

Summary
The ability of the brain to store and process information relies on changing the strength of connections between neurons. Synaptic facilitation is a form of short-term plasticity that enhances synaptic transmission for less than a second. Facilitation is a ubiquitous phenomenon thought to play critical roles in information transfer and neural processing. Yet our understanding of the function of facilitation remains largely theoretical. Here we review proposed roles for facilitation, and discuss how recent progress in uncovering the underlying molecular mechanisms could enable experiments that elucidate how facilitation, and short-term plasticity in general, contribute to circuit function and animal behavior.
Introduction
It is well established that synapses are regulated by their past activity on times scales of milliseconds to days and longer. Most attention has focused on long-term changes in synaptic strength. Even before Sherrington coined the term “synapse”, Ramón y Cajal suggested that the brain might store memories by modifying the connections between neurons (Sotelo, 2003). More than 70 years later, this hypothesis gained experimental support with the discovery of long-term potentiation (LTP) of synapses that lasts for hours or much longer. In subsequent years a growing consensus has been reached regarding the mechanisms that produce LTP (Nicoll, 2017). In contrast, even though short-term synaptic facilitation was discovered long before LTP, clarifying the mechanisms and determining the functions of facilitation has been elusive.
Synaptic transmission was first studied at the frog neuromuscular junction (NMJ), where one of the initial observations was that synaptic responses become much larger when presynaptic cells are activated by closely spaced stimuli (Eccles et al., 1941; Feng, 1940). When pairs of stimuli are used to study this phenomenon it is termed paired-pulse facilitation (Figure 1A-B), and facilitation generally leads to synaptic enhancement for more complex presynaptic activation (Figure 1C-D), provided stimuli are separated by tens to hundreds of milliseconds. Facilitation runs counter to the natural tendency of synapses to weaken during repeated activation, a phenomena known as depression that can result from depletion of the readily releasable pool (RRP) of synaptic vesicles and a decrease in neurotransmitter release (Zucker and Regehr, 2002). This suggests that facilitating synapses possess specialized mechanisms that boost neurotransmitter release even as the pool of available synaptic vesicles decreases.
Figure 1. Synaptic facilitation.
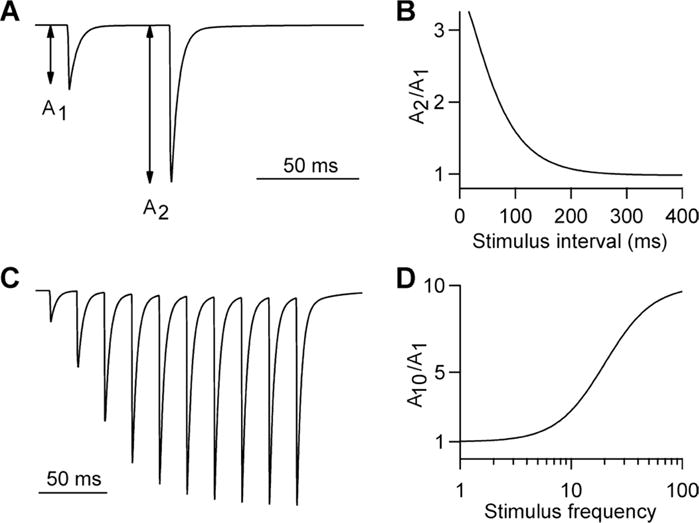
A) Illustrated excitatory postsynaptic currents evoked by a pair of pulses delivered at a 50 ms interval. At many synapses, facilitation increases the amplitude of the second response more than 2-fold.
B) Typical time course of paired-pulse facilitation
C) Illustrated facilitation produced by a 50 Hz train of 10 stimuli
D) Facilitation as a function of stimulus frequency
Synaptic facilitation has subsequently been observed throughout the nervous system, yet clarifying the mechanisms and functional roles of facilitation has been remarkably difficult. Recently, however, progress has been made in identifying proteins that mediate synaptic facilitation. This opens up the exciting possibility that manipulating these proteins could finally enable experimental tests of the effects of facilitation on circuit activity, behavior, and neuropsychiatric diseases. In this Review, we discuss the hypothesized roles of facilitation, and highlight the newly discovered mechanisms for facilitation that could allow the first direct tests of the functional importance of any form of short-term plasticity.
