

Abstract
Intrachromosomal amplification of chromosome 21 is a heterogeneous chromosomal rearrangement occurring in 2% of cases of childhood precursor B-cell acute lymphoblastic leukemia. These abnormalities are too complex to engineer faithfully in animal models and are unrepresented in leukemia cell lines. As a resource for future functional and preclinical studies, we have created xenografts from the leukemic blasts of patients with intrachromosomal amplification of chromosome 21 and characterized them by in-vivo and ex-vivo luminescent imaging, flow immunophenotyping, and histological and ultrastructural analyses of bone marrow and the central nervous system. Investigation of up to three generations of xenografts revealed phenotypic evolution, branching genomic architecture and, compared with other B-cell acute lymphoblastic leukemia genetic subtypes, greater clonal diversity of leukemia-initiating cells. In support of intrachromosomal amplification of chromosome 21 as a primary genetic abnormality, it was always retained through generations of xenografts, although we also observed the first example of structural evolution of this rearrangement. Clonal segregation in xenografts revealed convergent evolution of different secondary genomic abnormalities implicating several known tumor suppressor genes and a region, containing the B-cell adaptor, PIK3AP1, and nuclear receptor co-repressor, LCOR, in the progression of B-cell acute lymphoblastic leukemia. Tracking of mutations in patients and derived xenografts provided evidence for co-operation between abnormalities activating the RAS pathway in B-cell acute lymphoblastic leukemia and for their aggressive clonal expansion in the xeno-environment. Bi-allelic loss of the CDKN2A/B locus was recurrently maintained or emergent in xenografts and also strongly selected as RNA sequencing demonstrated a complete absence of reads for genes associated with the deletions.
Introduction
Xenograft models of leukemia have been used to address a number of important fundamental and translational research questions relating to: the nature of leukemia stem cells, clonal evolution and experimental therapies.1–8 Limiting dilution studies have demonstrated that leukemia-initiating cells are common, while fluorescence in situ hybridization (FISH), genomic arrays, analysis of immunoglobulin/T-cell receptor rearrangements, immunophenotype and drug response have suggested that the disease can be propagated through multiple generations of mice with high fidelity.
Intrachromosomal amplification of chromosome 21 (iAMP21) is an intriguing cytogenetic abnormality, defining a specific subgroup of approximately 2% of cases of childhood precursor B-cell acute lymphoblastic leukemia (B-ALL). Chromosome 21 genomic profiles, although highly variable, always involve amplifications, flanked by regions of normal copy number or deletion.9,10 We have shown that the oncogenic potential of chromosome 21 is optimized through a combination of catastrophic sequence reorganization, driven by chromothripsis, deletion and amplification, resulting from dicentric chromosome formation, breakage-fusion-bridge cycles and whole chromosome arm duplications.11 This mechanism has the potential to produce a near infinite number of alternative derivative chromosomes 21. The structure of the iAMP21 chromosome is stabilized by telomere acquisition or duplication, while a combination of protected amplified genes are postulated to confer an overall growth advantage, leading to the development of ALL. Several lines of evidence indicate that iAMP21 is a stable, primary genetic change: (i) among 530 patients, iAMP21 was reported as a sub-clonal abnormality in only a single case;12 (ii) the iAMP21 chromosome morphology remains consistent between cells in the same patient; and (iii) the same chromosome structure is observed at diagnosis and relapse.9 A range of specific secondary genetic abnormalities: CRLF2 activating rearrangements, X chromosome gain, deletions of RB1, ETV6, the long arm of chromosome 7 (7q) and 11q, and mutations of the RAS pathway frequently co-occur with iAMP21.9,12,13
This distinct iAMP21-ALL subgroup is clinically defined by older age (median 9 years), low white blood cell counts and a high risk of relapse on standard therapy.14–16 Intensive therapy significantly reduces the risk of relapse17,18 but associated morbidity highlights an urgent need for less toxic regimens. Development of rational targeted therapies requires understanding of the mechanism by which these rearrangements initiate leukemia. However the requisite tools for functional studies are lacking because no iAMP21-ALL cell lines exist and the complex nature of the abnormalities exclude their reproduction in engineered animal models. To address this shortfall, we transplanted primary leukemia cells from five iAMP21-ALL patients into NOD/LtSz-scid IL2Rg null (NSG) mice. In-vivo luminescent imaging, to track the physical development of ALL, was used to assess these xenografts as potential models for functional and pre-clinical studies. In addition, we characterized the disease morphology at the microscopic and ultrastructural levels and, as a first application, have performed extensive genomic analysis to investigate clonal heterogeneity of iAMP21-ALL, from which some intriguing findings have emerged.
Methods
Patients
Viable cells from children diagnosed with iAMP21-ALL, as previously defined,19 were provided by the Bloodwise Childhood Leukaemia Cell Bank. Ethical approval was obtained for all patients and informed consent was granted in accordance with the Declaration of Helsinki. Karyotypes and demographic details of the patients used to generate xenografts or as controls for histological analysis are presented in Online Supplementary Tables S1 – S3.
Xenografts and isolation of leukemia cells
Primografts were created by intrafemoral injection of patients’ cells into NSG mice, as previously described.1 Between 2×104 and 2×106 primograft bone marrow or spleen cells from each mouse were used in the same procedure to create secondary and tertiary xenografts (Online Supplementary Table S4). Xenografts were culled at end stage-disease as defined in the Online Supplementary Methods. Bone marrow cells flushed from femora and disaggregated spleens were passed through 40 μm cell strainers. Leukemic cells used for all experimental work were purified from spleen preparations by separation over Ficoll-Paque [density 1.077 g/mL] (G.E. Health Care, Buckinghamshire, UK). Proportions of human and mouse cells and immunophenotypes of the human leukemia fractions were determined by flow cytometry as described in the Online Supplementary Methods.
Lentiviral transduction, in-vitro culture and in-vitro, in-vivo and ex-vivo imaging of xenograft cells
Detailed procedures are provided in the Online Supplementary Methods.
Histopathology and transmission electron microscopy
Detailed procedures are provided in the Online Supplementary Methods.
Single nucleotide polymorphism arrays
DNA extraction and SNP6.0 array hybridization and analysis were performed, as previously described.13 To define regions of chromosome 21 copy number evolution, single nucleotide polymorphism copy number values were subtracted between secondary xenografts 2°1B and 2°1A. Copy number abnormalities (CNA) in immunoglobulin or T-cell receptor regions, those not involving coding gene regions, present in patients’ remission samples or listed in the Toronto Database of Genomic Variants, are not reported. Genomic positions are those in the Hg19 database.
