 alternative reads required to call a variant. Variants with a variant allele fraction of less than 0.05 were excluded to ensure consistency across both sequencing platforms. FLT3-ITD was identified by filtering aligned BAM files for which one side maps to exon 13, 14 or 15 of FLT3 and the other side is unmapped. These one-sided mapped reads were then scanned for the presence of duplications of at least 10 bp. Reads that contained duplications were realigned among themselves to make a final determination of FLT3-ITD status.18
Mutations were grouped into 7 pathways as follows:
DNA methylation: DNMT3A, IDH1, IDH2, and/or TET2
Epigenetic regulators: ASXL1, EZH2, BCOR, SETBP1, BCORL1, SH2B3, SETD2, and/or CREBBP
Transcription factors: CEBPA (single or double), RUNX1, ETV6, WT1, and/or PHF6
Cohesin complex: STAG2, PDS5B, RAD21, and/or SMC3
RAS pathway: KRAS, NRAS, ITD, FLT3, KIT, CBL, RIT1, PTPN11, and/or NF1
Spliceosome pathway: U2AF1, ZRSR2, PRPF40b, SRSF2, and/or SF3B1
We separately explored the association between morphology and genetics based on AML ontogeny as specified by Lindsley et al.,14 in which the presence of SRSF2, SF3B1, U2AF1, ZRSR2, ASXL1, EZH2, BCOR, or STAG2 mutations defined secondary type AML, the presence of an NPM1 mutation [unless any secondary-type mutation was present at >5% variant allele frequency (VAF)] implied de novo AML, and all other cases were designated as having a pan-AML mutation pattern.14
Subclonal analysis
The presence of a leukemia subclone was defined as having at least one mutation at a frequency of at least 10% less than the mutation with highest VAF, and with the sum of the highest mutation and subclonal mutation VAF over 55%.19
Statistical analysis
Fisher exact test and the Wilcoxon rank sum test were used to compare categorical and continuous variables, respectively. In the event that a continuous variable was compared across 3 or more groups, the Kruskall-Wallis test was used. The Kendall coefficient of concordance, W, with a correction for ties, was used to compare the 3 observers’ score of dysplasia percentages and individual dysplastic feature scores; Kendall W ranges from 0 (no agreement) to 1 (complete agreement).
The association between each degree of dysplasia in all three lineages was assessed by both continuous and categorical analyses. Individual dysplastic findings assessed by categorical analysis explored different cut offs (>0, ≥2, and ≥3) for each evaluable dysplastic feature. We explored the associations between age, WBC, peripheral blasts (%), BM cellularity (%), BM blasts (%), platelet count, and hemoglobin level, considering individual mutations present more than 5 patients (>3%), mutation pathways, molecular ontogeny, and subclone status. There were 29 planned analyses based on mutation data for each of the morphological measures and patients’ clinical characteristics; therefore, a false discovery rate adjustment (q-value) was made for the planned analyses within the respective outcome. All tests were two-sided and a q-value (FDR-adjusted P-value) <0.05 was considered significant
Event-free survival (EFS), defined as the time in months from the date of diagnosis to the date of disease progression or death, was explored in the subset of patients that received induction chemotherapy. Nominal P-values are presented for univariate analyses. Cox proportional hazards models with transplant as a time varying covariate were considered to evaluate the association of mutation and morphological findings with EFS. Mutation data and morphological findings underwent univariate analysis with transplant-adjusted Cox models. Multivariable Cox models were considered with mutations and morphological findings with a univariate P-value <0.2 and the backwards elimination method was used to select a final model retaining transplant and variables with a P-value <0.1.
