

Abstract
Nasopharyngeal carcinoma (NPC) is an epithelial malignancy, which is notorious among head-and-neck cancers with its metastatic feature. Epstein–Barr virus (EBV) infection plays a fundamental role in NPC development with the mechanism is not well understood. Here we demonstrate that EBV oncoprotein LMP1 drives EMT and metastasis of NPC by reactivating the adhesion molecule, cadherin 6 (CDH6), which normally occurs in embryogenesis with unknown role in NPC. CDH6 was found to be upregulated in LMP1-positive NPC tissues, and was identified as a target of the epithelium-specific miR-203. LMP1-activated NF-κB transcriptionally repressed the miR-203 expression by binding to the promoter region of miR-203 gene. CDH6 activation in turn induced EMT and promoted metastasis in NPC. CDH6 depletion, NF-κB inhibitor and miR-203 overexpression were able to impair the EMT effects. The miR-203 downregulation in NPC tissues was strongly associated with metastasis clinically. The CDH6 activator, Runt-related transcription factor 2 (RUNX2), was also activated by EBV in the event. For both CDH6 and RUNX2 are components at TGF-β downstream, CDH6 became a node protein for the interplay of multiple signalings including NF-κB and TGF-β. Therefore, the switch-on of miR-203 was important for nasopharyngeal epithelial cells to maintain normal phenotype. This study demonstrates that EBV has evolved sophisticated strategies by driving epithelial cells to obtain malignant features, particularly in NPC metastasis, providing novel biomarkers for the therapy and prognosis of EBV-associated NPC.
Subject terms: Head and neck cancer, Tumour virus infections
Introduction
The Epstein–Barr virus (EBV) is a member of the human γ-herpesvirus, which infects >90% of the world’s population. EBV has been implicated to be strongly associated with the development of several malignancies including Burkitt’s lymphoma and nasopharyngeal carcinoma (NPC)1. NPC, primarily of epithelial origin, is a type of metastatic head-and-neck neoplasm that is highly prevalent in southern China and some other areas of East Asia and Africa2. EBV infection is one documented etiological factor1. However, the mechanism of EBV acts in the development of NPC remains largely to be understood. Latent membrane protein 1 (LMP1) is known as the viral oncoprotein that is notable for its transforming potential. LMP1 activates cell signalings such as nuclear factor (NF)-κB, which is an important transcription factor involved in the cell transformation regulation and tumorigenesis of NPC3–5. Distant metastasis is still the dominant treatment failure of NPC, although the current chemotherapy and radiotherapy applications are effective.
Epithelial–mesenchymal transition (EMT) is a process that epithelial cells lose adhesion and cytoskeletal components, being concomitant with a gain of mesenchymal components and the initiation of a migratory phenotype. The critical step in invasion and metastasis is attributed to this process6. MicroRNAs (miRNA) are non-coding RNAs that suppress the expression of protein-coding genes through imperfect base pairing with the 3' untranslated regions (UTRs) of target mRNAs7–10. In viral infection, host miRNAs critically regulate defense mechanisms though largely unknown. In NPC, little is known about the interaction between EBV and host miRNA in EMT and metastasis process. We previously found the epithelium-specific miR-203 was downregulated by EBV-encoded LMP1 to promote the proliferation of epithelial cells11, but the specific mechanism about miR-203 downregulation remains unknown. The miRNA-203 is also involved in several aspects of tumorigenesis in a few other epithelial malignancies with unknown role in NPC metastasis12–14. As metastasis is still the main cause of death of NPC patients, understanding the metastasis mechanism is very important. This study would illustrate that miR-203 is targeted by EBV to mediate this important stage or process of NPC development.
In cancer progression, cells have to gain some key hallmarks to become fully neoplastic and finally malignant. This is a multistep process in which rewiring of signaling pathways and reactivation of some molecules may be involved15. Whether a tumor virus-like EBV has the function to help cancer cells acquire these features remains largely to be elucidated. Adhesion molecules like N-cadherin are active only in embryogenesis and may be reactivated during tumorigenesis. Cadherin 6 (CDH6, also K-cadherin) is a member of cadherin family with very limited description in cancers and unknown role in NPC. In this approach, CDH6 was first predicted to be a miR-203 target and detected to be overexpressed in NPC tissues, leading to an intensive study about it.
In the present study, LMP1-activated NF-κB inhibits the expression of the host “switch” miR-203 through binding to the promoter region of miR-203 encoding gene. This contributes to the reactivation of CDH6, which in turn induces EMT and promotes metastasis of NPC. The bone metastasis marker and CDH6 activator, Runt-related transcription factor 2 (RUNX2) is also involved in the event. Both CDH6 and RUNX2 are downstream effectors of transforming growth factor β (TGF-β) signaling pathway13,16. CDH6 becomes a node protein between NF-κB and TGF-β signalings, which are the most important in cancer progression. The results reveal a new mechanism deciphering the role of EBV in the etiology and treatment failure of NPC.
Results
CDH6 is reactivated in NPC as a putative target of LMP1-suppressed miR-203
CDH6 was predicted to be a target of miR-203 and validated to be overexpressed in the LMP1-positive NPC tissues (Fig. 1a, case 1). Corresponding to LMP1-negative expression, miR-203 was overexpressed, whereas CDH6 was negatively expressed (Fig. 1a, case 2). The quantitative PCR (qPCR) results with scatterplots showed that high expression levels in the immunohistochemistry (IHC) correlate with RNA expression and vice versa (Figs. 1b-e). Corresponding to the high expression of LMP1 in NPC tissues (n = 77) (p = 0.0027), the expression of miR-203 was downregulated (p = 0.0062), whereas the expression levels of CDH6 (p = 0.014) and RUNX2 (p = 0.008) were upregulated. RUNX2 is known as another target of miR-203 and an activator of CDH613,16. The correlation analysis showed similar results between each two factors of them (Figs. 1f-j and Supplemental Fig. 1a). LMP1 expression is negatively correlated with miR-203 expression (p = 0.0015), and positively with CDH6 (p < 0.0001) and RUNX2 (p = 0.003). The expression levels of CDH6 and RUNX2 are positively correlated (p = 0.0136) (Figs. 1a, e, i, j). In addition, the expression of both CDH6 and RUNX2 were abnormally at high levels in the three tissues from patients with bone metastasis (Supplemental Fig. 1b).
Fig. 1. The expression relationship of EBV/LMP1, miR-203, CDH6, RUNX2 in NPC tissues.

