

Abstract
Increasing recognition of the role of B cells in the adaptive immune response makes B cells an important therapeutic target in autoimmunity. Numerous current and developmental immunotherapies target B cells for elimination through recognition of cell-surface proteins expressed specifically on B cells, in particular CD19 and CD20. Similarities and differences in the function and expression of these two molecules predict some shared, and some distinct, pharmacological effects of agents targeting CD19 versus CD20, potentially leading to differences in the clinical safety and efficacy of such agents. Here, we review current knowledge of CD19 and CD20 function and biology, survey current and developmental therapies that target these molecules, and discuss potential differences in elimination of B cells by drugs that target CD19 versus CD20, with particular focus on the central nervous system autoimmune diseases multiple sclerosis and neuromyelitis optica. The principles and mechanisms herein discussed might also be relevant to a variety of other nervous system autoimmune disorders, including NMDA (N-methyl-D-aspartate) receptor encephalitis, transverse myelitis and myasthenia gravis.
Keywords: autoimmunity, B cells, CD19, CD20, experimental autoimmune encephalomyelitis, multiple sclerosis, neuromyelitis optica, neuromyelitis optica spectrum disorder, pharmacology, plasmablasts, plasma cells
Introduction
B lymphocytes, also known as B cells, are mononuclear cells of the lymphocyte subset. B cells are critical in host defense by providing the humoral immunity component of the adaptive immune system through the secretion of antibodies (Abs) as well as by interacting with T cells and by generation of chemokines/cytokines. Subsets of B cells, in particular plasma cells and plasmablasts, have long been recognized as the primary source of Abs.1 B cells have been implicated in the pathogenesis of a number of autoimmune diseases, including the central nervous system (CNS) disorders, multiple sclerosis (MS) and neuromyelitis optica spectrum disorder (NMOSD). The mechanisms by which B cells contribute to the pathophysiology of MS and NMOSD are incompletely understood, but available evidence points to multiple roles in induction of an autoimmune response. Consequently, monoclonal antibodies (mAbs) that deplete B cells are being investigated and used for the treatment of these diseases.
In this review, we provide an update on the current knowledge of CD19 and CD20 function and biology, and summarize current and developmental therapies that target these molecules. Moreover, we discuss potential differences in elimination of B cells by drugs that target CD19 versus CD20, with particular focus on the CNS autoimmune diseases MS and neuromyelitis optica.
B cells in CNS autoimmunity
B cells in MS
In 1942, Elvin Kabat was one of the first investigators to detect monoclonal spikes of immunoglobulin (Ig), later termed oligoclonal bands (OCBs), in the cerebrospinal fluid (CSF) of patients with MS.2 These bands are not present in serum, indicating that they originate in the CNS. It has been speculated that these oligoclonal Abs may trigger and perpetuate disease activity. However, molecular and cellular host and pathogenic targets of OCBs in MS have not yet been identified. Histopathological studies also showed an abundance of Ig in some MS lesions, further suggesting an aberrant humoral immune response against CNS antigens.3 Molecular analyses of B lymphocytes in MS lesions showed hypermutations, and suggest a compartmentalized expansion of antigen-specific B cell populations.4 Prineas and Wright first described lymphoid tissue in cerebral perivascular spaces (CPVS) of autopsy-derived brain tissue of MS patients.5 In some patients, these lymphoid structures display characteristics of germinal centers in secondary lymphoid organs, where B cells proliferate and differentiate, and where B cell receptor (BCR) hypermutation occurs.6 CPVS also likely represent the primary anatomical structure in which antigen presentation within the brain occurs. Hematopoietically derived myeloid cell subsets and B cells reside in these spaces. Magliozzi and colleagues showed more recently that B cell follicles also exist in the cerebral meninges of patients with MS,7 and Serafini and colleagues demonstrated that these follicular structures are sites of Epstein–Barr virus (EBV) latency.8 This is a potentially interesting observation, since immune responses against EBV have been associated with MS.9–11 However, other investigators who attempted to reproduce the findings by Serafini and colleagues were unable to do so.12
B cells may play roles in CNS inflammation beyond the production of Abs. B cells constitutively express major histocompatibility complex (MHC) class I13 and II14 molecules, and are capable of presenting antigens to CD8+ and CD4+ T cells, respectively. Li and colleagues showed that B cells of basal vertebrates are capable of phagocytosis.15 However, B cells of mammalian species are incapable of engulfing proteins to phagocytose and digest them. In contrast to myeloid cells, B cells are able to endocytose Ab-fixed proteins. The unique role of B cells as antigen presenting cells (APC) results from the expression of the high-affinity BCR, which recognizes soluble antigens.16 This endows B lymphocytes with superior antigen recognition capabilities and an ability to selectively present antigens. Furthermore, B cells can bestow antigen-selectivity to myeloid cells through Fc receptor-binding Ab (opsonization).
Clinical studies with anti-CD20 therapies indicate an important role of B lymphocytes as APCs and secretors of cytokines and chemokines. The substantial decrease in the number of CD20+ B cells after administration of the anti-CD20 mAb rituximab was associated with a rapid and significant decrease (>50% of pretreatment levels) in CD3+ T cells in the CSF of recipient MS patients. The reduction in T cells was thought to be the result of a diminished expression of the chemokines CXCL13 and CCL19, but likely also relates to a relative loss of antigen presentation and other trophic factors by B cells.17 The rapid beneficial effects on magnetic resonance imaging (MRI) of the brain 12 weeks after initiation of rituximab therapy18 suggests that the APC function of B cells, along with the expression of soluble inflammatory molecules, is crucial in the perpetuation of MS disease activity. In this study, the levels of CSF IgG were not affected by rituximab treatment.