Fluorescence in situ hybridization and multiplex ligation-dependent probe amplification
Dual color FISH on 100–200 interphase cells was performed using fluorescently labeled BAC probes hybridizing to the RUNX1 (RP11-773I18) and APP (RP11-66H5 and RP11-15D13) genomic regions or commercially available probes to the CDKN2A/B genomic region and chromosome 9 centromere (CytoCell, Cambridge, UK), as previously described.20 Multiplex ligation-dependent probe amplification (MLPA) was performed using the SALSA MLPA 335 kit (MRC-Holland, the Netherlands), as previously described.21
Analysis of RAS pathway mutations and RNA sequencing
Detailed procedures are provided in the Online Supplementary Methods.
Results
Development of xenografts and characterization by in-vivo and ex-vivo imaging
Of six primary and two relapsed cases of iAMP21-ALL transplanted into femora of NSG mice, five, including one relapsed case, developed ALL derived from the human cells, in one or more animals (Online Supplementary Table S1). The mean time to development of end-stage disease in primografts was 30 weeks and splenomegaly was seen in all engrafted animals. Secondary and tertiary xenografts were established in three and one cases, respectively, and all were assigned unique identifiers indicating passage number and patient of origin, for example 2°3e was one of several secondary xenografts derived from patient 3. Xenograft leukemia cells constituted between 40–92% of bone marrow and 23–53% and 79–99% of crude and purified spleen samples, respectively (Online Supplementary Table S4). Essentially all human cells isolated from xenografts expressed the B-cell markers CD19 and CD10 but analysis of CD34 and CD38 demonstrated considerable phenotypic divergence between mice (Online Supplementary Table S5 and Online Supplementary Figure S1).
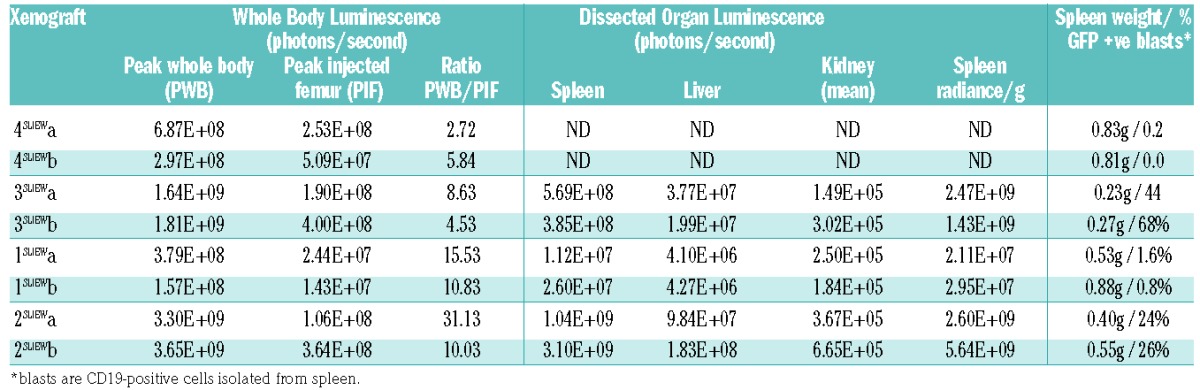
To investigate their potential for use in in-vivo and in-vitro functional studies, we transduced xenograft stocks from four iAMP21-ALL patients with the pSLIEW lentivirus vector that expresses luciferase and enhanced green fluorescent protein (EGFP).22 Three days after transduction, a total of 3×106 cells from each patient were transplanted by intra-femoral injection into two NSG mice each, here identified by the patients’ number followed by aSLIEW or bSLIEW. Less than 1% of transduced cells were EGFP-positive by fluorescence-activated cell sorting analysis at this time point (Online Supplementary Figure S2) or by fluorescence microscopy after 1 week of culture on MS-5 feeder layers (data not shown); nevertheless, by 2–4 weeks following transplantation, luminescent signals, clearly localized to the injected femora, were seen on whole body imaging of all mice. Leukemia spread to other bones and organs with noticeable variation in the strength of signal at some sites (Online Supplementary Figure S3). This variation was highlighted by measurement of luminescent signals from organs post-mortem and by analysis of the relationship between signal development at different sites over time (Figure 1A,B and Table 1). Signal variation in the spleen was shown to relate to the proportion of infiltrating blast cells that expressed EGFP rather than to overall tumor load (Figure 1C, Table 1 and Online Supplementary Table S4). Serial three-dimensional reconstructions of one xenograft showed dramatic increases in signals from the spleen and head between weeks 11–15 (Figure 1D).
Figure 1.

In xivo and ex-vivo imaging of xenografts. (A) Serial quantification of the luminescent signal from injected and contralateral femora and the whole body for each xenograft suggesting variations in the rate at which cells migrate from the site of injection and the degree to which different sites are infiltrated by transduced cells. For example, in 3a/bSLIEW the strength of signal from the contralateral femur lagged substantially behind that of the injected femur until weeks 10–13, while in 1a/bSLIEW and 2bSLIEW the two femora showed similar levels from week 2. (B) Example of ex-vivo imaging of dissected organs showing total luminescent readings for spleen, liver and kidney. Images are representative of six animals analyzed. (C) Examples of FACS analysis of cells isolated from the spleens of 3aSLIEW and 4aSLIEW, demonstrate marked contrast in the proportion of CD19-positive cells that are EGFP-positive. (D) A single example of serial three-dimensional reconstructions of luminescent signals in 2aSLIEW, 11 and 15 weeks after transplant. Arrows point to regions of the skull, spleen and a third site showing strong signal increase between these two time points. The skeleton projected for orientation is not derived from this animal.
Table 1.
In-vivo and ex-vivo luminescent imaging data and spleen weights for xenografts transplanted with iAMP21-ALL cells transduced with pSLIEW.