a The detection of EBER, LMP1, miR-203, CDH6 and RUNX2 in NPC tissues. IHC (for LMP1, RUNX2 and CDH6) and ISH (for EBER and miR-203) assays were performed on the serial sections from each case, respectively. Calibration bars correspond to 50 µm in panels. b-e The scatter diagrams for the detection of LMP1, miR-203, CDH6 and RUNX2 in NPC tissues by qPCR, respectively. A total of 77 NPC and 7 normal tissues were used. b The detection of LMP1. c The detection of miR-203. d The detection of CDH6. e The detection of RUNX2. f-j Correlations between every two factors of LMP1, miR-203, CDH6 and RUNX2 according to their expression levels by qPCR detection. f Correlations between LMP1 and miR-203. g Correlations between CDH6 and miR-203. h Correlations between LMP1 and CDH6. i Correlations between LMP1 and RUNX2. j Correlations between RUNX2 and CDH6. Two-tailed Spearman’s correlation analysis was carried out. The corresponding difference is considered significant when p < 0.05
The miR-203 directly targets CDH6
CDH6 was predicted as a target, which contained two potential binding sites at the 3'-UTR for miR-203 by softwares (Fig. 2a). To determine this, luciferase reporters were constructed (Supplemental Fig. 1) and the activity was examined. The result showed significantly decreased luciferase activity for both binding sites and not for their mutants (Fig. 2b). The use of miR-203 inhibitor promoted the expression of CDH6 and RUNX2 in HK-1 and 5-8F NPC cells (Figs. 2c-f). The results validated that CDH6 was the target of miR-203. Consistent with in vitro results, IHC or in situ hybridization (ISH) detections in xenografts generated from 293-EBV cells revealed the same expression relationship between miR-203 and CDH6/RUNX2, as well as LMP1 and miR-203 (Fig. 2g). The expression of the epithelial marker, E-cadherin, was also reduced when CDH6 was at high expression level (Fig. 2g).
Fig. 2. miR-203 directly regulates CDH6.

a Two potential miR-203 binding sites with base pairing at the CDH6 3'-UTR as shown were predicted by the software, TargetScan. b Luciferase assay for the miR-203 binding to CDH6 3'-UTR. The miR-203 mimics decreased luciferase activities compared with wild-type CDH6 3'-UTR, whereas it did not influence those using mutant CDH6 3'-UTR or scramble miRNA as a negative control (Ctrl). Results are means ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001. c-f The detection of miR-203, CDH6 and RUNX2 in 5-8F or HK-1 treated with miR-203 inhibitor. Ctrl, negative control. c The mRNA expression detection of miR-203 and CDH6 in 5-8F. d The mRNA expression detection of miR-203 and CDH6 in HK-1. e The expression detection of CDH6 and RUNX2 in 5-8F by western blotting. The right panel shows the mean gray values and their difference from three experiments. f The expression detection of CDH6 and RUNX2 in HK-1. The right panel shows the mean gray values and their difference from three experiments. g The detection of EBER, LMP1, miR-203, CDH6, RUNX2 and E-cadherin in xenograft sections derived from 293-EBV. IHC (for LMP1, CDH6, RUNX2 and E-cadherin) and ISH (for EBER and miR-203) assays were performed respectively. The lower panels showed the corresponding negative controls (Ctrl) using scramble miRNA, probe or phosphate-buffered saline (PBS; for antibody control). Calibration bars correspond to 50 µm in panels
CDH6 induces EMT and promotes invasion and metastasis of NPC
The CDH6 and miR-203 expression exhibited different levels in NPC cell lines with different metastatic abilities at both mRNA and protein levels (Fig. 3a and Supplemental Fig. 3). There was a significant difference of the expression of CDH6 and miR-203 between HK-1 and 5-8F cells. Based on the background of CDH6 expression levels, we selected the HK-1 with a lower level of CDH6 for overexpression analysis and 5-8F with higher level of CDH6 for small-interfering RNA (siRNA) analysis. In order to examine the role of CDH6 targeted by miR-203, we used three siRNAs to suppress CDH6 expression in 5-8F cells (Supplemental Fig. 4) and chose number 2 siRNA based on its best inhibition efficiency for the subsequent application. The expression vector, pCMV-CDH6, was used to increase the expression of CDH6 in HK-1 cells. In HK-1 cells with low level of CDH6 background, the ectopic expression of CDH6 inhibited the expression of epithelial marker (E-cadherin) and increase the expression of mesenchymal markers (N-cadherin, Vimentin, Slug and Snail) (Fig. 3b and Supplemental Fig. 5a). On the other hand, the deletion of CDH6 by siRNA in highly metastatic 5-8F cells showed opposite results (Fig. 3b and Supplemental Fig. 5b). In order to confirm that CDH6-elicited EMT was a prelude of tumor metastasis, more experiments were carried out. Transwell assays demonstrated that CDH6 significantly promoted the invasion and migration activities of NPC cells respectively (Figs. 3c-f).
Fig. 3. CDH6 promotes EMT, cell migration and invasion of NPC cells.

a The different expression levels of CDH6 and miR-203 in NPC detected by qPCR and western blotting. Results are means ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001. For the CDH6 detection by western blotting assay, the quantification analysis of band gray values from three experiments is showed in Supplemental Fig. 3. b Effect of CDH6 on the expression of EMT markers by western blotting analysis. Due to the different level of CDH6 expression background in the cells, overexpression of CDH6 (CDH6) and interference by si-CDH6 were used in HK-1 and 5-8F cells, respectively (the same below). “Ctrl” represents the empty vector control for the overexpression plasmid of CDH6 or the scramble sequenced siRNA control for si-CDH6 (the same below). The quantification analysis of band gray values from three experiments is showed in Supplemental Fig. 5. c, d Effect of CDH6 on cell migration measured by transwell assay without matrigel. c Effect of CDH6 on cell migration in HK-1 treated with overexpression of CDH6. d Effect of CDH6 on cell migration in 5-8F treated with si-CDH6 to inhibit the expression of CDH6. e, f Effect of CDH6 on cell invasion measured by transwell matrigel penetration assay. e Effect of CDH6 on cell invasion in HK-1. f Effect of CDH6 on cell invasion in 5-8F. Calibration bars correspond to 100 µm in panels c–f. Results are means ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001
The actin cytoskeleton of cells is an essential component of pseudopod and filopodia formation, which are associated with cancer invasion and metastasis17. As shown in Fig. 4a, CDH6 facilitated the formation of stress fibers (fluorescein isothiocyanate (FITC)-phalloidin stained F-actin), and the fiber amount was significantly reduced by CDH6 siRNA in 5-8F cells (Fig. 4b).
Fig. 4. CDH6 promotes the formation of F-actin fibers and induces the activation of RhoA/Rac2 signaling.