B cells in NMOSD
NMOSD is an autoimmune disorder of the CNS that predominantly targets the optic nerves, brainstem and the spinal cord.19,20 Once the diagnosis of NMOSD is established on the basis of clinical manifestations, AQP4 autoantibody testing or MRI imaging, or a combination of these,20 disease exacerbations are identified clinically and confirmed by MRI as indicated. Similar to relapsing-remitting MS (RRMS),21,22 the disease course of NMOSD is typically relapsing-remitting. Histopathological evaluations of biopsy and autopsy material identified perivascular IgM, IgG and complement (in particular the C9neo antigen) depositions, and immune cell infiltrate consisting of neutrophils, eosinophils and macrophages.23 CD3+ and CD8+ T cells and natural killer cells are also present.24 Lennon and colleagues identified NMOSD-IgG (also termed AQP4-IgG), an Ab that binds to aquaporin-4 (AQP4), in a subset of patients with NMOSD.25,26 AQP4 is critical for the transportation of water across the cell membrane of numerous cell types. Within the CNS, it is highly expressed in astrocytes.27,28 Many NMOSD lesions, regardless of age, disease stage and disease activity, display an extensive loss of AQP4 immunoreactivity.29–31 The pathogenicity of NMOSD-IgG has been demonstrated in animal models of NMOSD.32–34 Chihara and colleagues showed a positive correlation between the number of CD20−CD19intCD27highCD38highCD180− peripheral blood plasmablasts and the serum AQP4 Ab titers in NMOSD seropositive patients.35 The percentage of peripheral blood plasmablasts also correlated with the number of disease relapses. Interleukin (IL)-6 promoted the survival of blood plasmablasts, and their production of AQP4-IgG in vitro. AQP4-expressing plasma cells (PCs) have also been identified in the CSF of an NMOSD patient following relapse.36 More recently, Abs against other molecular targets, including myelin oligodendrocyte glycoprotein (MOG), have been identified in patients with NMOSD who are seronegative for AQP4-IgG, and mounting evidence suggests a role for some of these other autoantibodies in NMOSD pathogenesis.37,38
In summary, there is evidence to suggest that B cells play differential pathogenic roles in the CNS autoimmune diseases MS and NMOSD through multiple mechanisms including production of pathogenic autoantibodies, secretion of cytokines/chemokines, antigen presentation and T cell interactions, providing a strong rationale for the pursuit of B cell depletion as a therapeutic strategy in these diseases. The apparent beneficial effects of B cell depletion in MS and NMOSD, discussed later, further support the important role that B cells play in these diseases.
CD19 and CD20
The largely B cell-restricted expression of two cell surface markers, CD19 and CD20, provides an opportunity to selectively target B cells with immunotherapeutic cytotoxic agents. Here, we highlight the expression and functions of CD19 and CD20 and discuss their utility as molecular targets for B cell depletion.
CD19
The CD19 antigen is a type I transmembrane glycoprotein that belongs to the immunoglobulin Ig superfamily. It is expressed on early pro-B cells, late pro-B cells, memory B cells, plasmablasts and some plasma cells, the latter of which are the main cellular source of protective, highly target-specific Abs, but also of autoantigen-specific Abs.39–42 The overall expression increases approximately threefold during B cell maturation.
CD19 impacts B lymphocyte activation and differentiation through modulation of BCR signaling. CD19 is relevant in establishing optimized immune responses by modulating (1) antigen-independent B cell development, and (2) immunoglobulin-induced B lymphocyte activation. Deficiency in CD19 in experimental animals and humans results in impaired humoral responses, and an overall increased susceptibility to infection.42–44 In contrast, overexpression of a human (h)CD19 transgene under its endogenous promoter45,46 leads to an autoimmune disorder in the tight skin (TSK/+) mouse, a model of human systemic sclerosis (SSc).47 In this disease model, skin sclerosis was possibly the consequence of interleukin (IL-6) overexpression and auto-Ab secretion. The hCD19 transgenic mouse model has also been used to explore CD19 as a therapeutic molecular target. Two weeks after a single administration of anti-CD19 mAbs, the majority of mature B cells in the hCD19 mice were depleted, and serum IgM, IgG, and IgA Ab levels were significantly diminished.48
CD20
CD20 is an activated-glycosylated transmembrane phosphoprotein whose expression initiates somewhat later, and is lost somewhat earlier, during B cell development and differentiation than is CD19.49–53 CD20 is involved in B cell activation, differentiation, and calcium transport, and CD20 deficiency in humans results in a reduced capacity to mount a B cell response to T cell-independent antigens.54 Like CD19, CD20 appears to exert effects through interaction with BCR, and substantial evidence points to a biological role for CD20 in amplifying calcium signals that are transduced through the BCR during antigen recognition by immature and mature B cells.55 Expression of CD20 in Chinese hamster ovary (CHO) cells, for example, introduces a novel mechanism for calcium influx which is dependent on the cytoplasmic domain of CD20 responsible for association with lipid rafts, and in B cells a reduction of CD20 expression reduces calcium influx stimulated by BCR signaling.56
Several investigators recently showed that a subset of CD3+ T cells also stains positive for CD20, and that these cells can be depleted with anti-CD20 therapy.57–59 The therapeutic magnitude of this effect on clinical and paraclinical outcomes in human disease has yet to be determined.
CD19 and CD20 expression
Differential expression of CD19 and CD20 on late-stage B cells, specifically plasmablasts and plasma cells, predicts potential differences in the outcome of therapeutic targeting of B cells via these proteins. It has long been accepted that while both CD19 and CD20 expression are eventually lost from terminally differentiating plasma cells, CD19 expression persists on plasmablasts and some plasma cells after CD20 expression has been lost (Figure 1).42,60–62 Recent immunophenotypic flow cytometry analyses of both the CD19+ and CD19– B cell subsets in human blood and lymphoid tissues demonstrates that CD19+ plasma cells are the major immunoglobulin-secreting subset in peripheral blood, bone marrow, spleen and tonsils, with the CD19+ subset approximately threefold more abundant than the CD19− subset in bone marrow and spleen (Figure 2) (Groves et al., submitted). Importantly, these Ig-secreting cells were found to lack CD20 expression, with the exception of low-abundance CD20+ cells in tonsils. Furthermore, since IgG Abs to vaccine antigens, such as influenza, tetanus, measles, mumps and rubella (MMR) and polio, are produced by both CD19+ and CD19− subsets, and CD19− long-lived Ab-producing plasma cells,66,67 these results suggest that CD19-targeted depletion of B cells may leave previously established humoral immunity intact, while eliminating a main source of pathogenic autoantibodies.
Figure 1.

Summary of CD19 and CD20 expression during B cell development. CD19 expression is observed on earlier-stage B cells than is CD20. In addition, CD19 expression is found to persist on late-stage antibody-secreting B cells (plasmablasts and plasma cells) after CD20 expression has been lost.
Figure 2.
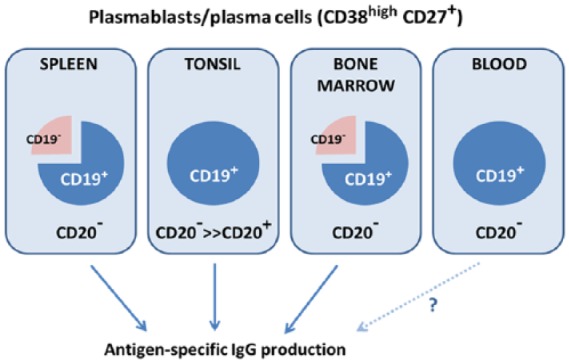
CD19+CD20− cells in lymphoid tissues provide immune memory. Late-stage (CD38highCD27+) B cells isolated from bone marrow, spleen, tonsil and blood are predominantly CD19+ and almost entirely CD20−. In lymphoid tissues, both the CD19+ and CD19− subsets produce antigen-specific IgG in response to prior viral exposure/vaccination. Analysis of antigen-specific IgG production by CD19+ cells from blood was not feasible, but the existence of such cells in primary and secondary lymphoid tissues suggests they also exist in circulation. Based on results from Groves and colleagues (Groves et al., submitted).