Results
Patients
A total of 168 cases of de novo AML without WHO-defined cytogenetic abnormalities were identified with a median age of 60.6 years (range 19.9–86.7). A total of 137 cases (81%) had a normal karyotype and 31 (18%) had an abnormal karyotype. The most common cytogenetic abnormalities were +8 (8 cases), +11 (4 cases), and +13 (2 cases); other trisomies were seen in 8 cases, chromosome losses in 3 cases, ring chromosome in 1 case, and balanced or unbalanced translocations in 5 cases. The individual mutations identified in more than 5 patients (>3% of the study cohort) were: NPM1 (n=72), DNMT3A (n=68), FLT3-ITD (n=44), TET2 (n=40), FLT3 (n=26), IDH1 (n=31), ASXL1 (n=30), NRAS (n=30), SRSF2 (n=28), IDH2 (n=27), RUNX1 (n=27), PTPN11 (n=20), WT1 (n=15), STAG2 (n=14), BCOR (n=10), BCORL1 (n=6), CEBPA (n=11, including 3 with double CEBPA mutations), KRAS (n=8), RIT1 (n=8), NF1 (n=7) and CBL (n=6). Mutations were grouped into pathways as follows: DNA methylation (n=120), RAS pathway (n=97), NPM1 (n=72), epigenetic regulators (n=50), transcription factors (n=51) and spliceosome (n=39). The Lindsley et al. AML ontogeny groupings were as follows: secondary type (n=60), de novo type (n=64), and pan-AML type (n=42); 2 cases could not be assigned to the AML ontogeny group because of missing information for one or more group-defining mutations.14 The Pappaemanuil et al. AML classification groupings were as follows: chromatin-spliceosome (n=60), NPM1 (n=69), CEBPA (n=3), other driver mutations (n=23), no mutations (n=5), IDH2R172 (n=6).26 Comparing the two groupings, the chromatin/spliceosome group was composed of 54 secondary and 6 pan-AML cases; the NPM1 group 64 de novo, 4 secondary, and 1 pan-AML; the other driver group 22 pan-AML and 1 secondary AML; the no mutation and IDH2R172 groups were all pan-AML. A total of 92 cases (55%) had a leukemia subclone.20 The 2016 WHO Classification categories (AML-MRC being defined as ≥50% dysplastic cells present in at least 2 lineages, in the absence of NPM1, double CEBPA, or RUNX1 mutations) included the following: AML-MRC (n=19), AML-NOS (n=50), AML-NPM1 (n=71), AML-RUNX1 (n=25), and AML-CEBPA (n=3).
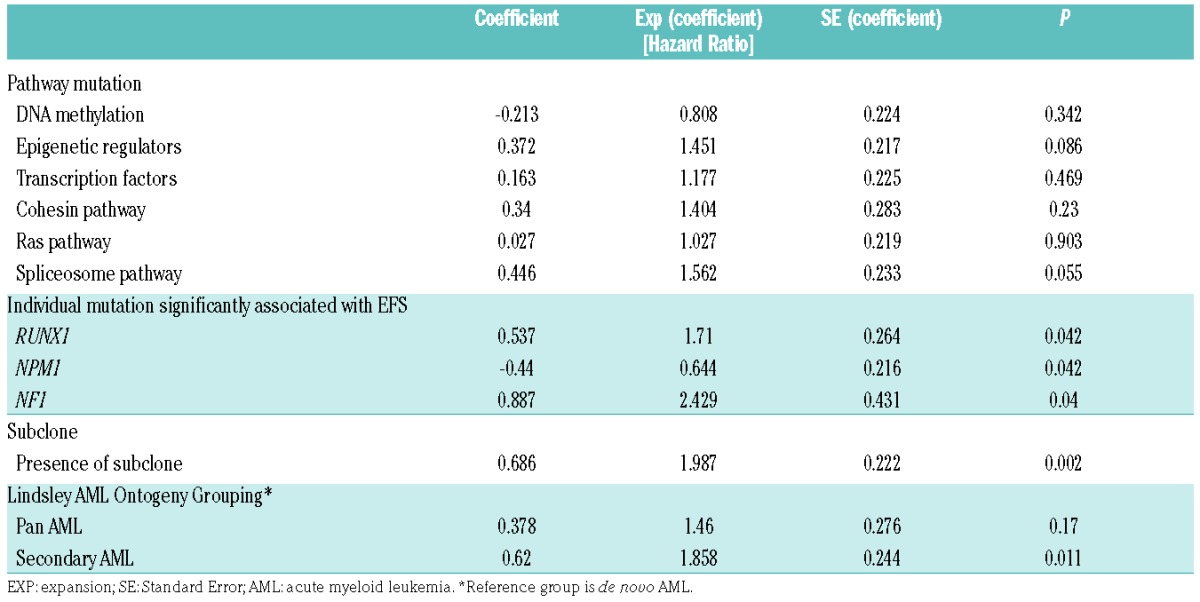
Table 1.
