

Abstract
Hereditary haemorrhagic telangiectasia (HHT), also known as Osler–Weber–Rendu syndrome, is a rare autosomal dominant vascular disorder. Patients with HHT may present with a wide spectrum of clinical manifestations, some considered to be life-threatening. We present the case of a 53-year-old male who presented with massive haemoptysis. Chest computed tomography scan was remarkable for a large anterior, left lower lobe arteriovenous malformation. The patient underwent a pulmonary angiogram with embolization of a large left lung arteriovenous malformation, which proved to be successful in controlling the bleeding.
INTRODUCTION
Hereditary hemorrhagic telangiectasia, also known as Osler–Weber–Rendu syndrome, is a rare autosomal dominant vascular disorder that affects ~1 in 5000 individuals [1]. Patients may present with a wide spectrum of clinical manifestations, some considered to be life-threatening [2]. The most common presentation is epistaxis, gastrointestinal bleeding, iron deficiency anemia and mucocutaneous telangiectasia [2]. In most cases, arteriovenous malformations are usually asymptomatic, however, on certain occasions a wide range of complications including high-output heart failure, portal hypertension, liver failure, hemoptysis, polycythemia, cerebral abscess and stroke may occur [3]. Patients with pulmonary arteriovenous malformation may also present with dyspnea, cyanosis and massive hemoptysis.
CASE REPORT
This is the case of a 53-year-old non-smoking male with history of anemia, blood transfusions and recurrent episodes of epistaxis who was presented to the emergency room with cough of ~2 weeks. Cough was mostly dry initially then became productive with evidence of blood in the sputum, to the amount of three cups daily. Other symptoms were minimal dyspnea and generalized weakness. The patient was born in the Caribbean, denied recent travel, exposure to sick contacts, weight loss and night sweats. Furthermore, a recent PPD was negative. He had neither previous episodes of hemoptysis nor any family history of hematologic or cutaneous problems. On physical examination, the patient was coughing up fresh bright red blood. Additionally, mucocutaneous telangiectasias were identified on the lips and tongue (Fig. 1). Lungs were essentially clear to auscultation. Hemoglobin was 5.4 g/dl, MCV of 69 fl, MCH of 19.8 pg and platelets level of 304 000. Metabolic and coagulation profiles were grossly unremarkable. Chest computed tomography scan was remarkable for a 2.5 × 3.7 × 4.0 cm smooth oval circumscribed anterior left lower lobe mass associated with bronchovascular structures, suggestive of a large arteriovenous malformation (AVM) (Fig. 2). No evidence of lung parenchymal infiltrates or lesions was identified. Patient was admitted to the intensive care unit for volume resuscitation with blood products. After resuscitation, the patient was transferred to the interventional radiology suite for a pulmonary angiogram with embolization of a large left lung AVM, which proved to be successful in controlling the bleeding (Figs 3–5). After immediate response following the procedure, patient was discharged 3 days later. We followed up the patient up to 3 months and he remained asymptomatic without associated complications or further episodes of hemoptysis. Follow-up brain magnetic resonance imaging, endoscopy and colonoscopy were negative for others AVMs.