Theorized roles of facilitation
The NMJ, in addition to being the synapse where facilitation was discovered, also provided early insights into the functional importance of facilitation. The crayfish was a particularly useful model, because crayfish muscles with known roles in behavior are innervated by NMJs with distinct forms of synaptic plasticity (Atwood and Karunanithi, 2002) (Figure 2A). In the crayfish abdomen, a deep layer of “phasic” muscles generates the fast powerful contractions required for swimming and escape responses, while a superficial layer of “tonic” muscles produces sustained and finely-graded contractions required for postural control (Atwood, 2008). Phasic NMJs produce large initial responses that generate immediate, powerful contractions, but synaptic responses and muscle contractions both fatigue rapidly with repeated activation (Bruner and Kennedy, 1970; Kennedy and Takeda, 1965a). In contrast, at the onset of activity tonic NMJs produce small responses and do not elicit contractions, but during sustained activation these synapses undergo profound frequency-dependent facilitation and produce graded tension (Kennedy and Takeda, 1965b). The use of parallel pathways with different synaptic plasticity appears to be a common strategy for muscle control in crustaceans (Bradacs et al., 1997; Sherman and Atwood, 1972), and mammals, where motor neurons that innervate fast and slow muscles exhibit differing firing properties (Hennig and Lomo, 1985) and short-term plasticity (Reid et al., 1999).
Figure 2. Functional consequences of facilitation.
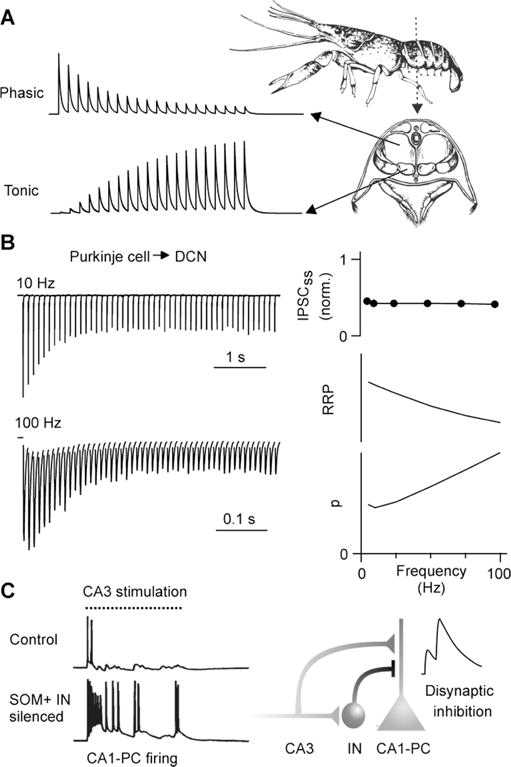
A) Illustration of differential short-term plasticity of NMJs in crayfish abdominal muscles. Phasic NMJs produce large initial responses, but depress rapidly. Initial responses from tonic NMJs are small, but facilitate markedly as a function of firing frequency.
B) (Left) IPSCs recorded from a cerebellar nuclear neuron evoked by stimulating Purkinje cell axons at different frequencies. (Right) Average steady-state responses (top) are maintained at high frequencies because depletion of the readily releasable pool of vesicles (RRP, middle) is offset by frequency-dependent facilitation of p (bottom). The size of the RRP and p were predicted by a model fit to data. Adapted from Turecek et al. (2016).
C) (Left) Prolonged stimulation of Schaffer collaterals normally elicits only a short burst of activity with place-field characteristics, but when interneurons are pharmacogenetically silenced CA3 cells fire for the entire stimulus duration. Adapted from Lovett-Barron et al. (2012). (Right) Excitatory synapses from CA3 impinge on CA1 pyramidal cells (CA1-PC), as well as on SOM+ interneurons (IN) that inhibit CA1-PC dendrites. As CA3 inputs facilitate during the course of stimulation, IN firing gradually increases. This leads to a net facilitation of disynaptic inhibition that allows interneurons to shut down pyramidal cell activity late in the train. Adapted from Bartley and Dobrunz (2015).
Facilitation counteracts depression
Depletion of the RRP becomes increasingly prominent when synapses are activated at high frequencies for prolonged periods, and consequently many synapses exhibit frequency-dependent depression. At some synapses, however, facilitation counteracts depression by increasing p in a frequency-dependent manner. This is thought to be the case in the avian auditory system where synapses that convey intensity information maintain their response amplitudes over a wide range of firing frequencies (MacLeod et al., 2007) and are thereby able to encode the absolute firing rate of auditory inputs (Sullivan and Konishi, 1984; Takahashi et al., 1984). A similar role for facilitation was observed in mice at cerebellar output synapses between Purkinje cells and the deep cerebellar nuclei (Turecek et al., 2016). Purkinje cells typically fire action potentials continuously at high rates in vivo, and can encode information by modulating their firing rates (Heck et al., 2013). At many synapses depletion is more prominent at high frequencies, and steady-state transmission decreases with firing frequency. But at PC to DCN synapses facilitation increases with firing frequency, thereby offsetting depletion. This leads to steady-state synaptic strength that is independent of frequency (Figure 2B).