Anti-CD19 therapies in autoimmunity
Numerous therapeutic agents that target CD19 have been, or are currently being, investigated in clinical studies for B cell-mediated hematological malignancies; these include AFM11, blinatumomab, MDX-1342, MOR208, SAR3419, and SGN-CD19A.68 Of particular interest to CNS autoimmunity, two agents that target CD19 are in clinical development in autoimmune diseases: XmAb5871 and MEDI-551 (inebilizumab).
XmAb5871
This is a humanized Fc engineered Ab attached to FcgRIIb and the BCR.69 It was previously shown that engagement of the low-affinity Ab receptor FcgRIIb downregulates B cell activation, and specifically suppresses only B cells recognizing cognate Ag, but does not cause B cell depletion.70 XmAb5871 already completed a phase Ib/IIa clinical trial for moderate-to-severe rheumatoid arthritis, and entered into phase II development for the treatment of IgG4-related disorders and systemic lupus erythematosus (SLE).
MEDI-551 (inebilizumab)
This is an afucosylated humanized IgG1 kappa anti-CD19 mAb.71,72 Among anti-CD19 therapies, MEDI-551 is the furthest along in development for NMOSD, and its pharmacological and biological properties will be outlined in more detail. The absence of fucose from MEDI-551 results in a 10-fold enhanced affinity of the mAb to FcγR IIIA (CD16), and mouse FcγRIV, and consequently increased antibody-dependent cellular cytotoxicity (ADCC) activity.71 The B cell-depleting activity of MEDI-551 was demonstrated in both in vitro and in vivo studies.71
SLE1 × hCD19 Tg mice, which spontaneously produce increasing levels of autoreactive Abs as they age, were used to assess some of the pharmacodynamic properties of MEDI-551. In these animals, MEDI-551 depletes B cells in the blood, bone marrow and spleens, including the splenic germinal center, rapidly and effectively.73 Total serum IgM and IgG titers diminished significantly, and anti-double stranded DNA (IgM and IgG), anti-histone (IgG), anti-nuclear (IgG), and anti-single stranded DNA (IgG and IgM) were significantly decreased. The ability of MEDI-551 to decrease the number of Ab-secreting cells appears compartment-specific, and was more substantial in the spleen than in bone marrow. This is likely the result of higher terminally differentiated CD19− plasma cell numbers in bone marrow.
The experimental autoimmune encephalomyelitis (EAE) animal model of MS also provided a strong rationale for development of MEDI-551 in MS and NMOSD. Chen and colleagues administered a single 10 mg/kg dose of MEDI-551 to human CD19 transgenic (hCD19Tg) mice, in which EAE was actively induced through vaccination with recombinant MOG protein (rhMOG1–125).74 Specifically, MEDI-551 was administered before the onset of EAE, or when EAE disease was established. The infiltration of leukocytes into the spinal cord is significantly reduced. In addition, MEDI-551 treatment reduced the numbers of short-lived and long-lived autoreactive CD138 plasma cells in the spleen and bone marrow, respectively. Interestingly, these investigators also showed that potentially protective CD1dhiCD5+ regulatory B cells were relatively resistant to depletion, and that myelin-specific Foxp3+ regulatory T cells (Tregs) were expanded. In a follow-up study, the same authors demonstrated that MEDI-551 ameliorates EAE more effectively than does antimurine CD20 mAb.75 MOG-specific Abs and short-lived and long-lived auto-Ab-secreting cells were essentially undetectable in MEDI-551-treated mice, but remained detectable in anti-CD20 mAb-treated mice. Residual disease severity in the CD20 mAb-treated animals positively correlated with the frequency of treatment-resistant plasma cells in the bone marrow, many of which were CD19+.
Clinical investigations of MEDI-551 confirm the pharmacodynamic activities observed in animal models. In a phase I, randomized, double-blind, placebo-controlled clinical trial that evaluated single intravenous (IV) doses of MEDI-551 from 0.1 mg/kg to 10 mg/kg in patients with SSc,76 MEDI-551 resulted in thorough and durable depletion of B cells, with longer duration of depletion associated with higher dose. Although not powered to demonstrate efficacy, the study provided some evidence of MEDI-551 clinical activity on diseased skin.
MEDI-551 has also been assessed in a phase I, randomized, double-blind, placebo-controlled clinical trial in patients with RRMS.77 MEDI-551 was administered IV at doses from 30 mg to 600 mg, or subcutaneously at doses from 60 mg to 300 mg, and achieved thorough depletion of circulating B cells at all doses tested. MEDI-551 exhibited an acceptable safety profile and reduced the mean number of cumulative new or enlarging MRI lesions over 24 weeks. Due to the small size of this study, however, any conclusions regarding efficacy of MEDI-551 in RRMS must be supported by further investigation.
In both of these phase I studies, depletion of plasma cells in treated patients was examined using a validated gene signature assay that provides a quantitative assessment of PCs in blood and tissue.78 In both SSc and RRMS patients, MEDI-551 achieved dramatic reduction of circulating plasma cells which paralleled that of total B cells, consistent with the direct targeting of PCs by MEDI-551.76,77
MEDI-551 is currently being investigated in patients with NMOSD in a phase II/III, multinational, double-blind, placebo-controlled clinical trial, the N-MOmentum study [ClinicalTrials.gov identifier: NCT02200770].79 In this study, efficacy is being assessed as a reduction in the risk of NMOSD attack in the first 6 months after treatment. In addition, pharmacodynamic effects on B cells (including plasmablasts and PCs), pathogenic autoantibody titers and other biomarkers will be examined to explore their relationship with clinical activity.