a Effect of CDH6 overexpression on the stress fiber formation in HK-1 cells by IF analysis using FITC-labeled phalloidin. Green, phalloidin staining (F-actin); blue, Hoechst (H) 33342 staining (nuclei). b Effect of CDH6 depletion by siRNA on the fiber integrity. The stress fibers were decreased and fractioned, due to the elasticity, the fractioned fibers were accumulated to both edges, near the nucleus and membrane. c Effect of CDH6 on the expression of RhoA and Rac2 detected by western blotting analysis. CDH6, the overexpression of CDH6; si-CDH6, the interference of siRNA-CDH6. Ctrl, the empty vector control for the CDH6 overexpression or scramble sequenced control for si-CDH6. Calibration bars correspond to 50 µm in panels a and b
RhoA/Rac2 signaling is related to the actin filament structure and contributes to cell migration18. The detection result showed that RhoA/Rac2 activity was inhibited by CDH6 in HK-1 cells stimulated by the CDH6 interference in 5-8F cells (Fig. 4c).
All these results showed that CDH6 regulated EMT and promoted metastasis of NPC.
LMP1-activated NF-κB transcriptionally inhibits miR-203 and thus activates CDH6
NF-κB is the main signaling activated by LMP13. We previously demonstrated that EBV induced miR-203 downregulation at the transcription level (pri-miRNA)11, but the specific mechanism remained unknown. The promoter of miR-203 encoding gene was predicted to be within 2000 bp at upstream of the transcription start site (TSS) of miR-20319. Bioinformatics analysis indicated that there were four putative NF-κB-binding sites inside the promoter region of miR-203. They were: –1300 to –1291(P1), –606 to –596 (P2), –175 to –166 (P3) and –17 to –8 (P4) (Fig. 5a). The region of 1452 bp from TSS was then cloned and identified by sequencing. Based on some preliminary experiments, four luciferase reporter plasmids with or without the P3 + P4 region were constructed for the binding detection (Figs. 5a, b). The P3 and P4 sites were combined as one because they were close to each other. The result showed that the P3 + P4 region was the active binding site for NF-κB (Fig. 5b). Chromatin immunoprecipitation (ChIP) assays also showed the binding activity of P3 + P4 region with NF-κB followed by PCR and qPCR detections (Figs. 5c, d). P1 and P2 sites did not have binding activity with NF-κB.
Fig. 5. NF-κB inhibits miR-203 by binding to its promoter region.

a Schematic diagram of the predicted binding sites for NF-κB on the miR-203 promoter region. P3 and P4 were combined as one site (P3 + P4) for the detection because they are close to each other. Different miR-203 promoter region fragments as indicated were inserted into the vector pGL3-enhancer for the luciferase activity assay. b Luciferase assay for the promoter binding activity of NF-κB. Luciferase activity was measured at 24 h post-transfection of the pGL3E- plasmids. The pGL3-enhancer vector served as a negative control. Ctrl, the empty vector (pCMV3); pCMV3-p65, the overexpression vector of NF-κB (pCMV3-p65). All the fragments containing the P3 + P4 site showed NF-κB (p65) binding activity, and the one (939bp) not containing P3 + P4 did not show binding activity. c, d ChIP assay was performed using the antibody against NF-κB p65 with IgG as a control in 293 or 293 with p65-overexpressing cells. Ctrl was the empty vector (pCMV3), and the plasmid pCMV3-p65 was constructed for NF-κB overexpression. The PCR amplification positions containing the p65-binding sites are indicated. Based on the result from c, the P1 site with negative activity was neglected in d. e Different protein expression levels of CDH6 and RUNX2 corresponding to the different levels of activated NF-κB (p-p65) in the cells containing EBV genomes (p2089 and its derivative). 293-BAC was a negative control. f Different mRNA expression levels of miR-203 and CDH6 in the cells containing EBV genomes (p2089 and its derivative). 293-BAC is a negative control. g-i CTAR1 and CTAR2 in LMP1 are known to be responsible for miR-203 downregulation and CDH6 activation via NF-κB. The 293 cells were transfected with LMP1 and the mutant plasmids, the expression of the indicated molecules were detected by western blotting or qPCR. g The detection of LMP1, p-p65, RUNX2 and CDH6 expression by western blotting. h The detection of miR-203 by qPCR. i The detection of CDH6 using qPCR. Results are means ± SD, n = 3. *p < 0.05, ***p < 0.001
As LMP1/NF-κB had been identified to be the major factors responsible for the miR-203 downregulation, the miR-203 and CDH6 expression were detected in the EBV-positive cells (C2089 and C22) and EBV-negative cells (293-BAC) with different expression levels of LMP1/NF-κB (Figs. 5e, f and Supplemental Fig. 6)11,20. The result verified that levels of miR-203 expression negatively related to CDH6 and LMP1/NF-κB expression. As LMP1 C-terminal activating regions (CTAR), CTAR1 and CTAR2, are responsible for NF-κB activation of LMP13, LMP1 CTAR deletion mutants, including LMP1-∆CTAR1, LMP1-∆CTAR2 and LMP1-∆CTAR3, were constructed and used to assess the expression relationship (Figs. 5g-i). The result showed that LMP1-∆CTAR1 and LMP1-∆CTAR2 were contributed to the downregulation expression of miR-203 and the upregulation expression of CDH6. All the results indicated that LMP1-activated NF-κB inhibited miR-203 expression and thus activated CDH6.
Caffeic acid phenethyl ester (NF-κB inhibitor) eliminates the effects of CDH6-mediated EMT in EBV-positive cell lines
Chemical NF-κB inhibitors can inhibit the phosphorylation of NF-κB, thus blocking NF-κB activation and nuclear entry21. As LMP1 could drive the CDH6 activation via NF-κB, we used NF-κB inhibitor for a further verification. In C666-1 cells, the NF-κB was stranded in the cytoplasm after treatment by the caffeic acid phenethyl ester (NF-κB inhibitor, Fig. 6a), indicating that the activity of NF-κB was inhibited. Further, the NF-κB inhibitor increased miR-203 and reduced CDH6/RUNX2 expression in EBV-positive cells, thereby eliminating the effect of LMP1/NF-κB on EMT (Figs. 6b-e).
Fig. 6. Caffeic acid phenethyl ester (NF-κB inhibitor) restores miR-203 expression and eliminates effects of LMP1/NF-κB on EMT in EBV-positive cell lines.