Temporal filtering
Synapses preferentially transmit certain patterns of presynaptic activity as a consequence of their short-term plasticity (Abbott and Regehr, 2004). Synapses with a low initial p transmit single action potentials unreliably, often failing to elicit any response in the postsynaptic cell. However, facilitation can raise p dramatically during bursts of activity to allow synapses to strongly influence postsynaptic cells. In this way facilitating synapses act as high-pass filters (Atluri and Regehr, 1996) (Figure 1D) and play a special role in encoding burst activity that is a common feature of neural circuits (Lisman, 1997). In contrast to facilitating synapses, depressing synapses are low-pass filters that preferentially signal the onset of activity after periods of quiescence (Abbott et al., 1997; Rose and Fortune, 1999). Synapses where facilitation and depression are both present can act as band pass filters (Dittman et al., 2000). In addition, a single neuron can contact multiple postsynaptic partners using synapses with different forms of short-term plasticity, suggesting that short-term plasticity is tuned in a target-dependent manner (Bao et al., 2010; Maccaferri et al., 1998; Markram et al., 1998; Reyes et al., 1998; Rozov et al., 2001; Scanziani et al., 1998; Sherman and Atwood, 1972). Short-term plasticity could also be important for the induction of long-term forms of plasticity. For facilitating synapses, increased neurotransmitter release during bursts could account for the observation that presynaptic bursts are more effective than low-frequency firing at inducing long-term potentiation (Dunwiddie and Lynch, 1978; Huerta and Lisman, 1995).
An example of high-pass filtering imposed by facilitation is provided by medial olivocochlear (MOC) efferents that inhibit cochlear outer hair cells (OHCs). OHCs increase acoustic sensitivity by mechanically amplifying vibrations produced by sound (Ashmore, 2008; Brownell et al., 1985). To maintain acoustic sensitivity over a wide dynamic range, and to avoid cochlear damage caused by runaway amplification, MOC afferents increase their firing rate with sound intensity (Brown et al., 1998). MOC synapses have a very low initial p, but gradually facilitate in a frequency-dependent manner during sustained firing (Ballestero et al., 2011; Goutman et al., 2005). This allows OHCs to ignore spontaneous or low-frequency MOC activity, while scaling inhibition of OHCs as a function sound intensity.
Information transfer at synapses
Facilitation is widely believed to play an important role in determining how synapses transmit information (Abbott and Regehr, 2004; Deng and Klyachko, 2011). Information theory provides a framework for assessing how reliably information encoded by presynaptic firing is conveyed to postsynaptic neurons (Zador, 1998). Because short-term plasticity regulates the strength of inputs as a function of past activity, dynamic synapses convey information about both the mean firing rate and the temporal history of presynaptic spikes (Fuhrmann et al., 2002). Information theory has been applied to understand the role of synaptic depression in circuits (Abbott et al., 1997; Cook et al., 2003) but the role of facilitation is less clear. In general, facilitating synapses transmit information most effectively at high frequencies (9-70 Hz) (Fuhrmann et al., 2002; Rotman et al., 2011). Facilitation appears to be crucial to information transfer in the hippocampus during high frequency burst firing, as it has been shown that information transfer remains intact during bursts even when syt1 is removed and fast synchronous release is replaced by a slow form of facilitating release (Xu et al., 2012). However, information will be lost if facilitation drives the postsynaptic cell to fire at its maximum firing rate. Thus it has been proposed that circuits match the properties of presynaptic facilitation to the dynamic range of postsynaptic neurons in order to maintain optimal coding (Scott et al., 2012).
Circuit dynamics
The contributions of short-term plasticity become more complex in neuronal circuits where both excitation and disynaptic inhibition are important. The interaction between facilitation and feed-forward inhibition may play a fundamental role in encoding spatial information in the hippocampus. Place cells in the CA3 and CA1 regions signal an animal’s location within its environment by firing high-frequency spike bursts when the animal enters a given location, known as a place-field (O’Keefe and Dostrovsky, 1971). CA3 pyramidal cells send excitatory synapses to CA1 pyramidal neurons, and also to local SOM+ interneurons that provide feedforward inhibition (Alger and Nicoll, 1982; Losonczy et al., 2002; Lovett-Barron et al., 2012). Thus CA3 inputs drive a sequence of excitation followed by inhibition (Pouille and Scanziani, 2001). The two inputs exhibit opposing plasticity: excitation facilitates while the feed-forward inhibition depresses (Bartley and Dobrunz, 2015). In principle the opposing plasticity of excitation and inhibition would act as a simple high-pass filter. However, CA3 inputs must fire multiple times before they facilitate enough to drive action potentials in the interneurons that inhibit the CA1 cell dendrites and terminate spike bursts (Figure 2C) (Bartley and Dobrunz, 2015; Lovett-Barron et al., 2012; Pouille and Scanziani, 2004; Royer et al., 2012). The dynamic properties of the excitation/inhibition balance allows the circuit to act in a switch-like manner to amplify short bursts of high frequency activity, and selectively transmit signals with place-field-like characteristics (Klyachko and Stevens, 2006; Moreno et al., 2016).