Anti-CD20 therapies in MS
Rituximab
The observations made in clinical trials with anti-CD20 mAbs has provided the strongest evidence to date to support a pathogenic role for B cells in MS and NMOSD. Rituximab is a chimeric IgG1 mAb that binds to the B lymphocyte surface antigen CD20.80 It is generally thought that rituximab depletes B lymphocytes via ADCC and complement-dependent cytotoxicity (CDC) mechanisms. There is also some evidence that rituximab depletes B cells within CPVS, either directly, or more likely through preventing repopulation from peripheral compartments.81 Work by Montalvao and colleagues suggests Ab-dependent cellular phagocytosis of B cells by Kupffer cells in the liver as the principal underlying mechanism of B cell reduction in mice.82 The efficacy of rituximab was first studied in a phase II clinical trial.18 The primary outcome was the total number of gadolinium-enhancing (Gd+) lesions on brain MRI. Rituximab was superior to placebo in achieving this outcome. There were no new detectable lesions between weeks 12 and 48 (endpoint for the study) postinfusion. Rituximab was also tested in patients with primary-progressive MS (PPMS). In a placebo-controlled phase II trial, in which the primary endpoint was prevention of confirmed disease progression defined as an increase in the Expanded Disability Status Scale (EDSS) sustained for 12 weeks.83 This endpoint was not reached. However, a preplanned subgroup analysis demonstrated that patient age and the presence of Gd+ lesions on a baseline MRI may be predictors of therapeutic responsiveness. This analysis informed a phase III clinical trial of ocrelizumab for patients with PPMS, which led to its approval (see below).
Ocrelizumab
Results similar to those with rituximab were recently demonstrated with ocrelizumab, a humanized recombinant anti-CD20 mAb.84 Its biological effects on B cells appear to be mediated primarily by ADCC rather than CDC.85 A phase II, randomized, placebo-controlled, multicenter trial in patients with RRMS tested low-dose ocrelizumab (600 mg) on day 1 and 15, high-dose ocrelizumab (2000 mg) on day 1 and 15 or interferon beta (IFNβ)-1a once a week versus placebo.84 At week 24, the number of CD+ lesions on brain MRI was diminished by 89% in the low-dose, and by 96% in the high-dose treatment groups. Both groups were significantly more effective than IFNβ-1a. The annualized relapse rate was significantly reduced in both ocrelizumab treatment groups compared with the placebo or IFNβ-1a groups. In two identical phase III trials, 821 and 835 patients with RRMS were randomized to receive ocrelizumab at a dose of 600 mg every 24 weeks or subcutaneous interferon beta-1a (IFNβ-1a; Rebif®) at a dose of 44 µg three times weekly for 96 weeks.86 The annualized relapse rate was significantly lower with ocrelizumab than with IFNβ-1a in both trials. All other clinical and paraclinical outcomes favored ocrelizumab over IFNβ-1a. Ocrelizumab also recently became the first agent to be approved for patients with PPMS based on the results from a phase III trial that randomly assigned 732 patients in a 2:1 ratio to receive IV ocrelizumab (600 mg) or placebo every 24 weeks for at least 120 weeks and until a prespecified number of confirmed disability progression events had occurred.87 Ocrelizumab was significantly superior to placebo in preventing disability progression confirmed at 12 weeks, the primary outcome. In the entire clinical trial program, ocrelizumab was relatively well tolerated.
Ofatumumab
This is a fully human IgG1 anti-CD20 mAb88,89 that depletes B lymphocytes predominantly through CDC. Ofatumumab has a higher avidity to CD20 than rituximab, and it adheres to an additional antigenic epitope.90 In a phase II study that enrolled 38 RRMS patients, participants received two doses of either 100, 300 or 700 mg ofatumumab, or placebo. At week 24, there was a significant reduction of Gd+ lesions on brain MRI in the treatment groups.91 A phase III clinical trial that compares the efficacy and safety of ofatumumab against teriflunomide in patients with RRMS is ongoing [ClinicalTrials.gov identifier: NCT02792218].
Ublituximab
Another humanized anti-CD20 mAb, this is currently in phase II clinical development for patients with RRMS [ClinicalTrials.gov identifier: NCT02738775].
Anti-CD20 therapies in NMOSD
Rituximab
This agent is currently considered a first-line therapy for patients with NMOSD by many providers, despite the fact that at this time, the evidence supporting the use of rituximab in NMOSD is mostly class IV with no prospective randomized controlled studies reported to date. Data from numerous small, uncontrolled studies suggest that rituximab has a beneficial effect on the relapse rates in NMOSD.92–95 However, the treatment responses have been heterogeneous and relapses on rituximab treatment are not infrequently reported. The reasons for rituximab treatment failure are not fully known. One possible explanation for rituximab failure could be limitations of rituximab in targeting pathogenic CD19+/CD20− plasmablasts and plasma cells. In fact, in rheumatoid arthritis patients treated with rituximab, peripheral CD19+/CD20− plasmablasts/plasma cells are continuously detected despite essentially complete (>99%) and durable (up to 6 months) depletion of earlier-stage CD19+/CD20+ B cells,62 and similar observations have been made in other autoimmune diseases including SLE and MS (Table 1). Alternatively, pro- and anti-inflammatory cytokine networks, which likely play a role in the occurrence of attacks, may respond differently to B cell depletion therapy in different patients.
Table 1.
The experience with Rituximab in human autoimmune disorders.
| RTX-treated patient population (tissue) | Key finding | Reference |
|---|---|---|
| Rheumatoid arthritis patients (peripheral blood) | Persistence of IgA-secreting CD19+ CD20− plasmablasts, despite effective depletion of total B cells. Some patients retain expression of IgA-rheumatoid factor. | Mei and colleagues62 |
| Idiopathic thrombocytopenic purpura patients who failed rituximab (spleen) | Persistence of anti-GpIIb/IIIa IgG-producing CD19+ CD20-plasma cells | Mahevas and colleagues (2013)63 |
| Warm autoimmune hemolytic anemia patients who failed rituximab (spleen) | Persistence of anti-RBC antibody-producing CD19+ plasma cells | Mahevas and colleagues ()64 |
| Systemic lupus erythematosus patients (peripheral blood) | Persistence of CD19+/CD20− plasma cells, despite effective depletion of total B cells | Anolik and colleagues (2004) |
| Relapsing/remitting multiple sclerosis patients (cerebrospinal fluid) | No change in number of CD19+/CD138+ plasma cells despite effective depletion of total B cells in cerebrospinal fluid | Piccio and colleagues (2010)65 |
It is interesting to note the variable and often minor reduction in AQP4-IgG titers in NMOSD patients treated with rituximab,94,96 mirroring the inconsistent effects of anti-CD20 therapy on potentially pathogenic immunoglobulin levels in other autoimmune diseases.97–99 It remains to be determined whether CD19-targeted agents affect pathogenic autoantibody levels in autoimmune disease patients.
Safety
Currently, no long-term safety data are available to adequately assess the impact of long-term CD19 Ab-mediated cell depletion in patients with CNS autoimmune disorders. Further clinical trials and accumulative experience over time are needed to establish the safety profile of anti-CD19 therapies in CNS autoimmune diseases. Some potential concerns of anti-CD19 therapies were recently articulated by Mei and colleagues.62 Weakening of protective adaptive humoral immune responses and host defense against pathogens and neoplasms may occur, and may not be restricted to the CNS compartment. Acquired adaptive immunity against vaccines may be diminished. The differential effects of anti-CD19 therapies on effector cells and regulatory cell populations is incompletely understood, and requires further study. Also, compartment-specific effects of anti-CD19 mAbs have not been fully explored.