a NF-κB localization detected by IF assay in C666-1 cells when treated with Caffeic acid phenethyl ester. DMSO was the negative control. Green, p65; blue, H33342 staining (nuclei). In-NF-κB represents NF-κB inhibitor (the Caffeic acid phenethyl ester). b Effect of Caffeic acid phenethyl ester on the expression of miR-203 and CDH6 mRNA in C666-1 cells. Real-time qPCR was performed for the detection. c Effect of Caffeic acid phenethyl ester on the expression of EMT markers, CDH6 and RUNX2 detected by western blotting analysis. d Effect of Caffeic acid phenethyl ester on the expression of miR-203 and CDH6 in the EBV-positive cells line, C2089. e Effect of Caffeic acid phenethyl ester on the expression of EMT markers, CDH6 and RUNX2. Calibration bars correspond to 20 µm in panel a. Results are means ± SD; n = 3. *p < 0.05, **p < 0.01
The miR-203 reverses and LMP1 promotes the CDH6-induced EMT in NPC cells directly
In order to ensure that the CDH6-induced EMT could be inhibited by miR-203 directly, the miR-203 overexpression was performed in NPC cell lines. The result showed that EMT was inhibited accompanied with the expression inhibition of CDH6 and RUNX2 (Figs. 7a, b). Reversely, LMP1 overexpression promoted EMT, as well as the expression of CDH6 and RUNX2 (Figs. 7c, d). In addition, we also verified that the overexpression of miR-203 in NPC cells revealed a suppressive role on cell migration (Supplemental Figs. 7a and b). And, the overexpression of LMP1 also showed a promotion role on invasion and migration of NPC cells (Supplemental Figs. 7c-d). When the expression of LMP1 was reduced by siRNA in C2089, the EMT of cells was inhibited (Supplemental Fig. 8). These results showed that miR-203 reversed and LMP1 promoted the CDH6-induced EMT in NPC.
Fig. 7. The miR-203 inhibits CDH6-induced EMT and LMP1 promotes EMT in NPC cells.

a, b The effect of miR-203 overexpression on the expression of CDH6, RUNX2 and EMT markers in NPC cells. The 5-8F and HK-1 were transfected with pMIR-GFP-miR-203 or pMIR-GFP-NC, respectively, at 36 h post-transfection, proteins and RNA were collected and used for the detections by western blotting or qPCR, respectively. Ctrl, the empty vector (pMIR-GFP-NC); miR-203, the overexpression of miR-203 (pMIR-GFP-miR-203). Results are means ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001. c, d The effect of LMP1 on the expression of CDH6, RUNX2 and markers of EMT in NPC 5-8F and HK-1 cells. At 36 h post-transfection with LMP1 or control plasmids, proteins and RNA were collected and used for the detections by western blotting or qPCR, respectively. Ctrl, the empty vector (pEGFP-C1); LMP1, the overexpression of LMP1 (pEGFP-C1-LMP1). Results are means ± SD; n = 3. *p < 0.05, **p < 0.01, ***p < 0.001
The miR-203 downregulation is strongly correlated with NPC metastasis clinically
The analysis of gene expression omnibus (GEO) data (n = 121, GSE70970) indicated that the miR-203 downregulation in NPC tissues was associated with clinical metastasis, including lymph nodal and distant metastasis (Figs. 8a, b). Low miR-203 level was strongly associated with distant-relapse-free survival especially within the first 5 years (p = 0.0003) (Fig. 8c). This result was consistent with the clinical discipline that the 5-year survival is an important indicator of therapy efficiency. In addition, high miR-203 expression increased disease-free survival (p = 0.0415) (Fig. 8d). The results suggested that the expression level of miR-203 became a good biomarker for the prognosis and therapy of NPC.
Fig. 8. miR-203 downregulation correlates with NPC metastasis clinically.

a-d The analysis of GEO data (n = 121, GSE70970) indicated that the miR-203 downregulation in NPC tissues was associated with local lymphatic metastasis (N stage 0 to 3) a, distant metastasis b and c and disease-free survival (DFS) d. Chi-square test was used for a and b, and Gehan–Breslow–Wilcoxon test was used for c and d
Discussion
NPC is notorious for its metastatic feature among head-and-neck malignancies and is closely associated with EBV infection1,2. The metastasis at multiple sites such as neck lymph nodes and distant organs including bone, liver and intracranial invasion, is a common event1,22. The relationship and mechanism among NPC, EBV and metastasis remain poorly defined. In the present approach, we have discovered specific mechanism regarding this respect. LMP1-activated NF-κB transcriptionally suppresses the expression of miR-203, which expresses in epithelial cells relatively specifically11,23. The miR-203 acts as a “switch” to simultaneously restrain CDH6 and CDH6 activator, RUNX2. But, miR-203 can be reversed by EBV-LMP1 to trigger EMT, leading to NPC metastasis (summarized in Fig. 9). CDH6 and RUNX2 become novel mesenchymal markers in EBV-associated NPC. The results also reveal a mechanism that LMP1 drives the crosstalk among NF-κB, TGF-β and RhoA/Rac signaling pathways, with CDH6 acting as a node among them. This approach demonstrates that EBV has evolved ingenious abilities in helping cells to obtain multiple cancer features.
Fig. 9. Schematic for the mechanism of EBV LMP1-triggered and CDH6-mediated EMT in NPCviamiR-203 inhibition.
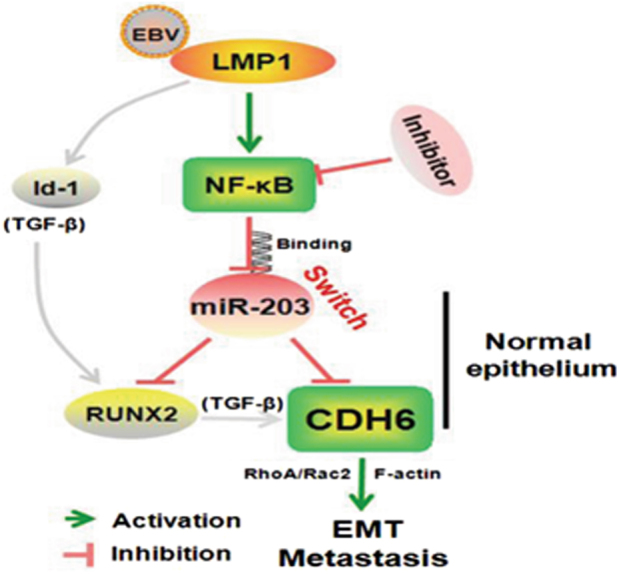
In normal epithelia, the expression of miR-203 turns off the expression of both CDH6 and CDH6 activator, RUNX2, for both CDH6 and RUNX2 are targets of miR-203. In NPC, due to EBV infection, the EBV-encoded oncoprotein LMP1 activates NF-κB, which further inhibits the expression of miR-203 through binding at the miR-203 promoter region, resulting in the reactivation of CDH6. NF-κB inhibitor can reverse the miR-203 downregulation of LMP1. The LMP1 partner, Id1, as well as miR-203 targeted RUNX and CDH6 are reported TGF-β downstream components that can be sequentially activated (in gray lines). The NPC cells endowed with CDH6 reactivation undergo EMT, in which RhoA/Rac2 signaling and F-actin fiber integrity were involved, leading to invasion and metastasis of NPC
LMP1 is an oncoprotein of EBV and expresses in most cells of EBV-associated NPC3,24,25. NF-κB, which has central functions in inflammation and cancer development5,26, is the main pathway activated by LMP127. Besides the transforming ability, LMP1 has been noticed for its induction of invasion and metastasis recently28–30. A meta-analysis has also revealed that LMP1 expression is positively associated with metastasis in NPC31. A few reports have shown that LMP1 regulates some transcriptional factors (TFs) to promote EMT in NPC with the exact mechanism unknown32,33. In the present study, we found that CDH6 reactivation by LMP1 induced EMT. The CDH6 has been limitedly described in cancers. Our results first assessed that CDH6 was reactivated by a virus CDH6 is a type 2 cadherin, which occurs during embryogenesis and is limited to express in some tissues such as kidney34. CDH6 has distinct functions compared with E-cadherin in tubulogenesis35 and functions like the mesenchymal marker N-cadherin in this study. This aberrant reactivation of CDH6 represented a hallmark of adhesion molecules in invasive carcinomas14,36. Our results were consistent with the study of Gugnoni et al.37. Similarly, the TF RUNX2 was also upregulated by LMP1. As RUNX2 can activate CDH616, this kind of doubled activation by EBV-LMP1 would be an effective confrontation to host. In the NPC distant metastasis post-treatment, bone is the most common metastatic site. Here we verified that the bone-effector RUNX2, as well as CDH6, were abnormally at high levels in NPC tissues from the patients suffering bone metastasis (Supplemental Fig. 1).