Morphology and ultrastructure of bone marrow and central nervous system reveals patient-specific heterogeneity, including evidence for systemic bone marrow niche destruction
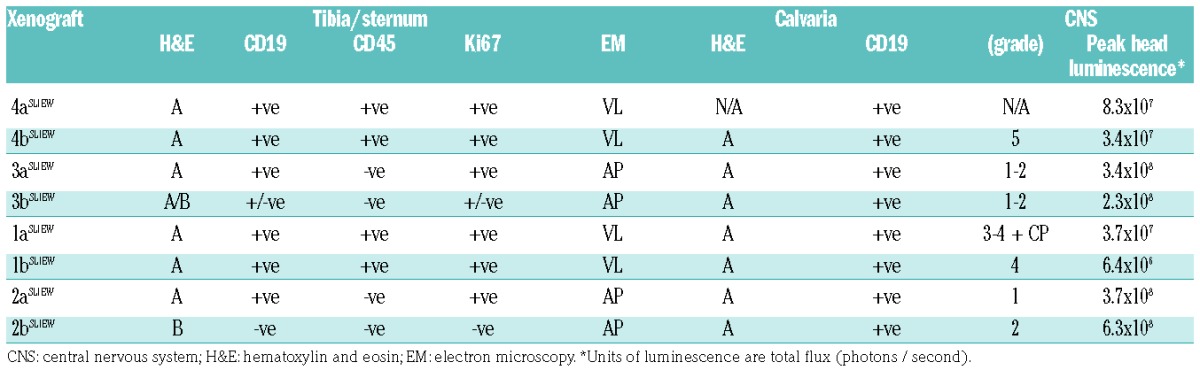
To investigate bone marrow morphology and central nervous system (CNS) involvement in the iAMP21-ALL models, we examined sections through tibiae, sterna and heads of the mice engrafted with SLIEW-transduced cells. Control NSG bone marrow stained with hematoxylin and eosin resembled that of a wild-type mouse23 and was negative for anti-human CD19, CD45 and Ki-67 staining (Figure 2A and Online Supplementary Figure S4). In xenografts, two types of morphology were seen, both of which differed from controls; type A, which closely resembled that of iAMP21-ALL patients’ trephines (Figure 2B,G, Online Supplementary Figures S4 and S5 and Online Supplementary Table S2) and type B, which although abnormal, clearly differed from the patients’ trephines (Figure 2C,G and Online Supplementary Figure S4). Cells in type A but not type B sections were actively cycling and of human origin as indicated by staining with anti-human Ki67, CD19 and CD45 antibodies (Figure 2D and Online Supplementary Figure S4). Individual sections presented with either type A or B morphology only with the exception of one sternal segment in which both types co-existed (Figure 2D). There was relatively sharp demarcation between the A and B type areas, suggesting that the iAMP21 ALL cells were organized into massive clumps that did not diffuse easily within the lumen.
Figure 2.

Histological sections of bone marrow from controls and xenografts transplanted with SLIEW transduced cells. (A) Control NSG mouse: hematoxylin and eosin (H&E) stained femur showing heterogeneous cell types, abundant megakaryocytes and vascular structures. (B) H&E stained femur from 4aSLIEW showing tightly packed homogeneously stained cells and an absence of megakaryocytes and vascular structures (image representative of six animals showing only morphology type A). (C) H&E stained femur from 3bSLIEW showing heterogeneous cell types but in comparison with controls, loss of cellularity and organization, absence of vascular structures, reduced numbers of megakaryocytes and presence of small darkly stained cells or cellular fragments (image representative of two animals showing morphology type B). (D) The only example seen of co-existence of A and B type morphology in a sternal segment of 3bSLIEW: first left panel; H&E stained whole section with box marking the region shown in the second left panel. Second left panel; H&E stained detail of a single sternum segment displaying both A and B type morphologies. Middle panel; whole sections stained with anti-human CD19 and Ki-67 antibodies, arrowheads indicate three regions corresponding to high resolution images in the right hand panels. Right hand panels; high resolution images of anti-CD19 and Ki-67 staining from regions 1, 2 and 3. Anti-CD19 and Ki67 stained human leukemia cells remain tightly packed (region 2) with little diffusion to adjacent areas of acellular marrow (region 3). Anti-Ki67 and CD19 staining demonstrating that the proportion of cycling human cells are reduced in region 2 compared with region 1 suggesting a microenvironment less favorable for leukemia cell growth. (E) Examples of TEM images of xenograft tibia sections. In 4aSLIEW and 1aSLIEW cells appear homogeneous compared with controls (Online Supplementary Figure S6) and have a high nuclear to cytoplasmic ratio (VL morphology). 3aSLIEW and 2aSLIEW displaying evidence of cell death and characteristics of apoptosis, such as chromatin clumping and nuclear fragmentation (AP morphology). Images are representative of four animals each showing VL and AP morphology. (F) Examples of skull and brain sections from 3bSLIEW (left panel) and 1bSLIEW (right panel) showing heavy infiltration of the calvaria in both cases (arrow 1), light (3bSLIEW) and heavy (1bSLIEW) infiltration of the meninges, respectively (arrow 2). Images are representative of seven animals. (G) Two examples of H&E stained patients’ trephines. The left hand panel is from patient 1 for comparison with the leukemia cells from the same patient in a mouse (1aSLIEW) tibia section shown in (B). Images are representative of seven iAMP21-ALL patients’ trephines analyzed. Scale bars are; (A, B, C) two right hand panels of (D and G) - 50 μm, far left hand and two middle panels of (D) – 1 mm, second from left panel of (D and F) 200 μm and (E) - 10 μm.
We performed transmission electron microscopy of decalcified sections of different tibiae or forelimb bones from each mouse. Xenograft ultrastructure always differed from that of wild-type controls and, as with bright field microscopy, two distinct categories could be identified (Figure 2E and Online Supplementary Figure S6). The first was termed viable leukemia (VL), as mitotic figures were present and cells appeared normal, although homogeneous by comparison with controls. There were more connections and less extracellular space between cells in preparations from 4a/bSLIEW compared with controls and 1a/bSLIEW. The second category, equivalent to histological type B, was termed apoptotic (AP), as no mitotic figures were present and cells were depleted in number with classical signs of apoptosis, in the form of condensed chromatin localized to the periphery of the nucleus.24
We also examined sections through the skulls and CNS of seven of the eight xenografts (Figure 2F and Online Supplementary Figure S7), revealing calvaria in all cases, packed with homogeneously stained cells resembling the A type morphology of tibial sections. CNS involvement ranged from small foci of leukemia cells to heavy meningeal infiltration, extending into the choroid plexus in one case. Comparison of CNS histological grades for each xenograft with bone marrow histopathology and transmission electron microscopy data (Table 2) showed heavy CNS involvement only in 1a/bSLIEW and 4a/bSLIEW, correlating with tibia marrow histological type A and transmission electron microscopy type VL. We infer the proportion of transduced cells infiltrating the CNS varied between mice because luminescent signals from the head failed to correlate with histological grade (Table 2).