Anti-CD20 therapy has been in use for at least a decade, with many thousands of patients treated, and appears to have a favorable safety profile. For example, in a long-term safety follow-up study of a large cohort of rheumatoid arthritis patients treated with one or more courses of rituximab, including more than 1200 patients followed for over 5 years,100 rates of all adverse events in the rituximab group were similar to those in the placebo group and were highest in the first 6 months after first exposure, in part due to infusion-related reactions which occurred primarily at the first infusion. The most frequent serious infectious events were lower respiratory tract infection, predominantly pneumonia. Serious opportunistic infections, tuberculosis, de novo and reactivated hepatitis B, and malignancy were rare. The US Food and Drug Administration (FDA) recently implemented changes to the prescribing information of rituximab and ofatumumab to add new Boxed Warning information about the potential risk of hepatitis B virus (HBV) infection reactivation.101 The FDA had previously issued a warning that two patients died of progressive multifocal leukoencephalopathy (PML) due to reactivation of JC virus in the CNS after being treated with rituximab for SLE.99
Overall, potential concerns about the increased risks of infection and malignancy from long-term immunosuppression may apply to both anti-CD19 and anti-CD20 mAb treatment. This may especially be true after long-term therapy in patients who have received other immunoregulatory agents. Long-term pharmacovigilance appears a prudent measure to capture any safety signals.
Discussion
The scientific rationale for B cell depletion in CNS autoimmunity is established, and there is clear evidence for clinical benefit of CD20-targeted agents (ocrelizumab, rituximab) in RRMS. Furthermore, rituximab is often used ‘off-label’ in NMOSD on the basis of data from numerous uncontrolled clinical studies. One important goal of B cell-depleting therapies in these diseases is the elimination of autoantibody-producing cells. In the case of CD20-targeted agents, this goal is achieved, at least partially, by preventing the de novo generation of plasma cells from their precursors, since CD20 expression is generally not present on autoantibody-producing cells. One potential downside of an anti-CD20 treatment approach is the incomplete reduction of plasma cells and plasmablasts. In MS and in other autoimmune diseases, there are ample data regarding the persistence of CD19+ PCs that produce pathogenic autoantibodies following treatment with the CD20-targeted agent rituximab (Table 1). CD19-targeting agents, on the other hand, deplete plasmablasts and plasma cells76–78 due to the retention of CD19 on many of these later-stage cells, and thus these agents may potentially be more effective at eliminating production of pathogenic autoantibodies. However this assumption needs to be tested in a clinical setting, and whether this translates into additional clinical benefit beyond that seen with CD20-targeted agents remains to be seen. Other desirable pharmacological effects of eliminating B cells, such as disrupting the aberrant proinflammatory cytokine networks that perpetuate inflammation in MS and NMOSD, are likely to be similar between CD19- versus CD20-targeted agents due to the shared expression of both markers during much of B cell development. For the same reason, a combination of anti-CD19 and anti-CD20 directed therapies would likely have no additive or synergistic effect.
A potential limitation of both CD19- and CD20-targeted therapies is that B cells that reside in target tissues may be less affected by this approach. Thus, elimination of immunological memory against one or multiple autoantigens may be incomplete. Questions remain regarding the understanding of the exact compartment(s) in which pathogenic B cells in NMOSD or MS reside, or the degree to which depletion of B cells in these compartment(s) can be achieved by therapeutic mAbs. Certainly, some agents are able to deplete B cells in diseased tissue; inebilizumab, for example, has been shown to deplete plasma cells in affected skin in SSc patients.78 Moreover, a key unanswered question pertains to the source of pathogenic Abs in CNS autoimmunity. One possibility is that autoantibodies are produced outside the CNS – for example, in circulation or lymphoid tissues, gaining access to the CNS at locations of compromised blood–brain barrier.103 Alternatively, peripheral AQP4-IgG may bind to traces of CNS antigen that drain to the periphery via the newly recognized CNS lymphatic system, resulting in the activation of CNS-autoreactive T cells.104 On the other hand, the existence of OCBs and abundant plasmablasts in the CSF of patients with MS, and the existence of AQP4-IgG-producing B cells in the CNS around the time of attack in NMOSD patients,35,36 suggests that intrathecal autoantibody production may play a role in these diseases, and that depletion of B cells within the CNS may be required for benefit. Of note, CNS-resident plasmablasts in both MS and NMOSD have been demonstrated to be CD19+CD20−,35,75 suggesting the possibility of direct depletion by CD19-targeted, but not CD20-targeted, agents.
There is strong biological plausibility to support the hypothesis that CD19-directed agents may be effective in the treatment of human CNS autoimmune disorders. However, there are also some potential concerns that deserve further study. Blair and colleagues recently reported human CD19+CD24hiCD38hi regulatory B cells105 that suppressed Th1 cell differentiation in vitro. Furthermore, CD19+CD138high plasma cells express high levels of the cytokines IL-10 and IL-35, which promote tissue repair and suppress T and B cell memory responses in vivo.106 Depletion of all B lymphocyte subsets might disrupt B cell homeostasis, and might create a milieu permissive to autoimmunity in some individuals.
Finally, there may be pharmacological limitations regarding the magnitude of B cell depletion in the CNS compartment for anti-CD19 and anti-CD20 therapies. In human patients, the penetration of therapeutic Abs into the CSF is approximately 0.1% of serum levels.107
Conclusion
The anti-CD20 mAbs ocrelizumab and rituximab have demonstrated clinical benefit in MS, and rituximab has been used ‘off-label’ in NMOSD patients. Data from anti-CD20 mAbs prove the utility of the B cell depletion as an effective mechanism in treatment of autoimmune disorders. Given the important role of autoantibodies in a number of these diseases, additional benefit may be gained by directly targeting the cells that produce the pathogenic autoantibodies – namely, plasmablasts and plasma cells. CD19-targeted therapies in clinical development in NMOSD and other autoimmune diseases may be a step forward in more completely correcting an aberrant adaptive immune response in these diseases.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Conflict of interest statement: The authors declare that there is no conflict of interest.
Contributor Information
Thomas G. Forsthuber, Department of Biology, University of Texas at San Antonio, TX, USA
Daniel M. Cimbora, Medimmune, Gaithersburg, MD, USA
John Nolan Ratchford, Medimmune, Gaithersburg, MD, USA.
Eliezer Katz, Medimmune, Gaithersburg, MD, USA.