The transforming growth factor β (TGF-β) pathway is as important as NF-κB in cancer development and has been found to be involved in the invasion and metastasis in NPC38,39. No report has shown about the downstream of TGF-β and its interplay with other pathways such as NF-κB in NPC. CDH6 and RUNX2 are recently identified as downstream targets of TGF-β15. Besides, Id1 is another target of TGF-β and it can also activate CDH6 through the mediation of RUNX240,41. It has been reported that Id1 can be activated by LMP1 in NPC as an LMP1 binding partner42,43. The miR-203 is the master switch in normal epithelial cells and is recognized and reversed intensively by EBV. In view of all these reported factors and our results, EBV has developed perfect strategies targeting the terminal effector of CDH6 to promote invasion and metastasis of NPC (Fig. 9).
TFs are common reported transactivators of cellular genes, and here we showed the other side of NF-κB as it inhibited miR-203 gene expression. Actually, the inhibiting function of TFs on genes is not rare seen. NF-κB was also reported to act as a repressor of target genes by others44. So far, along with others, we have shown that miR-203 is a negative regulator in NPC and its downregulation is involved in several processes of NPC development, including proliferation11, invasion and metastasis (the present study) and radiotherapy resistance45. It is these studies that reveal miR-203 to be a perfect biomarker for the progression and therapy of NPC at different stages.
In summary, the reactivation of CDH6 is first found to be associated with EBV infection. EBV LMP1 has evolved impeccable strategies to drive EMT and metastasis in NPC by inhibiting the host switch, miR-203. In the event, several pro-metastatic features are involved, including the reactivation of the adhesion molecule and the interplay of multiple pathways with CDH6 as a common node protein. NF-κB inhibitor, CDH6 interference and miR-203 are able to similarly restrain the EMT effect. The novel biomarkers are attractive targets for the designing of improved diagnostic and therapeutic strategies of invasive EBV-associated NPC. This study reveals novel etiological mechanism of EBV in the development of NPC.
Materials and methods
Cell lines
NPC cells were early gifts kindly provided by the laboratories that established them and were maintained in our laboratory46–48. HK-1 was a non-metastatic cell line derived from well-differentiated NPC tissue, and 5-8F was a highly metastatic one. Both HK-1 and 5-8F were EBV-negative. C666-1 was an EBV-positive NPC cell line48. The human embryonic kidney HEK293 or 293 was ATCC origin and used for the latent infection of the whole EBV genome (p2089)20,49,50, resulting in the cell lines, C2089, C22 as described by us11. The 293-BAC cell line was established by us and used for the negative control with only BAC-based vector harboring in HEK293 cells20,51. These cell lines were grown in Dulbecco’s modified Eagle’s medium (Gibco, California, USA) supplemented with 10% fetal calf serum (FBS).
Plasmid constructs and the resultant cell lines
The plasmids of LMP1 (pcDNA3.1-LMP1 and pEGFP-C1-LMP1) were constructed using conventional cloning methods and confirmed by DNA sequencing. The empty vectors (pcDNA3.1 and pEGFP-C1) were purchased from Invitrogen Inc. (California, USA). The T4 polynucleotide kinase kit (Thermoscientific, Massachusetts, USA) was used to constructed the CTAR deletion mutants for LMP1 (pcDNA3.1-∆CTAR1, pcDNA3.1-∆CTAR2 and pcDNA3.1-∆CTAR3). The pEGFP-C1-LMP1 harbored the full-length LMP1 of NPC origin with its sequence reported as previously (NPC4)52, which was stably transfected into HEK293 cells resulting in 293-LMP1 cell line. The 293-control harbors only the vector. The pMIR-GFP-miR-203 expression vector with puromycin selection was purchased from the Vigenebio Inc. (Shangdong, China). The stably transfected resultant HEK293 cell line was 293-miR-203 (293-NC is the corresponding control with pMIR-GFP-scramble). The pCMV3-p65 and pCMV3-CDH6 overexpression vectors were products of the Sino Biological Inc. (Beijing, China).
The promoter of miR-203 gene was predicted and analyzed by the online softwares, Softberry, Promoter2.0, PromoSe and Promoter Scan. The fragments of different size within the promoter region were inserted into the luciferase reporter vector, pGL3-enhancer (pGL3E-), a product of Promega Inc. (Wisconsin, USA). Four resultant plasmids were: pGL3E-1452bp, 869bp, 533bp and 939bp (Fig. 5a). The sequences of the primer pairs were shown in Supplemental Table S2.
The basic information about hsa-miR-203 (NR_029620.1) was collected from the miRBase. Human CDH6 (K-cadherin, NM_004932.3) contained two putative miR-203 binding sites at the 3'-UTR predicted by softwares, TargetScan and Pictar. The primers for the binding sites (including the mutants) amplification for the luciferase assay were shown in Supplemental Table S2. Restriction sites of SpeI and HindIII were used for the insertion into the dual-luciferase vector, pMIR.
All other primers for real-time qPCR, ChIP, and siRNA-CDH6 sequences were shown in Supplemental Table S1. Number 2 among the three siRNAs was selected for the knockdown of CDH6 based on preliminary experiments of efficiency detection.