Facilitation in working memory
Facilitation is also proposed to play a critical role in working memory, which involves the short-term storage of information for subsequent manipulation and decision-making. Working memory has long been attributed to persistent activity in recurrent networks in areas like the prefrontal cortex. Early electrophysiological recordings in non-human primates engaged in delayed-response tasks identified cells in the PFC that increase their firing during the delay period, but return to baseline activity after a response is executed (Fuster and Alexander, 1971). This elevated firing was initially hypothesized to be the neural basis of working memory (Goldman-Rakic, 1995). However, the idea that working memory is stored by elevated firing has recently been challenged. Many prefrontal cortex neurons do not fire persistently during the delay period, but instead exhibit dynamic patterns of activity along with periods of inactivity (Sreenivasan et al., 2014; Stokes, 2015; Stokes et al., 2013). This inspired models of working memory where information is stored not by absolute firing frequency, but by temporarily restructuring functional connectivity (Barak and Tsodyks, 2007; Deco et al., 2010; Mongillo et al., 2008; Sugase-Miyamoto et al., 2008). According to these models, information about the initial stimulus is stored by facilitating synapses between reciprocally-connected prefrontal cortex cells. The information can be retrieved by a subsequent sweep of activity through the network (Itskov et al., 2011; Mongillo et al., 2008).
It has been difficult to experimentally test the theorized functions of facilitation described above. To evaluate how facilitation influences neural circuits and behavior, it is desirable to have a means of selectively eliminating facilitation, preferably at a defined subset of synapses. This may become possible in the near future as clarification of the molecular mechanisms of facilitation provides targets for manipulation (Jackman et al., 2016; Nanou et al., 2016). Discovering these mechanisms could finally make it possible to test theories regarding the functional roles of facilitation that will help to clarify how neural circuits perform computations.
Proposed mechanisms of facilitation
Facilitation is an increase in the number of vesicles released
Facilitation is observed at many synapses with a low initial probability of release (p), whereas synapses with a high initial p usually exhibit use-dependent depression. This is thought to be a consequence of having a finite number of vesicles in a readily releasable pool (RRP). If initial p is high and a large fraction of vesicles fuse during the first action potential, the RRP becomes depleted and synaptic responses are depressed until the RRP is replenished. The importance of the initial p was first demonstrated at the NMJ, which is transformed from depressing in high external calcium when p high, to facilitating in low external calcium when p is low. The dependence of short-term plasticity on the initial p has made the measurement of facilitation a widely-used approach to detect changes in the initial p. However, this dependence of facilitation on the initial p makes it difficult to distinguish between an actual change in the mechanism of facilitation, and a change in initial p.
The first mechanistic question that was addressed was whether facilitation was a consequence of an increase in the number of vesicles released or an increase in quantal responses (the response to an individual vesicle). Intracellular recordings were used to detect quantal events under conditions where p was reduced by raising external magnesium and lowering external calcium (Del Castillo and Katz, 1954). Paired-pulse facilitation and facilitation during stimulus trains were found to reflect increased vesicle fusion, because there was a decrease in failures (trials in which presynaptic action potentials did not produce a postsynaptic response), an increase in the number of quanta released per stimulus, and quantal size did not change. However, these experiments did not determine whether the increase in vesicle fusion was caused by an increase in p for vesicles in the RRP, or an increase in the size of the RRP.
Although facilitation is a presynaptic phenomenon, postsynaptic contributions must also be considered. In rare circumstances short-term enhancement of synaptic strength can arise from the relief of polyamine block of calcium permeable AMPA receptors (see (Rozov and Burnashev, 1999)). For many synapses, when p is high multivesicular release occurs at a single active zone and leads to high occupancy of postsynaptic receptors and a sublinear relationship between the number of vesicles that fuse and the postsynaptic response (Wadiche and Jahr, 2001). This can decrease the amplitude of facilitation because p is higher for facilitated responses (Christie and Jahr, 2006; Foster et al., 2005).
Presynaptic calcium signaling and facilitation
Understanding calcium signaling within presynaptic boutons has been vital to evaluating the role of calcium in facilitation. It is now known that calcium can act on different spatial and temporal domains. When an action potential invades a presynaptic bouton calcium channels open, which leads to a large calcium signal of tens to hundred of micromolar that lasts a long as the calcium channels are open (<1 ms) (Augustine et al., 1991; Simon and Llinas, 1985; Yamada and Zucker, 1992). Calcium signals drop off significantly with distance from an open channel due to diffusion and capture by calcium buffers (Neher, 1998). If docked vesicles are located in the immediate vicinity of open calcium channels, this large brief calcium signal (Calocal) activates fast low-affinity calcium sensors such as synaptotagmin 1. The resulting vesicle fusion is primarily short-latency and short-lived because Calocal is so brief and synaptotagmin 1 has such rapid kinetics. But calcium also rapidly diffuses and equilibrates with calcium-binding proteins within presynaptic boutons, which gives rise to a residual calcium signal (Cares) that is initially tens to hundreds of nanomolar, and persists for tens to hundreds of milliseconds until the calcium is extruded from the presynaptic bouton.