Transplant-adjusted univariate analysis of factors affecting event-free survival (EFS).
Mutation association with morphology
Specific dysplastic features were scored on a scale of 0–4 (Figure 1). A summary of the specific dysplastic features in each lineage and the Kendall coefficient of concordance for each lineage is shown in Online Supplementary Table S1. The median score for all 3 observers was used for all analyses. The distribution of dysplasia scores by pathway mutation and individual mutation is presented in Figure 2. Distribution of subclones by mutations is shown in Figure 3.
Figure 1.

Examples of typical morphological dysplastic features found in de novo acute myeloid leukemia (AML). Megakaryocytes often show separated lobes (A) and small size micromegakaryocytes (B). Dysplastic changes in myeloid cells, including hypogranular cytoplasm and abnormal nuclear lobation (C). Dysplastic erythroid cells are shown with irregular nuclear contors (D).
Figure 2.

The distribution of dysplasia scores by mutational pathway, subclone status and individual mutation. (A) Erythoid lineage dysplasia scores with mutation pathways and individual mutations. (B) Myeloid dysplasia scores. (C) Megakaryocyte dysplasia scores. (D) Dysplasia scores based on Lindsley et al. molecular ontogeny group.14
Figure 3.

Presence of mutations in de novo acute myeloid leukemia (AML). Analyzed by presence of a subclone.
When evaluating the overall degree of dysplasia in each lineage according to mutational pathways, we found that cohesin mutations (q=0.046) were associated with greater overall megakaryocytic dysplasia, while RAS pathway mutations were marginally associated with greater megakaryocytic dysplasia. Evaluating mutations individually indicated that STAG2 and RIT1 were marginally associated with greater overall megakaryocyte dysplasia (q=0.064 and q=0.056, respectively) and STAG2 also marginally associated with greater overall myeloid lineage dysplasia (q=0.052). No association was detected between Lindsley et al. molecular ontogeny groupings or the presence of a subclone with the overall dysplasia in any lineages. RAS pathway mutations (q=0.006) and specifically an FLT3-ITD (q=0.006) were significantly associated with non-evaluable erythroid lineage due to a lack of significant erythropoiesis in the leukemic BM.
Next, the associations between evaluable specific dysplastic features in each lineage, with individual mutations, pathways, AML genetic ontogeny grouping and subclones were explored. The presence of any (score >0) megakaryocytes with separated nuclear lobes was associated with RAS pathway mutations (q=0.0425) and NPM1 mutations (q=0.022). Frequent megakaryocytes with separated nuclear lobes (score ≥3) were associated with cohesin pathway (q=0.010), and STAG2 mutations in particular (q=0.010). No significant associations were detected when considering a score cut off of 2 or over for megakaryocytes with separated lobes. A lack (0 score) of erythroid nuclear irregularities was associated with epigenetic regulator mutations (q=0.035).