Invasion and migration assays
These assays were performed for the analysis of cell invasion and migration capacity, using transwell inserts with 8 µm pores (Corning, New York, USA) in 24-well plates. A total of 1×105 cells in 200 µl of serum-free medium were added to the upper transwell chamber ready with (for invasion assay) or without (for migration assay) matrigel (BD Biosciences, New York, USA). The lower chamber contained complete medium with 20% FBS. Cells were incubated for another 24 h at 37°C. Then non-migrated cells in the upper chamber were removed with a cotton swab, and cells migrated through the upper transwell chamber were fixed with methanol, stained with hematoxylin. Five random fields per well were counted at ×20 magnification under a microscope and the average cell number was calculated. The experiments were repeated for three times.
Wound-healing assay
The cells were seeded in a six-well plate, 8 h later, the plasmids, or si-CDH6 or the controls, were transfected into the cells severally. A p10 pipet tip was used to gently create a scratch in the cell monolayer when these cells grown to 90% confluence. Wound width was measured with an ocular ruler to ensure that all wounds were the same width at the beginning of each experiment. Images were captured at 0, 24 and 48 h after the wounding, respectively.
Luciferase reporter assay
The luciferase pMIR-report system was used to determine the binding between miR-203 and CDH6 3'-UTR. The promoter activity of miR-203 was evaluated by another luciferase reporter system, pGL3-enhancer (pGL3E-). One μg of specific plasmids were co-transfected into 2×105 HEK293 cells for each well in a 24-well plate using Lipofectamine 3000 (Invitrogen Inc.). The luciferase activity of was measured at 24 h post-transfection by using a Dual-Luciferase Reporter Assay Kit (Promega Inc.). Relative luciferase activity was calculated as the ratio of firefly to renilla luciferase activities.
ChIP assay
ChIP assay was performed using the EZ ChIP Chromatin Immunoprecipitation Kit (Millipore) according to the manufacturer’s instructions. DNA–protein complexes were pulled down from HEK293 cells transfected with the plasmid pCMV3-p65 using the NF-κB antibody. Normal rabbit IgG (Santa Cruz, California, USA, sc-2027) was used as a control antibody. Precipitated DNA was then subjected to DNA agarose gel electrophoresis and qPCR analysis using specific primers (Supplemental Table S2).
Western blotting analysis
Western blotting was performed as previously described49. Briefly, cells were lysed with RIPA buffer containing a protease inhibitor phenylmethanesulfonyl fluoride (PMSF) to obtain the protein. Equivalent amounts of denatured proteins were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis using a 10% gel and transferred onto a polyvinylidene difluoride membrane (Millipore). The membrane was incubated with a primary antibody followed by a second antibody for the chemiluminescence detection. The specific antibodies in this approach were: the LMP1 monoclonal antibody (mAb) (DAKO Lifetech, Glostrup, Denmark, M0897), anti-p-NF-κB (p65) antibody (CST, Chicago, USA, #3033), anti-NF-κB antibody (rabbit mAb, Millipore, 06-418), anti-CDH6 antibody (mouse mAb, Millipore, MAB2013), anti-RUNX2 rabbit mAb (Abcam, Cambridge, UK, AB23981), the antibodies of EMT makers (anti-E-cadherin, anti-Vimentin, anti-N-cadherin, anti-Slug and anti-Snail antibody, CST, Chicago, USA, #9782), anti-GAPDH antibodies (Proteintech, Chicago, USA, 10494-I-AP) and β-actin antibodies (Proteintech, 66009-I-Ig).
Clinical tissue samples and xenografts samples
The NPC biopsies, normal or non-neoplastic nasopharynx epithelium specimens were obtained from the Affiliated Tumor Hospital of Central South University and the Second Xiangya Hospital of Central South University. The biopsy samples used in this study had been submitted for histopathological diagnosis as poorly differentiated or undifferentiated NPCs and embedded in paraffin. Approval of the research was consented in the ethics committee of the host institution.
Xenografts were previously generated from 293-EBV cells in nude mice and embedded in paraffin as described49. Serial paraffin sections were used for IHC assay to detect LMP1, CDH6 and RUNX2, and for ISH to detect EBER and miR-203.
ISH for EBER and miR-203 detections
ISH was performed in tissue specimens using specific oligonucleotide probes. The probe of EBER-1 (Boster Inc., Wuhan, China) and miR-203 (Invitrogen Inc.) probes were digoxigenin-labeled at the 3′ terminus. The miR-203 probe sequences were listed in Supplementary Table 1. The tumor specimens were formalin-fixed, paraffin-embedded and sectioned serially. ISH were performed using an enhanced sensitive ISH detection kit (Boster Inc.) according to the manufacturer’s instruction. The stained sections were observed under a microscope. A scramble probe was used as a control compared with the specific probe.
IHC analysis
IHC was performed based on a manufactured kit (Boster Inc.). A polyclonal anti-LMP1 antibody and anti-RUNX2 rabbit mAb was obtained from Abcam Chemicals (Cambridge, UK, AB23981). A monoclonal anti-E-cadherin was from the MXB Biotechnologies (Fujian, China, MAB0738). And anti-CDH6 antibody was purchased from Millipore (MAB2013). The stained sections were observed under a microscope. The antibody diluent (phosphate-buffered saline) was used as control at the same step of first antibody addition.
NF-κB inhibitor treatment
Caffeic acid phenethyl ester (Selleck Chemicals, Texas, USA, S7414) is a specific inhibitor of NF-κB activation. Dimethyl sulfoxide (DMSO) was used to dissolve the inhibitor. EBV-positive cells were cultured in medium containing 4 µM of NF-κB inhibitor. After 24 h of treatment, the cellular proteins and total RNA were extracted respectively and subjected to the analysis.
Real-time quantitative PCR (RT-qPCR)
RNA isolation and amplification were performed as described previously20. Next, 1 µg of total RNA sample was reversely tran-scribed into complementary DNA using the First Strand cDNA Synthesis Kit (Thermoscientific). The miR-203 primers for the qPCR reaction were ordered from Rib Bio, and the sequences of all primers were seen in Supplementary Table 2.
Immunofluorescence (IF) assay
IF assay was according to a previous study53 using a fluorescent microscope (BX53, Olympus, Japan). Staining of F-actin stress fibers was performed with FITC-phalloidin (Sigma, Massachusetts, USA, p5282), as described by Wang et al.53. Nucleus was stained using Benzimide H33342.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, California, USA). The differences between groups were analyzed using the Student's t-test or one-way analysis of variance test. The data were expressed as the means ± SD. The experiments were repeated independently three times, showing similar results. And each experiment was performed in triplicate.The p-values < 0.05, 0.01 or 0.001 were considered to indicate statistical significance with different degrees, respectively.