It is exceedingly difficult to measure Calocal because it is so short-lived and localized. As a result, much of what we know about Calocal is based on models, simulations and ultrastructure (Chen et al., 2015; Fogelson and Zucker, 1985; Simon and Llinas, 1985). The calcium-dependence of release has often been studied using caged calcium to rapidly elevate calcium throughout presynaptic terminals (Heidelberger et al., 1994). This approach suggested that at the calyx of Held vesicle fusion is triggered by a calcium increase of 25 μM lasting several hundred microseconds (Schneggenburger and Neher, 2000). Cares is readily measured with fluorometric calcium indicators, and it is typically tens to a few hundred nanomolar (Atluri and Regehr, 1996; Brenowitz and Regehr, 2007; Scott and Rusakov, 2006).
One of the leading ways of studying the role of calcium in synaptic transmission is to introduce exogenous buffers into presynaptic boutons (Augustine et al., 2003). The introduction of calcium buffers into presynaptic terminals can affect Calocal and Cares. A simple equation can be used to estimate how an exogenous buffer will affect the steady-state Calocal signal that develops near the mouth of an open Ca2+ channel within microseconds of the channel opening (Naraghi and Neher, 1997; Neher, 1998),
| (1) |
where ICa is the calcium current through an open channel, F is the faraday constant, DCa is the diffusion coefficient of free calcium, r is the distance from an open channel, and λ is the length constant defined by,
| (2) |
where kon is the buffer’s on-rate, and [B] is the concentration of free buffer. In general, high concentrations of fast calcium buffers are most effective at reducing Calocal. This is why fast buffers such as BAPTA are generally effective at reducing Calocal and decreasing fast synaptic transmission. Even slow buffers such as EGTA can reduce Calocal, with EGTA most effective if the release site is some distance away from open calcium channels. The kinetics of calcium buffers also influences their effect on residual calcium. Fast buffers such as BAPTA reduced the amplitude of Cares and slow the decay of Cares (Neher and Augustine, 1992; Tank et al., 1995). The effects of slow buffers such as EGTA are more complex. During the time in which EGTA slowly binds calcium, Cares decays rapidly (Atluri and Regehr, 1996). After the initial rapid decay, calcium equilibrates with EGTA and there is a slow decay phase of Cares.
Manipulation of presynaptic calcium levels with calcium buffers provided the first indication that facilitation was produced by Cares. At the crayfish neuromuscular junction, facilitation produced by a burst of presynaptic activity was strongly attenuated when Cares was rapidly lowered by optically uncaging the caged calcium chelator diazo-2 (Kamiya and Zucker, 1994). At the granule cell to Purkinje cell synapse, the introduction of EGTA into granule cell presynaptic terminals accelerated the decay of Cares and paired-pulse facilitation (Atluri and Regehr, 1996). The acceleration in the decay of facilitation occurred even when the EGTA introduced into the presynaptic terminal did not affect the initial p and thus did not significantly reduce Calocal. These studies suggested that Cares. produces facilitation at these synapses, but the target of Cares and the mechanism of facilitation remained unclear.
The residual calcium hypothesis
Katz and Miledi (1968) proposed that facilitation could be a consequence of a larger calcium signal available for the second of two closely spaced stimuli (Figure 3A). In the late 1960’s little was known about the time course of calcium entry evoked by an action potential, or the amplitude and duration of calcium signals. Moreover, concepts such as local and global calcium signals were not fully developed. Despite these limitations, Katz and Miledi hypothesized that calcium could persist in nerve terminals following an initial stimulus and could lead to a larger calcium signal and more release by a second action potential. The observation that small increases in calcium entry could produce large changes in synaptic strength (Dodge and Rahamimoff, 1967) was crucial to the formulation of the residual calcium hypothesis of facilitation. Subsequent studies quantified the extent of facilitation under a variety of experimental conditions and modeled facilitation while taking into account the known calcium dependence of release (Barrett and Stevens, 1972; Magleby, 1973; Zengel and Magleby, 1982). These studies suggested that the observed facilitation was consistent with the residual calcium hypothesis, but critical evaluation of the residual calcium hypothesis was limited by a lack of information regarding presynaptic calcium signals.
Figure 3. Proposed mechanisms for facilitation.