Olaf Stüve, Neurology Section, VA North Texas Health Care System, Medical Service, Dallas, TX, USA; Department of Neurology and Neurotherapeutics, University of Texas Southwestern Medical Center, Dallas, TX, USA; Department of Neurology, Klinikum rechts der Isar, Technische Universität München, Germany.
References
- 1. Fagraeus A. The plasma cellular reaction and its relation to the formation of antibodies in vitro. J Immunol 1948; 58: 1–13. [PubMed] [Google Scholar]
- 2. Kabat EA, Moore DH, Landow H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. J Clin Invest 1942; 21: 571–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000; 47: 707–717. [DOI] [PubMed] [Google Scholar]
- 4. Lehmann-Horn K, Kronsbein HC, Weber MS. Targeting B cells in the treatment of multiple sclerosis: recent advances and remaining challenges. Ther Adv Neurol Disord 2013; 6: 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prineas JW, Wright RG. Macrophages, lymphocytes, and plasma cells in the perivascular compartment in chronic multiple sclerosis. Lab Invest 1978; 38: 409–421. [PubMed] [Google Scholar]
- 6. Kim S, Davis M, Sinn E, et al. Antibody diversity: somatic hypermutation of rearranged VH genes. Cell 1981; 27: 573–581. [DOI] [PubMed] [Google Scholar]
- 7. Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007; 130: 1089–1104. [DOI] [PubMed] [Google Scholar]
- 8. Serafini B, Rosicarelli B, Franciotta D, et al. Dysregulated Epstein–Barr virus infection in the multiple sclerosis brain. J Exp Med 2007; 204: 2899–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ascherio A, Munger KL. 99th Dahlem conference on infection, inflammation and chronic inflammatory disorders: Epstein–Barr virus and multiple sclerosis – epidemiological evidence. Clin Exp Immunol 2010; 160: 120–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bray PF, Bloomer LC, Salmon VC, et al. Epstein–Barr virus infection and antibody synthesis in patients with multiple sclerosis. Arch Neurol 1983; 40: 406–408. [DOI] [PubMed] [Google Scholar]
- 11. Sumaya CV, Myers LW, Ellison GW. Epstein–Barr virus antibodies in multiple sclerosis. Arch Neurol 1980; 37: 94–96. [DOI] [PubMed] [Google Scholar]
- 12. Willis SN, Stadelmann C, Rodig SJ, et al. Epstein–Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain 2009; 132: 3318–3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marshak A, Doherty PC, Wilson DB. The control of specificity of cytotoxic T lymphocytes by the major histocompatibility complex (AG-B) in rats and identification of a new alloantigen system showing no AG-B restriction. J Exp Med 1977; 146: 1773–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kappler JW, Marrack P. The role of H-2-linked genes in helper T-cell function: I. In vitro expression in B cells of immune response genes controlling helper T-cell activity. J Exp Med 1977; 146: 1748–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li J, Barreda DR, Zhang YA, et al. B lymphocytes from early vertebrates have potent phagocytic and microbicidal abilities. Nat Immunol 2006; 7: 1116–1124. [DOI] [PubMed] [Google Scholar]
- 16. Lesley JF, Kettman JR, Dutton RW. Immunoglobulins on the surface of thymus-derived cells engaged in the initiation of a humoral immune response. J Exp Med 1971; 134: 618–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cross AH, Klein RS, Piccio L. Rituximab combination therapy in relapsing multiple sclerosis. Ther Adv Neurol Disord 2012; 5: 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 2008; 358: 676–688. [DOI] [PubMed] [Google Scholar]
- 19. Cree BA, Goodin DS, Hauser SL. Neuromyelitis optica. Semin Neurol 2002; 22: 105–122. [DOI] [PubMed] [Google Scholar]
- 20. Wingerchuk DM, Banwell B, Bennett JL, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015; 85: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 1996; 46: 907–911. [DOI] [PubMed] [Google Scholar]
- 22. Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014; 83: 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 2002; 125: 1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saadoun S, Bridges LR, Verkman AS, et al. Paucity of natural killer and cytotoxic T cells in human neuromyelitis optica lesions. Neuroreport 2012; 23: 1044–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364: 2106–2112. [DOI] [PubMed] [Google Scholar]
- 26. Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005; 202: 473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Connolly DL, Shanahan CM, Weissberg PL. The aquaporins: a family of water channel proteins. Int J Biochem Cell Biol 1998; 30: 169–172. [DOI] [PubMed] [Google Scholar]
- 28. Pittock SJ, Weinshenker BG, Lucchinetti CF, et al. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006; 63: 964–968. [DOI] [PubMed] [Google Scholar]
- 29. Misu T, Fujihara K, Kakita A, et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 2007; 130: 1224–1234. [DOI] [PubMed] [Google Scholar]
- 30. Roemer SF, Parisi JE, Lennon VA, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007; 130: 1194–1205. [DOI] [PubMed] [Google Scholar]
- 31. Sinclair C, Kirk J, Herron B, et al. Absence of aquaporin-4 expression in lesions of neuromyelitis optica but increased expression in multiple sclerosis lesions and normal-appearing white matter. Acta Neuropathol 2007; 113: 187–194. [DOI] [PubMed] [Google Scholar]
- 32. Bradl M, Misu T, Takahashi T, et al. Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 2009; 66: 630–643. [DOI] [PubMed] [Google Scholar]
- 33. Bennett JL, Lam C, Kalluri SR, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol 2009; 66: 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Asavapanumas N, Ratelade J, Papadopoulos MC, et al. Experimental mouse model of optic neuritis with inflammatory demyelination produced by passive transfer of neuromyelitis optica-immunoglobulin G. J Neuroinflammation 2014; 11: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chihara N, Aranami T, Sato W, et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci USA 2011; 108: 3701–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bennett JL, Lam C, Kalluri SR, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol 2009; 66: 617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mader S, Gredler V, Schanda K, et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J Neuroinflammation 2011; 8: 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jarius S, Ruprecht K, Kleiter I, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 1: frequency, syndrome specificity, influence of disease activity, long-term course, association with AQP4-IgG, and origin. J Neuroinflammation 2016; 13: 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bradbury LE, Kansas GS, Levy S, et al. The CD19/CD21 signal transducing complex of human B lymphocytes includes the target of antiproliferative antibody-1 and Leu-13 molecules. J Immunol 1992; 149: 2841–2850. [PubMed] [Google Scholar]