Table 2.
Summary of histological and ultrastructural (EM) data for bone marrow and CNS of mice transplanted with iAMP21-ALL cells.
To investigate the relative incidence of the morphological types we examined bone marrow sections, stained with hematoxylin and eosin and anti-CD19, from 13 additional xenografts derived from seven B-ALL patients (Online Supplementary Tables S3 and S6). Among these cases, type B morphology was seen in two primografts, one derived from a relapsed iAMP21-ALL patient and the second from a case with high hyperdiploid ALL; areas of A type morphology were also seen in both (Online Supplementary Figure S8). Other xenografts displayed A type morphology either exclusively (Online Supplementary Figure S9) or infiltrating apparently normal mouse bone marrow (Online Supplementary Figure S10). Interestingly these latter cases supported our initial observation that the ALL cells may grow in clumps because CD19-positive cells formed distinct clusters.
Segregation of copy number abnormalities in xenografts implicates known and novel genes in the progression of acute lymphoblastic leukemia with intrachromosomal amplification of chromosome 21
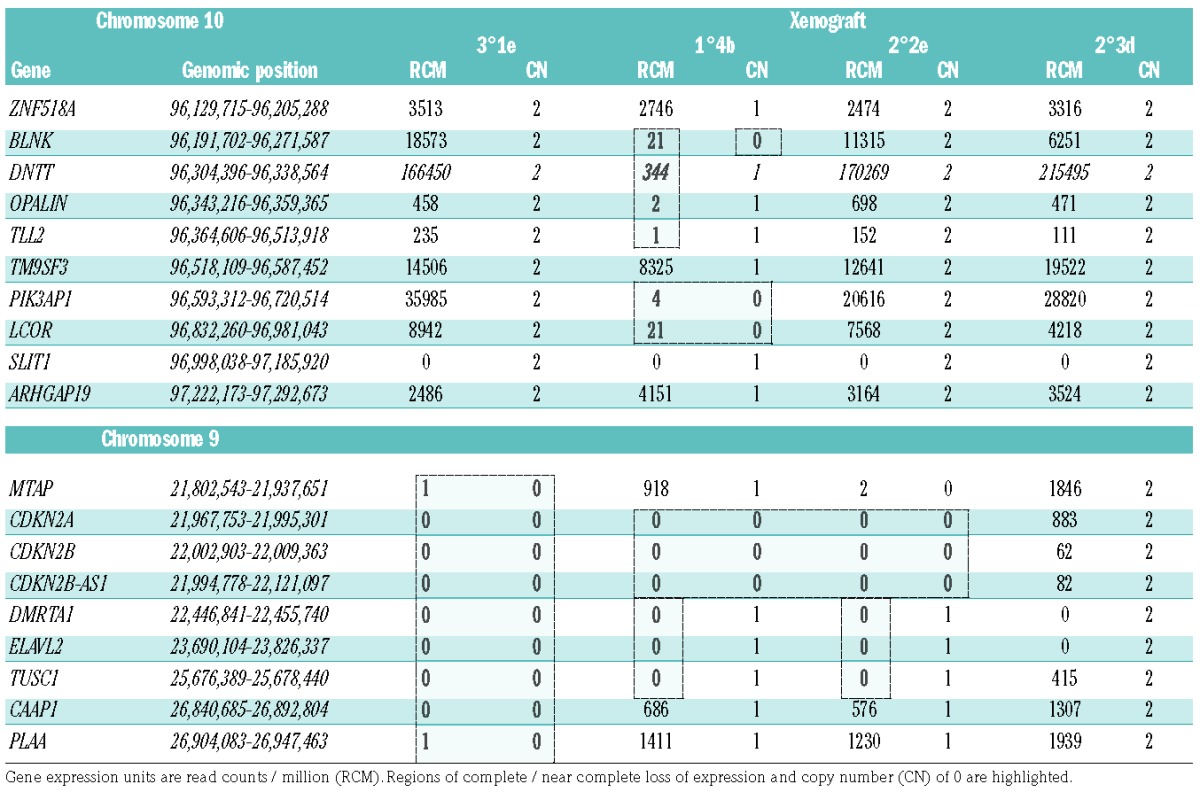
We used SNP6.0 array profiles to evaluate the genomic stability of iAMP21-ALL in 21 xenografts from five patients. Presentation and remission samples were available for all except patient 4. A core of three to 16 concordant CNA, involving coding gene regions, were clonal at presentation and retained in all xenografts (Online Supplementary Table S7). The existence of competing sub-clones and branching genomic evolution was demonstrated by discordant CNA, which occurred at a rate of between four and 12 (Figure 3 and Online Supplementary Table S7). Clonal trisomies or copy number neutral loss of heterozygosity, present in each patients’ sample, were lost after transplantation, while deletions and amplifications were typically sub-clonal and either lost or increased in level or newly emergent as sub-clones in xenografts. Exceptionally in xenografts of patient 1, a sharp transition in genomic architecture, involving clonal gain of CNA of three chromosomes, occurred. Chromosome 21 profiles usually remained unchanged across samples (Online Supplementary Figure S11), but interestingly in cells from patient 1, we observed structural evolution of the iAMP21 chromosome, involving a small region of copy number gain and nine distinct regions of loss of one or two copies (Figure 3A and Online Supplementary Table S8). Importantly the additional deletions did not affect two regions predicted to contain critical oncogenes11 but did re-define the proximal boundary of the region of highest level of amplification9 from 21:32,813,553–37 to 21:33,949,423. By FISH, we confirmed that the RUNX1 and APP gene regions were maintained at the same level of amplification and reduced in copy number from three to one, respectively. Additional rearrangements included bi-allelic deletion of the short arm of chromosome 9 (9p), resulting in homozygous loss of CDKN2A/B, as confirmed by MLPA (Online Supplementary Table S9), and mono-allelic deletion of 3p, involving the CMTM genes 6–8. SNP6.0 array and FISH provided no evidence of these CNA prior to their emergence in 2°1a. However, as previously reported, two reads in whole genome sequencing data were consistent with the presence of a minor clone carrying the chromosome 3 deletion in the patient’s cells at presentation.11
Figure 3.