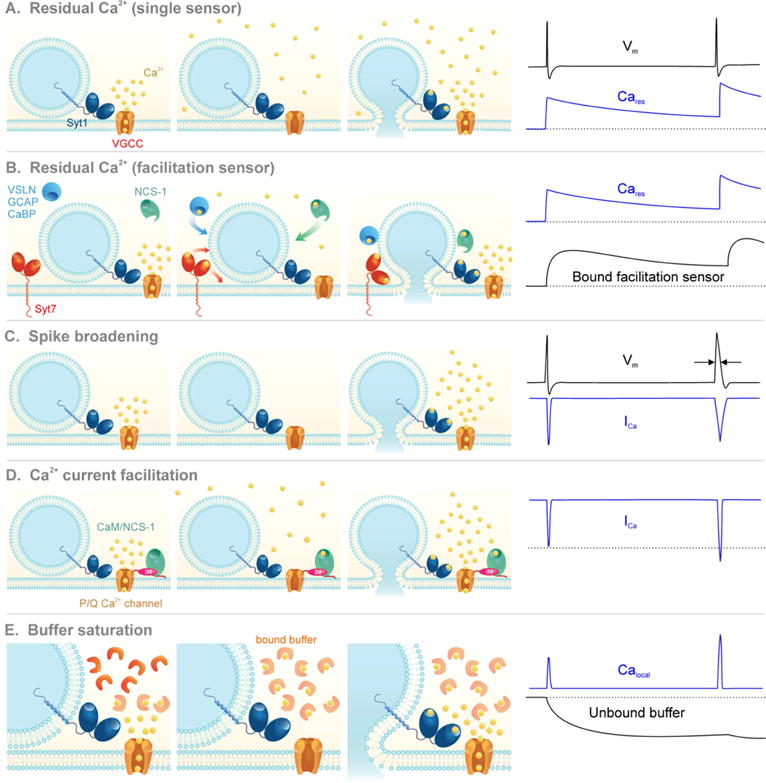
Illustration of the proposed mechanisms for facilitation. Voltage-gated Ca2+ channels (VGCC) open during the first action potential and allow Ca2+ influx (ICa). During the first action potential (AP) Ca2+ influx fails to fully activate Syt1 and trigger fusion.
A) The residual Ca2+ hypothesis: Cares from the first AP adds to Calocal during the second AP. Because the fusion sensor binds Ca2+ in a supralinear manner, a small increase in the Ca2+ signal drives a large increase in p.
B) Residual Ca2+ binds to a facilitation sensor: A high-affinity sensor, distinct from Syt1, binds to Cares in between APs and raises p by interacting with the fusion machinery.
C) Spike broadening: Inactivation of K+ channels during the first AP broadens the waveform of the second AP, prolonging the time-course and total amount of ICa.
D) Calcium current facilitation: Ca2+ from the first AP binds to NCS-1 or CaM, which interact with the IQ-like motif (IM) on the intracellular C-terminus of P/Q channels to increase ICa during the second AP.
E) Buffer saturation: During the first AP there is enough unbound Ca2+ buffer to capture incoming ions and decrease the Ca2+ signal sensed by Syt1 (Calocal). During the second AP the buffer becomes saturated and cannot constrain Calocal.
According to the simplest form of the residual calcium hypothesis, for the first stimulus release is proportional to (Calocal)4 and for the second release is proportional to (Calocal + Cares)4.
| (3) |
This formulation of the residual calcium hypothesis is based on a single calcium sensor such as synaptotagmin 1 (syt1) or syt2 acting as the sensor for both rapid transmission and facilitation. It is possible to evaluate the residual calcium hypothesis model of facilitation by determining the relative amplitudes of Calocal and Cares, and by measuring PPR. Cares, as measured with fluorometric calcium indicators, is typically 10’s to a few hundred nM, while Calocal is 25-100 μM (Zucker, 1996). Thus Cares/Calocal is ~ 0.01 at most synapses and would only produce a PPR of approximately 1.04, which could not account for significant facilitation. However, syt1 and syt2 possess multiple calcium binding sites, and it is possible that one of these sites has slow kinetics and a high affinity that contributes to facilitation. Although is difficult to exclude a contribution from such a mechanism because so little is known about Calocal for most synapses, it seems unlikely that such a mechanism could account for large facilitation.
The search for a second calcium sensor for facilitation
The great disparity between the calcium concentrations that trigger release and those that produce facilitation is inconsistent with the simplest form of the residual calcium hypothesis in which a single calcium sensor triggers release and produces facilitation (Yamada and Zucker, 1992). This prompted the hypothesis that facilitation is mediated by a second calcium sensor distinct from the one that triggers fusion. Experimental findings constrain the properties of such a sensor. First, it must respond to the submicromolar calcium signals that drive facilitation. This is most readily accomplished if the sensor has a high affinity for calcium. Second, it must not be saturated by the high local calcium that triggers release. Saturation is avoided if the sensor has slow kinetics or is located at a distance from calcium channels. Finally, it must be capable of enhancing p.