Mutation association with patients’ characteristics and other morphology
The age distribution differed by AML ontogeny groupings (q=0.002), where the median ages in the AML genetic ontogeny groups were 47.1 years [Interquartile Range (IQR 34.0 – 64.2)] for patients with pan-AML mutations only, 64.8 years (IQR 55.5–70.2) for patients with secondary AML type mutations, and 60.5 years (IQR) 51.5–67.3 for patients with de novo AML type mutations. Older age was also associated with spliceosome pathway mutations (q<0.0001), specifically SRSF2 mutation (q<0.0001). Patients with secondary-type AML mutations had the lowest median WBC (3.4×109/L; IQR 2.0–35.1) followed by pan-AML (16.2×109/L; IQR 3.4–43.7) and de novo AML had the highest median WBC (60.5×109/L; IQR 51.5–67.3, q=0.003). At presentation, a lower median WBC was found in AML with epigenetic regulator mutations (q=0.003), spliceosome mutations (q=0.015), ASXL1 (q=0.015) and BCOR (q=0.0029) mutations. A higher median WBC was found in AML with RAS pathway mutations (q<0.0001) and AML with NPM1 mutations (q=0.004).
Evaluation of BM showed that higher BM cellularity was associated with RAS pathway mutations (q=0.029), specifically FLT3-ITD (q=0.029), and NPM1 mutations (q=0.029). No significant differences in blast percentages in blood or BM were found in any of the mutations or mutation groupings.
Influence of mutations, including subclone analysis, and morphological findings on event-free survival
An analysis evaluating the association between EFS adjusted for receipt of allogeneic stem cell transplant (allo-SCT) and individual mutations, mutation pathways, presence or absence of subclones, and morphological dysplastic findings was performed in the 153 patients who were treated with standard induction chemotherapy. In this group, 75 patients (49%) received allo-SCT in first CR and 26 (17%) additional patients received allo-SCT after disease relapse.
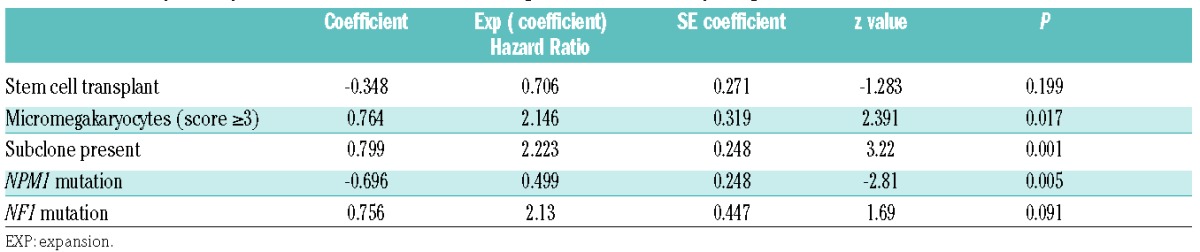
In transplant-adjusted univariate analysis, NPM1 mutation was associated with longer EFS (nominal P=0.042), while RUNX1 (P=0.042) and NF1 (P=0.04) mutations were associated with shorter EFS. Patients with secondary AML type mutations had shorter EFS (P=0.011) compared to patients with de novo AML type mutations (P=0.011). The presence of a subclone (see Figure 3) was also associated with shorter EFS (P=0.002). Among morphological features, the presence of frequent micromegakaryocytes (score ≥3) was associated with shorter EFS (P=0.018). Regarding the effect of the WHO Classification-defined AML-MRC category, there was no significant difference in EFS between AML-MRC versus all the other AML cases (P=0.941), nor was there any significant difference in EFS between AML-MRC versus AML-RUNX1 (P=0.253), AML-MRC versus combined AML-NPM1/AML-CEBPA (P=0.407), and AML-MRC versus AML-NOS (P=0.704). The final multivariable model indicated that micromegakaryocytes (score ≥3) and presence of a subclone were associated with shorter EFS while NPM1 mutation was associated with longer EFS; mutation in the RAS pathway gene NF1 was marginally associated with shorter EFS (Table 2).
Table 2.