Figure 1:
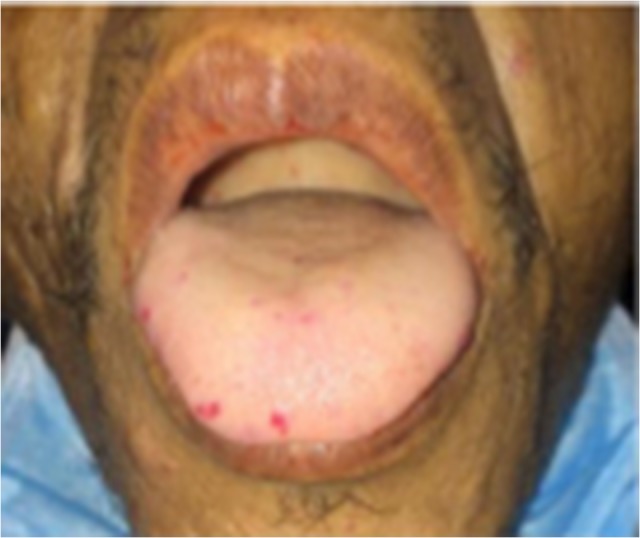
Multiple mucosal hemorrhagic telangiectasias, more prominent on the right lateral aspect of the tongue
Figure 2:
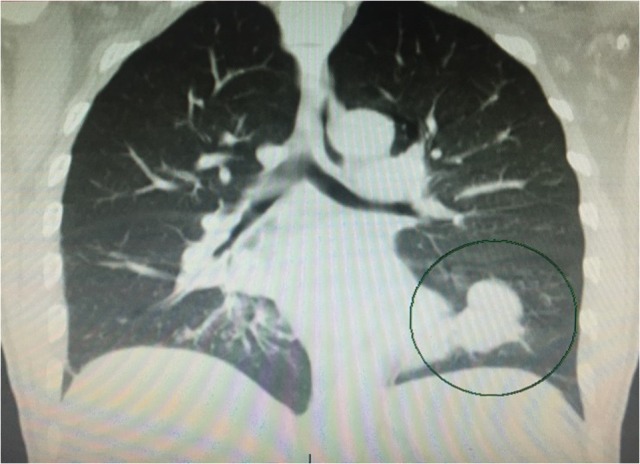
Chest computer tomography shows left lower lung pulmonary AVM (black circle)
Figure 3:
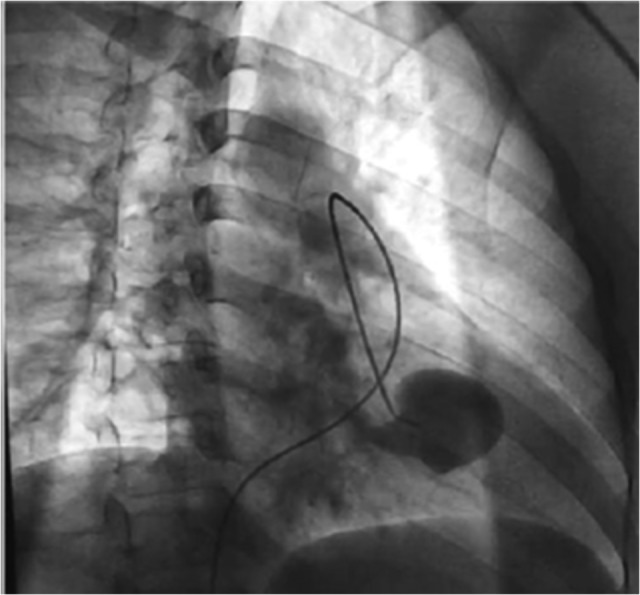
Pre-embolectomy fluoroscopy image showing afferent artery with the presence of aneurysm with feeding effect
Figure 5:
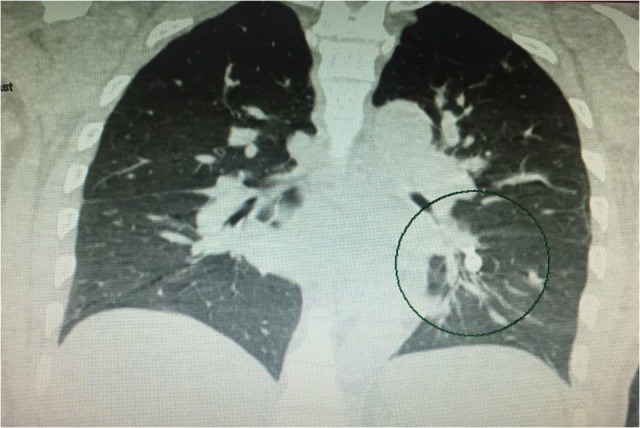
Chest computer tomography showing status post-embolectomy of pulmonary AVM (black circle)
Figure 4:
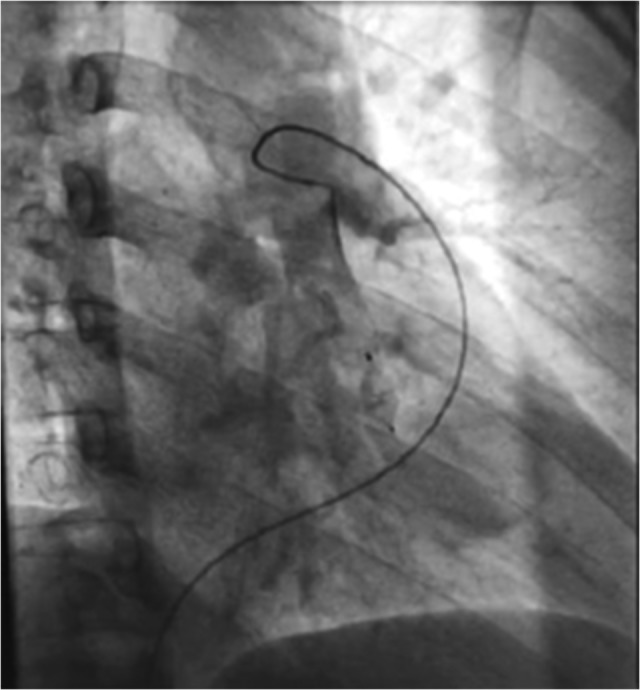
Post-embolectomy fluoroscopy image showing afferent artery without signs of perfusion or feeding
DISCUSSION
Our patient presented with massive hemoptysis with more than 500 ml of expectorated blood over a 24-h period. Most common causes of hemoptysis were ruled out and it was after a thorough physical examination along with the images that the diagnosis of Osler–Weber–Rendu (hereditary hemorrhagic telangiectasia (HHT)) was made. The diagnosis of this rare condition is made on the basis of the Curaçao criteria established in June 1999, which include the following factors: spontaneous and recurrent epistaxis, multiple mucocutaneous telangiectasia, visceral lesions and first degree family history [3]. This condition is an autosomal dominant hereditary disease that can present with sporadic mutation with varying penetrance and expression [4]. Even though many genes have been implicated with the disease pathogenesis, the most important ones codes for transmembrane glycoproteins expressed abundantly on vascular endothelial cells. They will mediate vascular remodeling by affecting the extracellular matrix production [4]. The main characteristics of the