It is a daunting task to identify the calcium sensor for facilitation, because there are many candidate calcium-binding proteins present in presynaptic terminals (Burgoyne, 2007; Craxton, 2010). Most of these proteins bind calcium with one or more E/F hand or C2 domains. The E/F hand domain proteins include the ubiquitously expressed CaM, NCS-1, visinin-like proteins (VSNLs) that include hippocalcin, neurocalcin d and VILIPs1-3, recoverin and guanylyl-cyclase activating proteins (GCAPs), KCHIP proteins that regulate potassium channels, and finally a much less-studied family of proteins known as calcium-binding proteins (CaBP). Many of these proteins have been implicated in the calcium-dependent regulation of calcium entry through voltage-gated calcium channels (see below), but they could also regulate other targets and increase p independent of changes in calcium entry. For most of these E/F hand proteins their possible role in regulating synaptic facilitation independent of regulating calcium entry has not been examined. NCS-1 was the first protein suggested to be a calcium sensor for facilitation that acts independently of regulating calcium signaling or the initial p. Expressing NCS-1 in cultured hippocampal cells transformed depressing synapses into facilitating synapses without altering calcium entry or initial p (Sippy et al., 2003). However, this facilitation decayed with a time constant of 14 ms, which is about 10 times faster than facilitation in wildtype animals, and they did not establish that facilitation in wildtype animals relies on NCS-1.
There are also many C2-domain-containing proteins within presynaptic terminals, many of which are known to be important for synaptic transmission (Corbalan-Garcia and Gomez-Fernandez, 2014). Most notable are the synaptotagmin (syt) isoforms syt1 and syt2, which act as the calcium sensors for synchronous vesicle fusion at most central synapses (Sudhof, 2013). Other C2-domain-containing proteins include classical PKCs, ferlins, DOC2, rabphillin, RIM1,2, Munc13, perforin and copine. Further studies are required to determine if any of these proteins act as calcium sensors for facilitation, although at present there is no evidence to support such a role.
Synaptotagmins are a large family of membrane-associated proteins that include eight calcium-sensitive isoforms (1-3,5-7,9,10) that are candidate calcium sensors for facilitation (Craxton, 2010; Sudhof, 2002). A tandem pair of C2 domains confers these isoforms with the ability to bind calcium, membranes, and SNARE complexes. The isoforms bind to phospholipids with different calcium affinities (Li et al., 1995) and different kinetics (Davis et al., 1999; Hui et al., 2005; Tucker et al., 2003). Syts 1, 2 and 9 act as low-affinity calcium sensors for rapid neurotransmitter release and consequently are not promising candidates to mediate facilitation (Xu et al., 2007). Syts 3, 5-7 are widely expressed in the brain (Mittelsteadt et al., 2009) but do not rescue fast transmission in syt1-deficient hippocampal cultures (Xu et al., 2007), suggesting they do not mediate fast synaptic transmission and instead perform some other function. Of all isoforms, syt 7 has the highest affinity for calcium (Sugita et al., 2002). In vitro assays found that the speed of disassembly of syt-liposome complexes was fast for syts 1-3, moderate for syts 5, 6, 9, 10, and far slower for syt7 (Brandt et al., 2012; Hui et al., 2005).
Syt7’s high calcium affinity, slow binding kinetics, and widespread distribution in the brain led to the hypothesis that syt7 could be a sensor for facilitation (Zucker, 1996; Zucker and Regehr, 2002). It was also suggested that syt7 could mediate a delayed form of synaptic transmission known as asynchronous release (Hui et al., 2005), where exocytosis persists at a low rate for tens to hundreds of milliseconds after an action potential (Atluri and Regehr, 1998; Best and Regehr, 2009; Goda and Stevens, 1994). However, initial tests found no role for syt7 in synaptic transmission. At the Drosophila NMJ, knockdown of syt7 affected neither synchronous release nor facilitation (Saraswati et al., 2007), and cultured cortical neurons from syt7 knockout mice also exhibited normal synaptic transmission (Maximov et al., 2008) (although facilitation was not explicitly examined).
The role of syt7 in facilitation was recently examined in brain slices at 4 synapses where syt7 is expressed and facilitation is prominent (Jackman et al., 2016). Paired-pulse facilitation was eliminated in syt7 KO mice at Schaffer collateral synapses, hippocampal mossy fiber synapses, corticothalamic synapses and lateral performant path synapses onto dentate granule cells (Figure 4). Synaptic enhancement during brief trains was also eliminated at all synapses with the notable exception of mossy fiber synapses, where spike broadening, and local buffer saturation have been shown to contribute to synaptic enhancement. At Schaffer collateral synapses, loss of syt7 did not affect the calcium-dependence of release, the initial p, calcium entry or residual calcium signals. In addition, viral expression of wildtype syt7 in presynaptic cells rescued facilitation in syt7 knockout animals, but expression of mutant syt7 with a calcium-insensitive C2A domain did not. Together these findings indicate syt7 is a calcium sensor for facilitation for many synapses in the brain.
Figure 4. Facilitation mediated by syt7.