Analysis of evolution of copy number abnormalities in xenograft models. Panels in blue boxes show heat maps of copy number from SNP6.0 arrays for chromosomal regions showing evidence of clonal genomic evolution in xenografts, P (presentation), R (remission), 1°,2° and 3° (primary, secondary and tertiary xenografts). Box-flow diagrams illustrate loss, gain or change in level of genomic abnormalities. White boxes; sample analyzed, gray boxes; sample not analyzed, red type; CN gain of one, purple; CN gain of two, light red; sub-clonal CN gain, black; CN loss of one, blue; CN loss of two, gray; subclonal loss, green CN-LOH. * indicates deletion was detected by whole genome sequencing but not by SNP6.0 array. (A) Patient 1 – including heat maps of 9p demonstrating gain of bi-allelic deletion of a region containing CDKN2A/B and focal mono-allelic deletion of a region of 3p containing the CMTM genes 6–8. Chromosome 21 heat maps illustrate a complex pattern of copy number gain and loss characteristic of iAMP21 and also demonstrate additional CN changes in one 2° and all 3° xenografts. The color-coded bar depicts regions of chromosome loss or gain in the derivative iAMP21, [der(iAMP21)]. Green box; chromosome 21 CN profiles before and after iAMP21 evolution and showing the position of probes used for FISH analysis. Upper black box; FISH image of metaphase and interphase cells showing multiple copies of the RUNX1 region and three copies of the APP region at presentation. Lower black box; FISH images showing three or one copy of the APP region in cells from the patient, a 1° and two 2° xenografts. (B) CNA discordant between patient 2 and xenografts including; loss of whole chromosome (WC) X, gain of an iso(Xp) and gain of a focal deletion of Xp involving the genes ZNF157 and ZNF41. (C) CNA discordant between patient 3 and xenografts including different deletions involving ETV6 one of which potentially resulted in a novel ETV6-BICD1 fusion gene. (D) Differences in copy number profiles between three 1° xenografts from patient 4. These included two different large mono-allelic deletions of chromosome 10 (1 and 2) both of which contained a focal bi-allelic deletion (3), present in all xenografts and involving PIK3AP1 and LCOR. A second bi-allelic deletion (4) was present in two xenografts only and involved BLNK. Other discordant CNA included complex rearrangements of 17q and 14q, focal deletions of 4q34.1, 16p13 and 22q13.1 and a focal amplification of 4q13.2. E. Sub-clonal deletions at 2p25.1, 3q13.3 and 12q24.1 present in patient 5 became dominant clones in the xenograft while a sub-clonal deletion of 3p21.3 was lost and sub-clonal deletions of 1q42.3 and 5q33.3 (marked by red arrow 1) were gained. In the patients’ relapse sample the 3p21.3 deletion became dominant while other sub-clonal abnormalities detected at presentation were lost. As with the xenograft a focal deletion of 5q33.1 (marked by red arrow 2) emerged at relapse. The two 5q deletions both resulted in loss of coding exons of EBF1. Genomic positions of breakpoints derived from SNP6.0 analysis and genes contained within focal CNA, for all patients and xenografts, are annotated in Online Supplementary Table S7.
Suggestive of convergent clonal evolution and highlighting the relevance of specific chromosomal regions to disease progression, several were targeted by different abnormalities segregated in xenografts from the same patient. Consistent with an oncogenic role for genes on Xp, patient 2 carried competing sub-clones marked by gain of a whole X chromosome or isochromosome Xp (Figure 3B). Whether emergence of a focal deletion of Xp, involving the zinc finger genes, ZNF157 and ZNF41, was related to the presence of the larger scale CNA remains unclear but they were unlikely to have been driven by CRLF2 overexpression, as genetic analysis ruled out rearrangement of this locus in the patient’s sample.25 In patient 3 large overlapping deletions of 12p, both involving ETV6, were segregated (Figure 3C). Although no patient’s material was available, differences in CNA involving the long arm of chromosome 10 were identified in xenografts from a relapse sample of patient 4 (Figure 3D). Strongly indicative of convergent evolution and hence a role in leukemia progression, the same focal bi-allelic deletion, involving PIK3AP1 (BCAP) and LCOR (C10orf21), was nested within two distinctly different, large, mono-allelic 10q deletions. Evidently of independent origin, as it was detected only in a single xenograft, one of the large deletions also harbored a second likely co-operating focal bi-allelic deletion that resulted in loss of BLNK. Lastly, passage of patient 5 cells in a primary xenograft resulted in concomitant loss of copy number neutral loss of heterozygosity of 12q, with progression from sub-clonal to clonal deletion of a region of 12q containing SH2B3 (Figure 3E). Comparison between relapse and the xenograft showed no overlap in progression of specific CNA, although interestingly the EBF1 gene was targeted by different deletions in the two samples.
Mutations affecting the RAS pathway drive clonal expansion
To investigate progression of mutations activating the RAS pathway previously identified in patient 113 we performed whole exome sequencing of selected and Sanger sequencing of all derived xenografts (Figure 4A). Interestingly, while an NF1 mutation remained clonal in all samples, two different mutations affecting NRAS and one of KRAS marked a dramatic clonal evolution. Remarkably the KRAS mutation, present as a dominant clone in both primary and one secondary xenograft was undetectable at a read depth of over 6000 in the presentation sample and also undetected in other xenografts which instead carried a dominant NRAS mutation detected in only 1% of reads at presentation. NRAS and FLIT3 mutations detected at presentation in patients 3 and 5, respectively, were clonal in xenografts while the FLIT3 mutation was lost at relapse (Figure 4B,C).
Figure 4.