Final transplant-adjusted multivariable model including mutational and morphological features
Discussion
To our knowledge, this is the first study to explore the relationship of specific dysplastic findings with mutation patterns in de novo AML. We found that among the broad spectrum of dysplastic morphologies evaluated in our study, only megakaryocyte morphology showed significant associations with mutation pattern or outcome. Interestingly, we found that one specific morphological feature, megakaryocytes with separated lobes, was correlated with cohesin pathway and NPM1 mutations but did not impact outcome, while another morphological feature, micromegakaryocytes, was associated with poor outcome, but did not correlate with any mutations.
The biological significance of morphological dysplasia in de novo AML is controversial, despite the fact that multilineage dysplasia (at least 50% dysplastic cells in at least 2 hematopoietic lineages) defines a subset of cases of AML with myelodysplasia changes that lack a defining cytogenetic abnormality or antecedent MDS.4 In our cohort of de novo AML cases, we found that cohesin pathway (and specifically STAG2) mutations were associated with greater overall megakaryocytic dysplasia and the specific finding of megakaryocytes with separated nuclear lobes. Mutations in cohesin pathway genes were present in 11% of all patients in our cohort; a higher rate than that reported by Thol et al. (5.9%) or in The Cancer Genome Atlas (2.5–3.5%). However, this rate might be influenced by the fact that our study included only AML cases without a history of a prior myeloid neoplasm and without MDS-associated cytogenetic abnormalities.2,17 Interestingly, Thol et al. and The Cancer Genome Atlas research network found a strong association between NPM1 mutations and mutations in cohesin genes,2,17 and in our study 6 (31%) of 19 cases with cohesin mutations also had NPM1 mutations, and the latter were also associated with frequent megakaryocytes with separated nuclear lobes. RAS pathway mutations were frequently seen in our cohort, being present in 58% of the cases and, similar to cohesin pathway and NPM1 mutations, were associated with megakaryocytes with separated nuclear lobes.
The underlying reasons for the association of cohesin pathway mutations with dysmegakaryopoiesis, and specifically separated megakaryocyte nuclei, are uncertain. Cohesin knockdown mice display a skewing in their stem cell compartment in the BM including myeloid hyperplasia, decrease in erythroid and megakaryocytic progenitors, and increase in nuclear size.21 Studies have also found that ASXL1 loss leads to MDS-like disease in mice and increased frequencies of dysplastic myeloid cells in BM.22 Li et al. suggested that ASXL1 binds to the cohesin complex and plays an essential role in maintaining normal chromatin separation during cell division, suggesting an overlapping molecular mechanism that underlies the pathogenesis of the myeloid disorders.22 Devillier et al. evaluated patients with AML-MRC and found that ASXL1 mutations were associated with a higher amount of dysgranulopoiesis, but not dyserythropoiesis or dysmegakaryopoiesis.13 Cho et al. found that spliceosome mutations were significantly found in AML-MRC, especially in cases with a preceding MDS or dysplasia.23 We did not find any associations between ASXL1 mutations and dysplasia, which could reflect our exclusion of cases with history of MDS or MDS-type cytogenetic abnormalities.