Electronic supplementary material
Acknowledgements
We thank Dr Wolfgang Hammerschmidt (GSF-National ResearchCenter for Environment and Health, Germany) for kindly providing us the Maxi-EBV system, which was used in the establishment of C2089, C22 and 293-BAC cell lines. We are grateful for the help of Songqing Fan in collecting clinical specimens. We also thank our colleagues, Drs Jian Ma, Wei Xiong, Ming Zhou, Xiaoling Li, Juanjuan Xiang, Minghua Wu and Xiayu Li, for sharing related information during our work. This work was supported by the National Natural Science Foundations of China (81372139 and 31670171), the National Key Research and Development Program of China (2017YFC1200204), Hunan Provincial Natural Science Foundations of China (2015JJ2149 and 2016JC2035), Innovation Foundations for Postgraduates and Undergraduates (CX2016B055 and CX20170534), and Programme of Introducing Talents of Discipline to Universities (111-2-12).
Competing interests
The authors declare no competing financial interests.
Footnotes
This article has been retracted. Please see the retraction notice for more detail: 10.1038/s41389-022-00439-x
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
10/17/2022
This article has been retracted. Please see the Retraction Notice for more detail: 10.1038/s41389-022-00439-x
Supplementary information
The online version of this article 10.1038/s41389-017-0005-7 contains supplementary material.
References
- 1.Young LS, Rickinson AB. Epstein-Barr virus. 40 years on. Nat. Rev. Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 2.Tang LL, et al. Global trends in incidence and mortality of nasopharyngeal carcinoma. Cancer Lett. 2016;374:22–30. doi: 10.1016/j.canlet.2016.01.040. [DOI] [PubMed] [Google Scholar]
- 3.Zheng H, Li LL, Hu DS, Deng XY, Cao Y. Role of Epstein-Barr virus encoded latent membrane protein 1 in the carcinogenesis of nasopharyngeal carcinoma. Cell. Mol. Immunol. 2007;4:185–196. [PubMed] [Google Scholar]
- 4.Ding L, et al. Latent membrane protein 1 encoded by Epstein-Barr virus induces telomerase activity via p16INK4A/Rb/E2F1 and JNK signaling pathways. J. Med. Virol. 2007;79:1153–1163. doi: 10.1002/jmv.20896. [DOI] [PubMed] [Google Scholar]
- 5.Chaturvedi MM, Sung B, Yadav VR, Kannappan R, Aggarwal BB. NF-κB addiction and its role in cancer: 'one size does not fit all'. Oncogene. 2011;30:1615–1630. doi: 10.1038/onc.2010.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugden B, et al. Epstein-Barr virus-encoded LMP2A induces an epithelial–mesenchymal transition and increases the number of side population stem-like cancer cells in nasopharyngeal carcinoma. PLoS. Pathog. 2010;6:e1000940. doi: 10.1371/journal.ppat.1000940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/S0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 8.Jia S, Zhai H, Zhao M. MicroRNAs regulate immune system via multiple targets. Discov. Med. 2014;18:237–247. [PubMed] [Google Scholar]
- 9.Zeng L, Cui J, Wu H, Lu Q. The emerging role of circulating microRNAs as biomarkers in autoimmune diseases. Autoimmunity. 2014;47:419–429. doi: 10.3109/08916934.2014.929667. [DOI] [PubMed] [Google Scholar]
- 10.Deng X, et al. The role of microRNAs in autoimmune diseases with skin involvement. Scand. J. Immunol. 2015;81:153–165. doi: 10.1111/sji.12261. [DOI] [PubMed] [Google Scholar]
- 11.Yu H, et al. Epstein-Barr virus downregulates microRNA 203 through the oncoprotein latent membrane protein 1: a contribution to increased tumor incidence in epithelial cells. J. Virol. 2012;86:3088–3099. doi: 10.1128/JVI.05901-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saini S, et al. Regulatory role of miR-203 in prostate cancer progression and metastasis. Clin. Cancer Res. 2011;17:5287–5298. doi: 10.1158/1078-0432.CCR-10-2619. [DOI] [PubMed] [Google Scholar]
- 13.Taipaleenmaki H, et al. Targeting of RUNX2 by miR-135 and miR-203 impairs progression of breast cancer and metastatic bone disease. Cancer Res. 2015;75:1433–1444. doi: 10.1158/0008-5472.CAN-14-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonkoly E, et al. MicroRNA-203 functions as a tumor suppressor in basal cell carcinoma. Oncogenesis. 2012;1:e3. doi: 10.1038/oncsis.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 16.Sancisi V, et al. Cadherin 6 is a new RUNX2 target in TGF-β signalling pathway. PLoS. ONE. 2013;8:e75489. doi: 10.1371/journal.pone.0075489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamazaki D, Kurisu S, Takenawa T. Regulation of cancer cell motility through actin reorganization. Cancer Sci. 2005;96:379–386. doi: 10.1111/j.1349-7006.2005.00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. Bioessays. 2007;29:356–370. doi: 10.1002/bies.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taube JH, et al. Epigenetic silencing of microRNA-203 is required for EMT and cancer stem cell properties. Sci. Rep. 2013;3:2687. doi: 10.1038/srep02687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuo L, et al. The copy number of Epstein-Barr virus latent genome correlates with the oncogenicity by the activation level of LMP1 and NF-kB. Oncotarget. 2015;6:41033–41044. doi: 10.18632/oncotarget.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loveridge CJ, MacDonald AD, Thoms HC, Dunlop MG, Stark LA. The proapoptotic effects of sulindac, sulindac sulfone and indomethacin are mediated by nucleolar translocation of the RelA (p65) subunit of NF-kB. Oncogene. 2008;27:2648–2655. doi: 10.1038/sj.onc.1210891. [DOI] [PubMed] [Google Scholar]
- 22.Genova P, et al. Solitary splenic metastasis from nasopharyngeal carcinoma: a case report and systematic review of the literature. World J. Surg. Oncol. 2016;14:184. doi: 10.1186/s12957-016-0941-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melar-New M, Laimins LA. Human papillomaviruses modulate expression of microRNA-203 upon epithelial differentiation to control levels of p63 proteins. J. Virol. 2010;84:5212–5221. doi: 10.1128/JVI.00078-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L, et al. Ubiquitination of MDM2 modulated by Epstein-Barr virus encoded latent membrane protein 1. Virus Res. 2007;130:275–280. doi: 10.1016/j.virusres.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 25.De Clercq E, Li G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang SH, et al. Vimentin, a novel NF-kappa B regulator, is required for meningitic Escherichia coli K1-induced pathogen invasion and PMN transmigration across the blood-brain barrier. PLoS. ONE. 2016;11:e0162641. doi: 10.1371/journal.pone.0162641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He Z, Xin B, Yang X, Chan C, Cao L. Nuclear factor-kappaB activation is involved in LMP1-mediated transformation and tumorigenesis of rat-1 fibroblasts. Cancer Res. 2000;60:1845–1848. [PubMed] [Google Scholar]