Analysis of mutations affecting the RAS pathway. The key to the analytical methods used are shown top right. Blue boxes show non-synonymous RAS pathway mutations identified. White boxes summarize the estimated variant allele (VA) frequencies and methods of analysis used for patients’ and xenograft samples. (A) In patient 1 an NF1 mutation was clonal at presentation and in all xenografts. By contrast mutations of NRAS and KRAS showed marked fluctuations in VA frequency; NRAS (1), present in the patient as a sub-clone, was detected in both 1° xenografts but not in blasts from 2° and 3° animals. NRAS (2), identified by high depth targeted sequencing in 1% of patients’ sample reads, was undetected by whole exome sequencing in primografts but emerged as a dominant clone in 2°2a and all derivative 3° xenografts. A mutation of KRAS, although undetected in more than 6000 reads by targeted sequencing of the patient’s sample, marked the dominant clone present in primografts and 2°2b. Sanger sequence traces illustrate the relationship between the NF1, NRAS (2) and KRAS mutations in the two 2° xenografts, traces shown for 2°1a are also representative of 1°1a and 1°1b, traces shown for 2°1b are also representative of 3°1a-g. (B) In patient 3 an NRAS mutation identified in the patient remained clonal in all 1° and 2° xenografts. The Sanger sequencing trace shown for 1°3a is representative of all xenografts. (C) In patient 5 a FLT3 itd, detected as a minor sub-clone by exome sequencing of the presentation sample, became dominant in the 1° xenograft as demonstrated by the generation of two distinct exon 14 polymerase chain reaction (PCR) amplicons of equal intensity (first lane bottom right). In contrast only a single PCR product was amplified from the relapse sample of this patient (second lane bottom right). Mutations detected in the patients’ presentation samples have been previously published.13
Transcriptional environment associated with deletions
Anticipating that xenograft preparations, in contrast to patients’ samples, would be free of non-leukemic human cells, we used them to investigate the transcriptional environment associated with bi-allelic deletions (Table 3). By comparison with non-deleted samples, and confirming clonal dominance of the chromosome 10 deletions, we saw marked reductions in the levels of transcription of PI3KAP1, LCOR and BLNK. Loss of the BLNK genomic region also resulted in silencing of DNTT and OPALIN. Three of the four sequenced xenografts carried bi-allelic 9p deletions affecting CDKN1A/B. Indicating strong pressure for clonal selection of bi-allelic loss, read counts within this region were reduced to zero in deleted cases. Interestingly two focal bi-allelic deletions were associated with silencing not only of the physically deleted genes but also of TUSC1, positioned more than 3 Mb away from the deletion boundaries.
Table 3.
The expression of genes in xenograft-derived iAMP21-ALL cells within and neighboring regions of bi-allelic deletion of chromosomes 10 and 9.
Discussion
Compared with genetically engineered animal models, xenografts bring several advantages to the study of ALL, not least, their potential to fully recapitulate the spectrum of genomic abnormalities that occur within individual patients of a given genetic sub-type. This is particularly relevant to iAMP21-ALL, in which the primary abnormality is structurally complex, unique to each patient and impossible to reproduce in engineered animal models. As there are no cell lines carrying iAMP21, the xenografts presented here represent an important resource for future functional and pre-clinical studies.
Highlighting the potential of lentiviral constructs integrated into xenograft cells, we demonstrated their in vivo expression. However we observed considerable temporal and spatial variation in signal development that, as demonstrated by analysis of spleen and CNS, was apparently related to heavy skewing of the ratios of transduced to non-transduced cells at specific sites. It seems likely that this skewing was caused largely by clonal expansion of small founder populations, particularly as tracking of specific genomic abnormalities demonstrated aggressive expansion of minor sub-clones in xenografts. As a consequence, accurate analysis of disease burden by in-vivo imaging, will in future require enrichment for EGFP-expressing cells prior to engraftment.
Unexpectedly, light microscopy and transmission electron microscopy together provided strong evidence that transplantation of NSG mice with iAMP21-ALL cells from two patients led to destruction of the bone marrow niche. As we saw no examples of similar morphology among iAMP21-ALL patients’ samples, it seemed likely that this phenomenon was xenograft-specific and a consequence of initiating a heavy leukemic burden at one site. Cells populating the affected areas, although damaged, were heterogeneous and showed little if any staining with human CD19 and CD45 suggesting they were of host origin. Histology was therefore consistent with destruction having occurred in areas of normal bone marrow before malignant cell infiltration suggesting systemic suppression of normal hematopoiesis, possibly through a mechanism such as cytokine scavenging, as recently reported to account for cytopenia in acute myeloid leukemia.26 The effect did not correlate with late stage disease, as the mean times between transplantation and culling were almost identical and spleen weights were greater and CNS infiltration heavier for mice with no evidence of niche destruction. Further analysis demonstrated that niche destruction is not restricted to the iAMP21-ALL subtype but is probably less common than suggested by our initial data.
Global analysis of genomes from the iAMP21-ALL patients and xenografts revealed a dynamic branching of genomic architecture, similar to that reported previously for B-ALL.4,6,7,27,28 However the rate of newly emergent CNA and their diversity in iAMP21-ALL xenografts suggested a leukemia-initiating cell compartment characterized by greater genetic heterogeneity compared with other B-ALL sub-types. Genomic arrays revealed an average of five CNA per transplanted iAMP21-ALL sample, while similar analysis defined only a single change among seven KMT2A-rearranged infant ALL samples engrafted into multiple mice.28 Additionally, among 12 BCR-ABL1-positive ALL samples, half showed no CNA discordance in xenografts.27 The iAMP21-ALL primografts also developed disease with a relatively long latency. Together with the older age of patients at diagnosis of iAMP21-ALL,12 these data suggest that the primary iAMP21 rearrangement confers only a moderate growth advantage, producing an indolent disease course over which diverse genetic sub-clones are sampled. As genetic diversity has been linked to clinical aggressiveness,29 this clonal heterogeneity of iAMP21-ALL may underlie the affected patients’ poor response to standard therapy.17