In multivariable analysis assessing the effects of individual mutations on outcome in our patient cohort, only NPM1 mutation was significantly associated with EFS. This finding validates the revision in the 2016 WHO Classification in which an NPM1 mutation supercedes the presence of multilineage dysplasia; cases with NPM1 mutation and multlineage dysplasia have been removed from the category of AML-MRC and are now retained in the prognostically favorable category of AML with mutated NPM1.4 We also found that the presence of a subclone (using criteria from Mossner et al.19) adversely affected outcome in the multivariable analysis. A subclone was most commonly seen in cases with DNA methylation (73 cases, 79%), RAS pathway (63 cases, 68%) and NPM1 (44 cases, 48%) mutations. Interestingly, although a subclone was associated with poor outcome in a multivariable analysis, it was not significantly associated with any specific mutation pattern, clinical features, or morphological features. Eisfield et al. investigated chronology of mutations during clonal evolution stratified by functional groups and showed that mutations in tumor suppressor genes, the cohesin complex or the spliceosome are commonly first mutational events.3 In the Papaemmanuil et al. study, mutations in DNMT3A, ASXL1, IDH1/2 and TET2 appeared to be acquired the earliest and were almost never found in isolation, while NPM1 mutations were usually secondary events.24 The only morphological features we identified to be associated with outcome was the presence of frequent (score ≥3) micromegakaryocytes; a finding that was independent of any mutations or mutation pattern. In MDS, Della Porta et al. also found that severity of megakaryocytic dysplasia correlated with outcome.12 Feng et al. found that micromegakaryocytes were independent prognostic factors and improved predictive accuracy of the International Prognostic Scoring System-revised (IPSS-R) in their study of 422 MDS patients.25 Our results suggest that micromegakaryocytes in de novo AML could represent a better marker for aggressive disease than the traditional WHO definition of multilineage dysplasia that takes into account dyserythropoiesis and dysgranulopoiesis, which did not affect outcome in our study. Our findings suggest that some cases currently diagnosed as AML-MRC on the basis of dyserythropoiesis and dysgranulopoiesis may not be truly biologically secondary disease and may not be appropriately classified with other AML-MRC cases defined by cytogenetics or history of MDS. Conversely, dysmegakaryopoiesis showed significant associations with specific mutations (when manifesting as forms with separated nuclear lobes) as well as shortened survival (when manifesting as micromegakaryocytes).
Our findings highlight the continued relevance of evaluating the background maturing hematopoiesis, specifically megakaryocytes, in de novo AML, even in the current era of detailed mutational analysis. The independent impact of micromegakaryocytes on outcome may reflect factors influencing megakaryocyte morphology that are not directly related to the leukemia mutation pattern, such as epigenetics, the BM microenvironment, or individual patients’ characteristics. The finding that a leukemia subclone adversely and independently impacted outcome implies that, not only the number and types of mutations, but also the clonal leukemia architecture should be taken into account in future AML risk stratification schemes.
Supplementary Material
Acknowledgments
The authors would like to thank Susan Wong for her editorial assistance with this work.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/4/626
References
- 1.Patel JP, Gönen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas Research Network, Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eisfeld AK, Mrózek K, Kohlschmidt J, et al. The mutational oncoprint of recurrent cytogenetic abnormalities in adult patients with de novo acute myeloid leukemia. Leukemia. 2017;31(10):2211–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. [DOI] [PubMed] [Google Scholar]
- 5.O’Donnell MR, Tallman MS, Abboud CN, et al. Acute Myeloid Leukemia, Version 3.2017, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2017;15(7):926–957. [DOI] [PubMed] [Google Scholar]
- 6.Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: Recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115(3):453–474. [DOI] [PubMed] [Google Scholar]