disease are the presence of abnormal capillary connections between arterioles and venules of the skin and mucosa, which arise as teleangiectasias [5]. This abnormal arteriovenous communication with focal dilatation of post-capillary venules often cause small capillary bleedings founded on skin and mucosal surfaces, particularly on the face, lips and nasopharynx [5]. Apart from small blood vessels AVMs, abnormalities can also affect communication between larger arteries and veins, which commonly occur in the pulmonary, hepatic and cerebral circulations and can lead to life-threatening bleeding. It is known that 15–30% of the patients with HHT develop pulmonary AVMs (PAVM’s) and about 70% of PAVMS’s are due to HHT [6]. The most common presenting features are those attributable to the PAVM’s itself. Features include dyspnea and hemoptysis in terms of parenchymal PAVM’s or endobronchial telangiectasias or hemothorax for subpleural PAVM’s [3]. Adults with HHT should be screened for PAVMs due to the high incidence [2]. Besides, there is evidence that treatment indeed reduces stroke and risk of brain abscess. Patients who have one or more PAVM with a feeding artery diameter from >2 to 3 mm, would benefit from treatment with embolotherapy rather than observation, as the former results in an immediate improvement in the radiographic appearance of PAVM’s, symptoms and oxygenation [3]. Over the long term, the rate of serious complications, especially neurologic events including stroke and cerebral abscess are reduced [7]. This case probably represents a sporadic mutation leading to Osler–Weber–Rendu disease due to a lack of history of hemoptysis, anemia or cutaneous disorders in his family. After 4 months from hospital discharge, the patient had no recurrence of hemoptysis.