- 28.Yoshizaki T, et al. Pathogenic role of Epstein-Barr virus latent membrane protein-1 in the development of nasopharyngeal carcinoma. Cancer Lett. 2013;337:1–7. doi: 10.1016/j.canlet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 29.Mohr CF, et al. The tumor marker Fascin is induced by the Epstein-Barr virus-encoded oncoprotein LMP1 via NF-κB in lymphocytes and contributes to their invasive migration. Cell. Commun. Signal. 2014;12:46. doi: 10.1186/s12964-014-0046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wasil LR, Shair KH. Epstein-Barr virus LMP1 induces focal adhesions and epithelial cell migration through effects on integrin-α5 and N-cadherin. Oncogenesis. 2015;4:e171. doi: 10.1038/oncsis.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Y, Wang Y, Zeng S, Hu X. LMP1 expression is positively associated with metastasis of nasopharyngeal carcinoma: evidence from a meta-analysis. J. Clin. Pathol. 2012;65:41–45. doi: 10.1136/jclinpath-2011-200198. [DOI] [PubMed] [Google Scholar]
- 32.Horikawa T, et al. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007;67:1970–1978. doi: 10.1158/0008-5472.CAN-06-3933. [DOI] [PubMed] [Google Scholar]
- 33.Horikawa T, et al. Epstein-Barr virus latent membrane protein 1 induces Snail and epithelial-mesenchymal transition in metastatic nasopharyngeal carcinoma. Br. J. Cancer. 2011;104:1160–1167. doi: 10.1038/bjc.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho EA, et al. Differential expression and function of Cadherin-6 during renal epithelium development. Development. 1998;125:803–812. doi: 10.1242/dev.125.5.803. [DOI] [PubMed] [Google Scholar]
- 35.Jia L, Liu F, Hansen SH, Ter Beest MB, Zegers MM. Distinct roles of cadherin-6 and E-cadherin in tubulogenesis and lumen formation. Mol. Biol. Cell. 2011;22:2031–2041. doi: 10.1091/mbc.e11-01-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat. Rev. Cancer. 2004;4:118–132. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- 37.Gugnoni M, et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene. 2017;36:667–677. doi: 10.1038/onc.2016.237. [DOI] [PubMed] [Google Scholar]
- 38.Cao S, et al. Upregulation of flotillin-1 promotes invasion and metastasis by activating TGF-β signaling in nasopharyngeal carcinoma. Oncotarget. 2016;7:4252–4264. doi: 10.18632/oncotarget.6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salma U, et al. Role of transforming growth factor-beta 1 and Smads signaling pathway in intrauterine adhesion. Mediat. Inflamm. 2016;6:4158287. doi: 10.1155/2016/4158287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ciarrocchi A, Piana S, Valcavi R, Gardini G, Casali B. Inhibitor of DNA binding-1 induces mesenchymal features and promotes invasiveness in thyroid tumour cells. Eur. J. Cancer. 2011;47:934–945. doi: 10.1016/j.ejca.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 41.Sancisi V, et al. RUNX2 isoform I controls a panel of proinvasive genes driving aggressiveness of papillary thyroid carcinomas. J. Clin. Endocrinol. Metab. 2012;97:e2006–2015. doi: 10.1210/jc.2012-1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lo AK, Dawson CW, Lo KW, Yu Y, Young LS. Upregulation of Id1 by Epstein-Barr virus-encoded LMP1 confers resistance to TGF-β-mediated growth inhibition. Mol. Cancer. 2010;9:155. doi: 10.1186/1476-4598-9-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun W, et al. Id-1 and the p65 subunit of NF-kB promote migration of nasopharyngeal carcinoma cells and are correlated with poor prognosis. Carcinogenesis. 2012;33:810–817. doi: 10.1093/carcin/bgs027. [DOI] [PubMed] [Google Scholar]
- 44.Liu S, et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell. 2010;17:333–347. doi: 10.1016/j.ccr.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qu JQ, et al. MiRNA-203 reduces nasopharyngeal carcinoma radioresistance by targeting IL-8/AKT signaling. Mol. Cancer Ther. 2015;14:2653–2664. doi: 10.1158/1535-7163.MCT-15-0461. [DOI] [PubMed] [Google Scholar]
- 46.Huang DP, et al. Establishment of a cell line (NPC/HK1) from a differentiated squamous carcinoma of the nasopharynx. Int. J. Cancer. 1980;26:127–132. doi: 10.1002/ijc.2910260202. [DOI] [PubMed] [Google Scholar]
- 47.Cai L, et al. Epstein-Barr virus-encoded microRNA-BART1 induces tumour metastasis by regulating PTEN-dependent pathways in nasopharyngeal carcinoma. Nat. Commun. 2015;6:7353. doi: 10.1038/ncomms8353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheung ST, et al. Nasopharyngeal carcinoma cell line (C666-1) consistently harbouring Epstein-Barr virus. Int. J. Cancer. 1999;83:121–126. doi: 10.1002/(SICI)1097-0215(19990924)83:1<121::AID-IJC21>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 49.Lu JH, et al. Epstein-Barr virus facilitates the malignant potential of immortalized epithelial cells: from latent genome to viral production and maintenance. Lab. Invest. 2010;90:196–209. doi: 10.1038/labinvest.2009.130. [DOI] [PubMed] [Google Scholar]
- 50.Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA. 1998;95:8245–8250. doi: 10.1073/pnas.95.14.8245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yu Z, et al. A precise excision of the complete Epstein-Barr virus genome in a plasmid based on a bacterial artificial chromosome. J. Virol. Methods. 2011;176:103–107. doi: 10.1016/j.jviromet.2011.06.015. [DOI] [PubMed] [Google Scholar]
- 52.Tang YL, et al. Genetic variations of EBV-LMP1 from nasopharyngeal carcinoma biopsies: potential loss of T cell epitopes. Braz. J. Med. Biol. Res. 2008;41:110–116. doi: 10.1590/S0100-879X2008000200006. [DOI] [PubMed] [Google Scholar]
- 53.Wang W, et al. Oxidored-nitro domain containing protein 1 (NOR1) expression suppresses slug/vimentin but not snail in nasopharyngeal carcinoma: inhibition of EMT in vitro and in vivo in mice. Cancer Lett. 2014;348:109–118. doi: 10.1016/j.canlet.2014.03.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.