- 40. Haas KM, Tedder TF. Role of the CD19 and CD21/35 receptor complex in innate immunity, host defense and autoimmunity. Adv Exp Med Biol 2005; 560: 125–139. [DOI] [PubMed] [Google Scholar]
- 41. Otero DC, Anzelon AN, Rickert RC. CD19 function in early and late B cell development: I. Maintenance of follicular and marginal zone B cells requires CD19-dependent survival signals. J Immunol 2003; 170: 73–83. [DOI] [PubMed] [Google Scholar]
- 42. Tedder TF. CD19: a promising B cell target for rheumatoid arthritis. Nat Rev Rheumatol 2009; 5: 572–577. [DOI] [PubMed] [Google Scholar]
- 43. Poe JC, Minard-Colin V, Kountikov EI, et al. A c-Myc and surface CD19 signaling amplification loop promotes B cell lymphoma development and progression in mice. J Immunol 2012; 189: 2318–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van Zelm MC, Reisli I, van der Burg M, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med 2006; 354: 1901–1912. [DOI] [PubMed] [Google Scholar]
- 45. Zhou LJ, Smith HM, Waldschmidt TJ, et al. Tissue-specific expression of the human CD19 gene in transgenic mice inhibits antigen-independent B-lymphocyte development. Mol Cell Biol 1994; 14: 3884–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Engel P, Zhou LJ, Ord DC, et al. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity 1995; 3: 39–50. [DOI] [PubMed] [Google Scholar]
- 47. Saito E, Fujimoto M, Hasegawa M, et al. CD19-dependent B lymphocyte signaling thresholds influence skin fibrosis and autoimmunity in the tight-skin mouse. J Clin Invest 2002; 109: 1453–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yazawa N, Hamaguchi Y, Poe JC, et al. Immunotherapy using unconjugated CD19 monoclonal antibodies in animal models for B lymphocyte malignancies and autoimmune disease. Proc Natl Acad Sci USA 2005; 102: 15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stashenko P, Nadler LM, Hardy R, et al. Characterization of a human B lymphocyte-specific antigen. J Immunol 1980; 125: 1678–1685. [PubMed] [Google Scholar]
- 50. Stashenko P, Nadler LM, Hardy R, et al. Expression of cell surface markers after human B lymphocyte activation. Proc Natl Acad Sci USA 1981; 78: 3848–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Loken MR, Shah VO, Dattilio KL, et al. Flow cytometric analysis of human bone marrow: II. Normal B lymphocyte development. Blood 1987; 70: 1316–1324. [PubMed] [Google Scholar]
- 52. Uchida J, Lee Y, Hasegawa M, et al. Mouse CD20 expression and function. Int Immunol 2004; 16: 119–129. [DOI] [PubMed] [Google Scholar]
- 53. Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol 2013; 131: 959–971. [DOI] [PubMed] [Google Scholar]
- 54. Kuijpers TW, Bende RJ, Baars PA, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest 2010; 120: 214–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Beers SA, Chan CH, French RR, et al. CD20 as a target for therapeutic type I and II monoclonal antibodies. Semin Hematol 2010; 47: 107–114. [DOI] [PubMed] [Google Scholar]
- 56. Li H, Ayer LM, Lytton J, et al. Store-operated cation entry mediated by CD20 in membrane rafts. J Biol Chem 2003; 278: 42427–42434. [DOI] [PubMed] [Google Scholar]
- 57. Palanichamy A, Jahn S, Nickles D, et al. Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol 2014; 193: 580–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Holley JE, Bremer E, Kendall AC, et al. CD20+ inflammatory T-cells are present in blood and brain of multiple sclerosis patients and can be selectively targeted for apoptotic elimination. Mult Scler Relat Disord 2014; 3: 650–658. [DOI] [PubMed] [Google Scholar]
- 59. Schuh E, Berer K, Mulazzani M, et al. Features of human CD3+CD20+ T cells. J Immunol 2016; 197: 1111–1117. [DOI] [PubMed] [Google Scholar]
- 60. Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol 2006; 6: 394–403. [DOI] [PubMed] [Google Scholar]
- 61. Perez-Andres M, Paiva B, Nieto WG, et al. Human peripheral blood B-cell compartments: a crossroad in B-cell traffic. Cytometry B Clin Cytom 2010; 78(Suppl. 1): S47–S60. [DOI] [PubMed] [Google Scholar]
- 62. Mei HE, Schmidt S, Dorner T. Rationale of anti-CD19 immunotherapy: an option to target autoreactive plasma cells in autoimmunity. Arthritis Res Ther 2012; 14(Suppl. 5): S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mahevas M, Patin P, Huetz F, Descatoire M, Cagnard N, Bole-Feysot C, et al. B cell depletion in immune thrombocytopenia reveals splenic long-lived plasma cells. The Journal of clinical investigation. 2013; 123(1): 432–42. Epub 2012/12/18. doi: 10.1172/JCI65689. PubMed PMID: 23241960; PubMed Central PMCID: PMC3533302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mahevas M, Michel M, Vingert B, Moroch J, Boutboul D, Audia S, et al. Emergence of long-lived autoreactive plasma cells in the spleen of primary warm auto-immune hemolytic anemia patients treated with rituximab. J Autoimmun. 2015; 62: 22–30. doi: 10.1016/j.jaut.2015.05.006. PubMed PMID: 26112660. [DOI] [PubMed] [Google Scholar]
- 65. Piccio L, Naismith RT, Trinkaus K, Klein RS, Parks BJ, Lyons JA, et al. Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch Neurol. 2010; 67(6): 707–14. doi: 10.1001/archneurol.2010.99. PubMed PMID: 20558389; PubMed Central PMCID: PMCPMC2918395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Arumugakani G, Stephenson SJ, Newton DJ, et al. Early emergence of CD19-negative human antibody-secreting cells at the plasmablast to plasma cell transition. J Immunol 2017; 198: 4618–4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bhoj VG, Arhontoulis D, Wertheim G, et al. Persistence of long-lived plasma cells and humoral immunity in individuals responding to CD19-directed CAR T-cell therapy. Blood 2016; 128: 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Naddafi F, Davami F. Anti-CD19 monoclonal antibodies: a new approach to lymphoma therapy. Int J Mol Cell Med 2015; 4: 143–151. [PMC free article] [PubMed] [Google Scholar]
- 69. Horton HM, Chu SY, Ortiz EC, et al. Antibody-mediated coengagement of FcγRIIb and B cell receptor complex suppresses humoral immunity in systemic lupus erythematosus. J Immunol 2011; 186: 4223–4233. [DOI] [PubMed] [Google Scholar]
- 70. Heyman B. Feedback regulation by IgG antibodies. Immunol Lett 2003; 88: 157–161. [DOI] [PubMed] [Google Scholar]
- 71. Herbst R, Wang Y, Gallagher S, et al. B-cell depletion in vitro and in vivo with an afucosylated anti-CD19 antibody. J Pharmacol Exp Ther 2010; 335: 213–222. [DOI] [PubMed] [Google Scholar]