Common causes of hemoptysis such as tuberculosis and malignancy among many others should always be excluded [8]. Physicians need to be aware of rare and unusual etiologies such as HHT. It is crucial to recognize the signs and symptoms of Osler–Weber–Rendu disease and its association with PAVM’s in order to provide appropriate management and treatment options. Few alternatives treatments are available for this disease, however, without proper recognition and identification by physicians, significant morbidity and mortality could result. Two studies showed that in an essentially unscreened and untreated population of HHT the median life expectancy was significantly lower than that patients without HHT. Patients with some HHT specific mutation died 3–7.1 years earlier than controls mainly related to neurological and hemorrhagic complications [9, 10].
HHT is a rare multisystemic disease, commonly manifested with recurrent epistaxis, mucocutaneous telangiectasia, iron deficiency anemia and hemoptysis, which may present abruptly [5]. Although many patients with HHT may be asymptomatic and unaware of the disease, life-threatening manifestations may occur [11]. It is crucial to establish the diagnosis as in this case to prevent possible complications. Its rapid recognition and management of specific vascular lesions as well as screening of asymptomatic individuals for PAVM’s will result in decrease rate of morbidity and mortality in this specific group.
CONFLICT OF INTEREST STATEMENT
None declared.
FUNDING
This case report did not receive any specific grant from any funding agency in the public, commercial or non-profit sector.
PATIENT CONSENT
A written informed consent was obtained from the patient for publication of the case.
GUARANTOR
Michael Cruz Caliz.
REFERENCES
- 1. Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev 2010;24:203 DOI:10.1016/j.blre.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 2. Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet 2011;48:73–87. DOI:10.1136/jmg.2009.069013. [DOI] [PubMed] [Google Scholar]
- 3. Lacombe P, Lacout A, Marcy PY, Binsse S, Sellier J, Bensalah M, et al. Diagnosis and treatment of pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia: an overview. Diagn Interv Imaging 2013;94:835 DOI:10.1016/j.diii.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 4. Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001;345:325–34. DOI:10.1056/NEJM200108023450503. [DOI] [PubMed] [Google Scholar]
- 5. Cottin V, Chinet T, Lavolé A, Corre R, Marchand E, Reynaud-Gaubert M, et al. Pulmonary arteriovenous malformations in hereditary hemorrhagic telangiectasia: a series of 126 patients. Medicine (Baltimore) 2007;86:1–17. DOI:10.1097/MD.0b013e31802f8da1. [DOI] [PubMed] [Google Scholar]
- 6. Nataraju KT, Mukherjee T, Doddaiah RPH, Nanjappa NG, Narasegowda L. A rare case of pulmonary arterio-venous malformation with recurrent anemia: hereditary hemorrhagic telangiectasia. Lung India 2015;32:384–8. http://doi.org/10.4103/0970-2113.159587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ference BA, Shannon TM, White RI Jr, Zawin M, Burdge CM. Life-threatening pulmonary hemorrhage with pulmonary arteriovenous malformations and hereditary hemorrhagic telangiectasia. Chest 1994;106:1387–90. [DOI] [PubMed] [Google Scholar]
- 8. Hirshberg B, Biran I, Glazer M, Kramer MR. Hemoptysis: etiology, evaluation, and outcome in a tertiary referral hospital. Chest 1997;112:440. [DOI] [PubMed] [Google Scholar]
- 9. de Gussem EM, Edwards CP, Hosman AE, Westermann CJ, Snijder RJ, Faughnan ME, et al. Life expextancy of parents with hereditary haemorrhagic telangiectasia. Orphanet J Rare Dis 2016;11:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Donaldson JW, McKeever TM, Hall IP, Hubbard RB, Fogarty AW. Complications and mortality in hereditary hemorrhagic telangiectasia: a population-based study. Neurology 2015;84:1886–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bergler W, Götte K. Hereditary hemorrhagic telangiectasias: a challenge for the clinician. Eur Arch Otorhinolaryngol 1999;256:10–5. [DOI] [PubMed] [Google Scholar]