Although each iAMP21 chromosome is unique with respect to the balance of regions amplified and deleted, within clinical trials patients are treated homogeneously.17 Our data support this approach, as they further confirm iAMP21 to be a primary abnormality, because the region identified as consistently amplified and spared from chromothripsis,11 was always retained. However in xenografts from one patient, we observed segregation of a structurally evolved iAMP21 chromosome which, together with other CNA, marked a clone that appeared to confer an exceptionally strong growth advantage. Structural evolution of iAMP21 has not been reported previously, although only few presentation/relapse pairs have been analyzed at the whole genome level and FISH is usually targeted only to the RUNX1 gene. This case demonstrates that even after stabilization of the iAMP21 chromosome evident at the time of diagnosis, these rearrangements can undergo further evolution, potentially influencing clinical features and treatment response. However this iAMP21 chromosome may be atypical, as it was reported to be a ring chromosome, which are known to be inherently unstable structures.30 It may be that other iAMP21 ring chromosomes have a tendency to further evolution, but this case was the only one included in this study. Whether this iAMP21 chromosomal evolution acted as a driver of leukemia progression remains uncertain, as it was co-selected with other abnormalities, including an NRAS mutation and bi-allelic loss of CDKN2A/B. Among the four other cases transplanted, three were affected by concordant or discordant CDKN2A deletions, two bi-allelic and one mono-allelic, detected by MLPA only and without apparent involvement of CDKN2B. Further suggesting strong selective pressure for loss of CDKN2A/B in the xenografts, as evidenced by RNA sequencing data, the bi-allelic deletions were all highly clonal. As deletion of this locus only occurs in about 12% of iAMP21-ALL patients,21 these observations support previous reports that CDKN2A/B loss is associated with rapid disease manifestation27 and is selected for in B-ALL xenografts,4 and are also in keeping with a xenograft-specific expression signature enriched for cell cycle genes.31 Alternative mutations of NRAS and KRAS were also strongly selected and both apparently cooperated with an NF1 mutation in xenografts. To our knowledge NF1 and RAS mutations have always been reported as mutually exclusive in B-ALL patients, although their co-occurrence in juvenile myelomoncytic leukemia has been associated with aggressive disease. In mouse models, a combination of NF1 deficiency and KRAS activating mutation reduced the latency of myeloid malignancy compared with either abnormality alone.13,32,33
Other chromosomal regions were strongly implicated in the progression of ALL, as targets of overlapping abnormalities segregated in different clones of xenografts. These included genes known to be involved in B-ALL; ETV6, SH2B3 and BLNK (SLP-65),34–36 as well as novel candidate tumor suppressor genes. Two distinct large deletions, selected in different xenografts, resulted in conversion to homozygosity of a micro-deletion involving PIK3AP1 and LCOR. PIK3AP1 encodes an adaptor protein linking the B-cell receptor and CD19 to activation of PI3K/Akt.37–39 A similar function in the transduction of pre-B-cell receptor signaling is likely and, although not previously implicated in childhood leukemia, focal deletions of PIK3AP1 have been reported in three cases of adult B-ALL.40,41 Combined with our evidence of strong selective pressure for its conversion from mono- to bi-allelic deletion, PIK3AP1 is an interesting candidate, possibly worthy of addition to the growing list of pre-B-cell receptor-related genes disrupted in childhood B-ALL.42 However a role for LCOR, which functions as a co-repressor of several nuclear hormone receptors,43 and has been reported to interact with methyltransferase complexes including polycomb repressive complex 2,44 should not be discounted.
In conclusion, we present the first successful xenografts of iAMP21-ALL and demonstrate their potential as experimental models for functional investigation of this poorly understood genetic subtype. These xenografts could also serve as models in pre-clinical trials or for personalized medicine, with the caveat that systemic niche destruction occurred in some cases. Tracking of CNA, to investigate genomic evolution in xenografts, revealed a surprisingly high rate of instability and examples of marked divergence in CN status of known leukemia driver genes. Further genomic screening of iAMP21-ALL xenografts is likely to reveal many more clonal abnormalities undetected in patients’ samples, augmenting data generated from clinical trial cohorts, as well as potentially guiding treatment in individual cases.
Supplementary Material
Acknowledgments
The authors would like to thank the Department of Cellular Medicine, Royal Victoria Infirmary, Newcastle-upon-Tyne NHS Foundation Trust for fixing and staining histological samples. Tracey Davey and Kath White, Electron Microscopy Research Services, Newcastle University for EM preparations and images and Clare Orange and Lynn Stevenson, University of Glasgow, for the brain histology and imaging. The brain histology slides were scanned by Glasgow University slide scanning and image analysis service at the QEUH. We would also like to thank the Bloodwise Childhood Leukaemia Cell Bank for providing primary childhood leukemia samples.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/103/4/634
References
- 1.Rehe K, Wilson K, Bomken S, et al. Acute B lymphoblastic leukaemia-propagating cells are present at high frequency in diverse lymphoblast populations. EMBO Mol Med. 2013;5(1):38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett DM, Seif AE, Carpenito C, et al. Noninvasive bioluminescent imaging of primary patient acute lymphoblastic leukemia: a strategy for preclinical modeling. Blood. 2011;118(15):e112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morisot S, Wayne AS, Bohana-Kashtan O, et al. High frequencies of leukemia stem cells in poor-outcome childhood precursor-B acute lymphoblastic leukemias. Leukemia. 2010;24(11):1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmitz M, Breithaupt P, Scheidegger N, et al. Xenografts of highly resistant leukemia recapitulate the clonal composition of the leukemogenic compartment. Blood. 2011;118(7):1854–1864. [DOI] [PubMed] [Google Scholar]
- 5.Terziyska N, Castro Alves C, Groiss V, et al. In vivo imaging enables high resolution preclinical trials on patients’ leukemia cells growing in mice. PLoS One. 2012;7(12): e52798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356–361. [DOI] [PubMed] [Google Scholar]
- 7.Patel B, Dey A, Castleton AZ, et al. Mouse xenograft modeling of human adult acute lymphoblastic leukemia provides mechanistic insights into adult LIC biology. Blood. 2014;124(1):96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Townsend EC, Murakami MA, Christodoulou A, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;29(4):574–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rand V, Parker H, Russell LJ, et al. Genomic characterization implicates iAMP21 as a likely primary genetic event in childhood B-cell precursor acute lymphoblastic leukemia. Blood. 2011;117(25):6848–6855. [DOI] [PubMed] [Google Scholar]