- 7.Miesner M, Haferlach C, Bacher U, et al. Multilineage dysplasia (MLD) in acute myeloid leukemia (AML) correlates with MDS-related cytogenetic abnormalities and a prior history of MDS or MDS/MPN but has no independent prognostic relevance: a comparison of 408 cases classified as “AML not otherwise specified” (AML-NOS) or “AML with myelodysplasia-related changes” (AML-MRC). Blood. 2010;116(15):2742–2751 [DOI] [PubMed] [Google Scholar]
- 8.Falini B1, Macijewski K, Weiss T, Bacher U, Schnittger S, Kern W, et al. Multilineage dysplasia has no impact on biologic, clinicopathologic, and prognostic features of AML with mutated nucleophosmin (NPM1). Blood. 2010;115(18):3776–3786. [DOI] [PubMed] [Google Scholar]
- 9.Díaz-Beyá M, Rozman M, Pratcorona M, et al. The prognostic value of multilineage dysplasia in de novo acute myeloid leukemia patients with intermediate-risk cytogenetics is dependent on NPM1 mutational status. Blood. 2010;116(26):6147–6148. [DOI] [PubMed] [Google Scholar]
- 10.Rozman M1, Navarro JT, Arenillas L, et al. Multilineage dysplasia is associated with a poorer prognosis in patients with de novo acute myeloid leukemia with intermediate-risk cytogenetics and wild-type NPM1. Ann Hematol. 2014;93(10):1695–1703. [DOI] [PubMed] [Google Scholar]
- 11.Weinberg OK, Pozdnyakova O, Campigotto F, et al. Reproducibility and prognostic significance of morphologic dysplasia in de novo acute myeloid leukemia. Mod Pathol. 2015;28(7):965–976. [DOI] [PubMed] [Google Scholar]
- 12.Della Porta MG, Travaglino E, Boveri E, et al. Rete Ematologica Lombarda (REL) Clinical Network. Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes. Leukemia. 2015;29(1):66–75. [DOI] [PubMed] [Google Scholar]
- 13.Devillier R, Mansat-De Mas V, Gelsi-Boyer V, et al. Role of ASXL1 and TP53 mutations in the molecular classification and prognosis of acute myeloid leukemias with myelodysplasia-related changes. Oncotarget. 2015;6(10):8388–8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindsley RC, Mar BG, Mazzola E, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alvarez Argote J, Dasanu CA. ASXL1 mutations in myeloid neoplasms: pathogenetic considerations, impact on clinical outcomes and survival. Curr Med Res Opin. 2017:1–7. [DOI] [PubMed] [Google Scholar]
- 16.Gibson CJ, Lindsley RC, Tchekmedyian V, et al. Clonal Hematopoiesis Associated With Adverse Outcomes After Autologous Stem-Cell Transplantation for Lymphoma. J Clin Oncol. 2017;35(14):1598–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinberg OK, Gibson CJ, Blonquist TM, et al. NPM1 mutation but not RUNX1 mutation or multilineage dysplasia defines a prognostic subgroup within de novo acute myeloid leukemia lacking recurrent cytogenetic abnormalities in the revised 2016 WHO classification. Am J Hematol. 2017;92(7):E123–E124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kluk MJ, Lindsley RC, Aster JC, et al. Validation and Implementation of a Custom Next-Generation Sequencing Clinical Assay for Hematologic Malignancies. J Mol Diagn. 2016;18(4):507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mossner M, Jann JC, Wittig J, et al. Mutational hierarchies in myelodysplastic syndromes dynamically adapt and evolve upon therapy response and failure. Blood. 2016;128(9):1246–1259. [DOI] [PubMed] [Google Scholar]
- 20.Thol F, Bollin R, Gehlhaar M, et al. Mutations in the cohesin complex in acute myeloid leukemia: clinical and prognostic implications. Blood. 2014;123(6):914–920. [DOI] [PubMed] [Google Scholar]
- 21.Mullenders J, Aranda-Orgilles B, Lhoumaud P, et al. Cohesin loss alters adult hematopoietic stem cell homeostasis, leading to myeloproliferative neoplasms. J Exp Med. 2015;212(11):1833–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li Z, Zhang P, Yan A, et al. ASXL1 interacts with the cohesin complex to maintain chromatid separation and gene expression for normal hematopoiesis. Sci Adv. 2017;3(1):e1601602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cho YU, Jang S, Seo EJ, et al. Preferential occurrence of spliceosome mutations in acute myeloid leukemia with preceding myelodysplastic syndrome and/or myelodysplasia morphology. Leuk Lymphoma. 2015;56(8):2301–2308. [DOI] [PubMed] [Google Scholar]
- 24.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng G, Gale RP, Cui W, et al. A systematic classification of megakaryocytic dysplasia and its impact on prognosis for patients with myelodysplastic syndromes. Exp Hematol Oncol 2016;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.