- 72. Stuve O, Cutter GR. Does natalizumab therapy benefit patients with multiple sclerosis? JAMA Neurol 2014; 71: 945–946. [DOI] [PubMed] [Google Scholar]
- 73. Gallagher S, Yusuf I, McCaughtry TM, et al. MEDI-551 treatment effectively depletes B cells and reduces serum titers of autoantibodies in mice transgenic for Sle1 and human CD19. Arthritis Rheumatol 2016; 68: 965–976. [DOI] [PubMed] [Google Scholar]
- 74. Chen D, Blazek M, Ireland S, et al. Single dose of glycoengineered anti-CD19 antibody (MEDI551) disrupts experimental autoimmune encephalomyelitis by inhibiting pathogenic adaptive immune responses in the bone marrow and spinal cord while preserving peripheral regulatory mechanisms. J Immunol 2014; 193: 4823–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Chen D, Ireland SJ, Davis LS, et al. Autoreactive CD19 +CD20− plasma cells contribute to disease severity of experimental autoimmune encephalomyelitis. J Immunol 2016; 196: 1541–1549. [DOI] [PubMed] [Google Scholar]
- 76. Schiopu E, Chatterjee S, Hsu V, et al. Safety and tolerability of an anti-CD19 monoclonal antibody, MEDI-551, in subjects with systemic sclerosis: a phase I, randomized, placebo-controlled, escalating single-dose study. Arthritis Res Ther 2016; 18: 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Agius M, Klodowska-Duda G, Sweeny S, et al. Safety and tolerability of MEDI-551 in patients with relapsing forms of multiple sclerosis: results from a phase I randomized, placebo-controlled escalating IV and SC dose study. In: European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS). Barcelona, Spain, 2015. [Google Scholar]
- 78. Streicher K, Morehouse CA, Groves CJ, et al. The plasma cell signature in autoimmune disease. Arthritis Rheumatol 2014; 66: 173–184. [DOI] [PubMed] [Google Scholar]
- 79. Cree BA, Bennett JL, Sheehan M, et al. Placebo-controlled study in neuromyelitis optica: ethical and design considerations. Mult Scler 2016; 22: 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stuve O, Cepok S, Elias B, et al. Clinical stabilization and effective B-lymphocyte depletion in the cerebrospinal fluid and peripheral blood of a patient with fulminant relapsing-remitting multiple sclerosis. Arch Neurol 2005; 62: 1620–1623. [DOI] [PubMed] [Google Scholar]
- 81. Martin M, Cravens PD, Winger R, et al. Depletion of B lymphocytes from cerebral perivascular spaces by rituximab. Arch Neurol 2009; 66(8): 1016–20. [DOI] [PubMed] [Google Scholar]
- 82. Montalvao F, Garcia Z, Celli S, et al. The mechanism of anti-CD20-mediated B cell depletion revealed by intravital imaging. J Clin Invest 2013; 123: 5098–5103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Hawker K, O’Connor P, Freedman MS, et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol 2009; 66: 460–471. [DOI] [PubMed] [Google Scholar]
- 84. Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing-remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet 2011; 378: 1779–1787. [DOI] [PubMed] [Google Scholar]
- 85. Genentech. Ocrelizumab [data on file]. South San Francisco (CA), 2003. [Google Scholar]
- 86. Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2017; 376: 221–234. [DOI] [PubMed] [Google Scholar]
- 87. Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017; 376: 209–220. [DOI] [PubMed] [Google Scholar]
- 88. Rommer PS, Dudesek A, Stuve O, et al. Monoclonal antibodies in treatment of multiple sclerosis. Clin Exp Immunol 2014; 175: 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Coiffier B, Lepretre S, Pedersen LM, et al. Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: a phase 1–2 study. Blood 2008; 111: 1094–1100. [DOI] [PubMed] [Google Scholar]
- 90. Zhang B. Ofatumumab. mAbs 2009; 1: 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sorensen PS, Lisby S, Grove R, et al. Safety and efficacy of ofatumumab in relapsing-remitting multiple sclerosis: a phase 2 study. Neurology 2014; 82: 573–581. [DOI] [PubMed] [Google Scholar]
- 92. Cree BA, Lamb S, Morgan K, et al. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005; 64: 1270–1272. [DOI] [PubMed] [Google Scholar]
- 93. Jacob A, Weinshenker BG, Violich I, et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol 2008; 65: 1443–1448. [DOI] [PubMed] [Google Scholar]
- 94. Pellkofer HL, Krumbholz M, Berthele A, et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 2011; 76: 1310–1315. [DOI] [PubMed] [Google Scholar]
- 95. Kim SH, Kim W, Li XF, et al. Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol 2011; 68: 1412–1420. [DOI] [PubMed] [Google Scholar]
- 96. Jarius S, Aboul-Enein F, Waters P, et al. Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 2008; 131: 3072–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cambridge G, Leandro MJ, Edwards JC, et al. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum 2003; 48: 2146–2154. [DOI] [PubMed] [Google Scholar]
- 98. Vallerskog T, Gunnarsson I, Widhe M, et al. Treatment with rituximab affects both the cellular and the humoral arm of the immune system in patients with SLE. Clin Immunol 2007; 122: 62–74. [DOI] [PubMed] [Google Scholar]
- 99. Stohl W, Gomez-Reino J, Olech E, et al. Safety and efficacy of ocrelizumab in combination with methotrexate in MTX-naive subjects with rheumatoid arthritis: the phase III FILM trial. Ann Rheum Dis 2012; 71: 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. van Vollenhoven RF, Fleischmann RM, Furst DE, et al. Longterm safety of rituximab: final report of the rheumatoid arthritis global clinical trial program over 11 years. J Rheumatol 2015; 42: 1761–1766. [DOI] [PubMed] [Google Scholar]
- 101. www.fda.gov/Drugs/DrugSafety/ucm366406.htm (accessed 20 February 2018).
- 102. www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm126519.htm (accessed 20 February 2018).
- 103. Platt MP, Agalliu D, Cutforth T. Hello from the other side: how autoantibodies circumvent the blood–brain barrier in autoimmune encephalitis. Front Immunol 2017; 8: 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kinzel S, Weber MS. The role of peripheral CNS-directed antibodies in promoting inflammatory CNS demyelination. Brain Sci 2017; 7: E70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Blair PA, Norena LY, Flores-Borja F, et al. CD19 +CD24hiCD38hi B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity 2010; 32: 129–140. [DOI] [PubMed] [Google Scholar]
- 106. Shen P, Roch T, Lampropoulou V, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014; 507: 366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Rubenstein JL, Combs D, Rosenberg J, et al. Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood 2003; 101: 466–468. [DOI] [PubMed] [Google Scholar]