- 10.Strefford JC, van Delft FW, Robinson HM, et al. Complex genomic alterations and gene expression in acute lymphoblastic leukemia with intrachromosomal amplification of chromosome 21. Proc Natl Acad Sci USA. 2006;103(21):8167–8172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Y, Schwab C, Ryan SL, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508(7494):98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrison CJ, Moorman AV, Schwab C, et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): cytogenetic characterization and outcome. Leukemia. 2014;28(5):1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan SL, Matheson E, Grossmann V, et al. The role of the RAS pathway in iAMP21-ALL. Leukemia. 2016;30(9):1824–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson HM, Broadfield ZJ, Cheung KL, et al. Amplification of AML1 in acute lymphoblastic leukemia is associated with a poor outcome. Leukemia. 2003;17(11): 2249–2250. [DOI] [PubMed] [Google Scholar]
- 15.Moorman AV, Richards SM, Robinson HM, et al. Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21). Blood. 2007;109(6):2327–2330. [DOI] [PubMed] [Google Scholar]
- 16.Attarbaschi A, Mann G, Panzer-Grumayer R, et al. Minimal residual disease values discriminate between low and high relapse risk in children with B-cell precursor acute lymphoblastic leukemia and an intrachromosomal amplification of chromosome 21: the Austrian and German acute lymphoblastic leukemia Berlin-Frankfurt-Munster (ALL-BFM) trials. J Clin Oncol. 2008;26(18):3046–3050. [DOI] [PubMed] [Google Scholar]
- 17.Moorman AV, Robinson H, Schwab C, et al. Risk-directed treatment intensification significantly reduces the risk of relapse among children and adolescents with acute lymphoblastic leukemia and intrachromosomal amplification of chromosome 21: a comparison of the MRC ALL97/99 and UKALL2003 trials. J Clin Oncol. 2013;31(27):3389–3396. [DOI] [PubMed] [Google Scholar]
- 18.Heerema NA, Carroll AJ, Devidas M, et al. Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk Children’s Oncology Group studies: a report from the Children’s Oncology Group. J Clin Oncol. 2013;31(27):3397–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrison CJ, Haas O, Harbott J, et al. Detection of prognostically relevant genetic abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: recommendations from the Biology and Diagnosis Committee of the International Berlin-Frankfurt-Munster study group. Br J Haematol. 2010;151(2):132–142. [DOI] [PubMed] [Google Scholar]
- 20.Schwab C, Harrison CJ. Acute lymphoblastic leukaemia. Methods Mol Biol. 2011;730:99–117. [DOI] [PubMed] [Google Scholar]
- 21.Schwab CJ, Chilton L, Morrison H, et al. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica. 2013;98(7):1081–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bomken S, Buechler L, Rehe K, et al. Lentiviral marking of patient-derived acute lymphoblastic leukaemic cells allows in vivo tracking of disease progression. Leukemia. 2013;27(3):718–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Travlos GS. Normal structure, function, and histology of the bone marrow. Toxicol Pathol. 2006;34(5):548–565. [DOI] [PubMed] [Google Scholar]
- 24.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Russell LJ, Capasso M, Vater I, et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood. 2009;114(13):2688–2698. [DOI] [PubMed] [Google Scholar]
- 26.Rauch PJ, Ellegast JM, Widmer CC, et al. MPL expression on AML blasts predicts peripheral blood neutropenia and thrombocytopenia. Blood. 2016;128(18):2253–2257. [DOI] [PubMed] [Google Scholar]
- 27.Notta F, Mullighan CG, Wang JC, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469(7330):362–367. [DOI] [PubMed] [Google Scholar]
- 28.Bardini M, Woll PS, Corral L, et al. Clonal variegation and dynamic competition of leukemia-initiating cells in infant acute lymphoblastic leukemia with MLL rearrangement. Leukemia. 2015;29(1):38–50. [DOI] [PubMed] [Google Scholar]
- 29.Park SY, Gonen M, Kim HJ, Michor F, Polyak K. Cellular and genetic diversity in the progression of in situ human breast carcinomas to an invasive phenotype. J Clin Invest. 2010;120(2):636–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guilherme RS, Meloni VF, Kim CA, et al. Mechanisms of ring chromosome formation, ring instability and clinical consequences. BMC Med Genet. 2011;12:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clappier E, Gerby B, Sigaux F, et al. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J Exp Med. 2011;208(4):653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caye A, Strullu M, Guidez F, et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet. 2015;47(11):1334–1340. [DOI] [PubMed] [Google Scholar]
- 33.Cutts BA, Sjogren AK, Andersson KM, et al. Nf1 deficiency cooperates with oncogenic K-RAS to induce acute myeloid leukemia in mice. Blood. 2009;114(17):3629–3632. [DOI] [PubMed] [Google Scholar]
- 34.Moriyama T, Metzger ML, Wu G, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 2015;16(16):1659–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez-Garcia A, Ambesi-Impiombato A, Hadler M, et al. Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood. 2013;122(14):2425–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jumaa H, Bossaller L, Portugal K, et al. Deficiency of the adaptor SLP-65 in pre-B-cell acute lymphoblastic leukaemia. Nature. 2003;423(6938):452–456. [DOI] [PubMed] [Google Scholar]
- 37.Inabe K, Kurosaki T. Tyrosine phosphorylation of B-cell adaptor for phosphoinositide 3-kinase is required for Akt activation in response to CD19 engagement. Blood. 2002;99(2):584–589. [DOI] [PubMed] [Google Scholar]
- 38.Okada T, Maeda A, Iwamatsu A, Gotoh K, Kurosaki T. BCAP: the tyrosine kinase substrate that connects B cell receptor to phosphoinositide 3-kinase activation. Immunity. 2000;13(6):817–827. [DOI] [PubMed] [Google Scholar]
- 39.Castello A, Gaya M, Tucholski J, et al. Nck-mediated recruitment of BCAP to the BCR regulates the PI(3)K-Akt pathway in B cells. Nat Immunol. 2013;14(9):966–975. [DOI] [PubMed] [Google Scholar]
- 40.Okamoto R, Ogawa S, Nowak D, et al. Genomic profiling of adult acute lymphoblastic leukemia by single nucleotide polymorphism oligonucleotide microarray and comparison to pediatric acute lymphoblastic leukemia. Haematologica. 2010;95(9):1481–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Safavi S, Hansson M, Karlsson K, Biloglav A, Johansson B, Paulsson K. Novel gene targets detected by genomic profiling in a consecutive series of 126 adults with acute lymphoblastic leukemia. Haematologica. 2015;100(1):55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eswaran J, Sinclair P, Heidenreich O, et al. The pre-B-cell receptor checkpoint in acute lymphoblastic leukaemia. Leukemia. 2015;29(8):1623–1631. [DOI] [PubMed] [Google Scholar]
- 43.Fernandes I, Bastien Y, Wai T, et al. Ligand-dependent nuclear receptor corepressor LCoR functions by histone deacetylase-dependent and -independent mechanisms. Mol Cell. 2003;11(1):139–150. [DOI] [PubMed] [Google Scholar]
- 44.Alekseyenko AA, Gorchakov AA, Kharchenko PV, Kuroda MI. Reciprocal interactions of human C10orf12 and C17orf96 with PRC2 revealed by BioTAP-XL cross-linking and affinity purification. Proc Natl Acad Sci USA. 2014;111(7):2488–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.