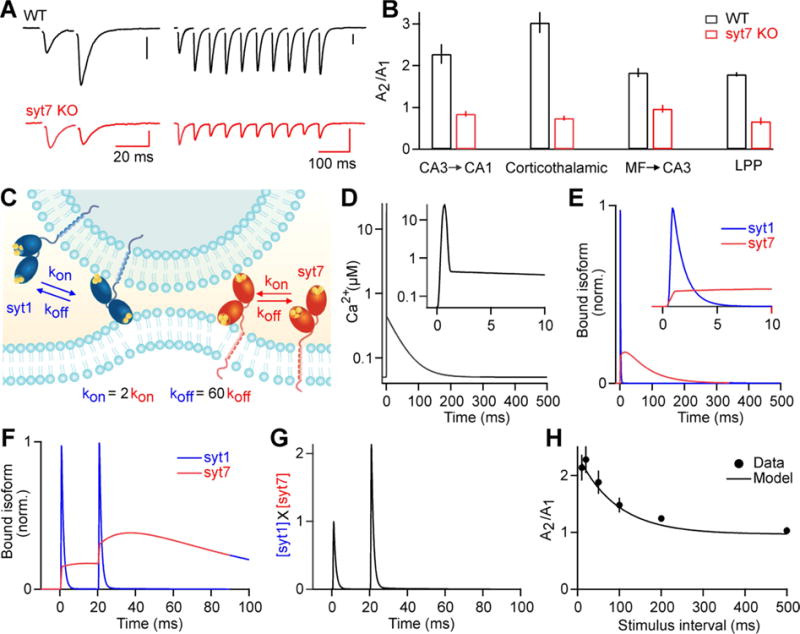
A) EPSCs recorded from CA1 pyramidal cells in WT and syt7 KO mice. Vertical scale bars 100 pA. Adapted from Jackman et al. (2016).
B) Paired-pulse ratios at 20 ms intervals, from hippocampal Schaffer collaterals (CA3 ➔ CA1), corticothalamic synapses, hippocampal mossy fibers (MF ➔ CA3), and hippocampal lateral perforant path synapses (LPP) from WT and syt7 KO mice.
C) In the presence of Ca2+, the C2A domain of syt1 binds to phospholipids twice as fast as the C2A domain syt7, and unbinds ~60-fold faster than syt7 (Brandt et al., 2012).
D) Simulated Ca2+ signal at a release site following an action potential. During the action potential, local Ca2+ rises to 25 μM briefly (FWHM = .34 ms, Sabatini and Regehr, 1998)). After the action potential, a residual Ca2+ signal of 400 nM decays back to resting Ca2+ (50 nM) with a 40 ms time constant (Brenowitz and Regehr, 2007).
E) Simulated phospholipid binding of both syt1 and syt7 during a single action potential, normalized to the maximal binding produced by a train of 10 action potentials at 100 Hz.
F) Phospholipid binding in response to a pair of action potentials.
G) The multiplied fraction of bound syt1 and bound syt7 ([syt1]X[syt7]), normalized to the peak initial response.
H) Paired-pulse ratios at different stimulus intervals recorded from Schaffer collateral synapses (adapted from Jackman et al. (2016)), along with the facilitation predicted by [syt1]X[syt7].
(E-H) Phospholipid binding was simulated using parameters from stopped-flow experiments of syt association with PIP2-containing target membrane liposomes (Brandt et al., 2012). Kd for Ca2+ was 41 μM and 1.5 μM, with a cooperativity of 1.9 and 2.8 for syt1 and syt7 respectively. For comparison to physiological recordings, binding kinetics were adjusted for temperature (34° C) (Hui et al., 2005). Koff 900 s−1 and 15 s−1 and Kon 415 s−1 and 190 s−1, for syt1 and syt7 respectively.
It is surprising that syt7’s role in facilitation was not revealed sooner. Syt7 was proposed as a candidate sensor for facilitation based on the high affinity for calcium (Zucker, 1996; Zucker and Regehr, 2002) and syt7 KO mice became available in 2003 (Chakrabarti et al., 2003). However, previous studies focused on the roles of syt7 other aspects of synaptic transmission and saw no effect on facilitation. Initially, it was questioned whether conflicting observations were the result of differences between the two syt7 knockout mouse lines used in various studies: syt7 mRNA has been detected in one line (Chakrabarti et al., 2003), but not the other (Maximov et al., 2008). However, neither line expresses syt7 protein, and direct comparisons have detected no significant differences in synaptic properties between neurons from the two lines (Bacaj et al., 2015; Bacaj et al., 2013).
In addition to its role in facilitation, syt7 is a multifunction protein that responds to submicromolar Cares signals to control multiple aspects of synaptic transmission and release. Syt7 mediates asynchronous release during prolonged high-frequency stimulation (Wen et al., 2010). Similarly, cultured hippocampal neurons from syt1 KO mice display prominent asynchronous release that is mediated by syt7 (Bacaj et al., 2015; Bacaj et al., 2013; Li et al., 2017). Syt7 interacts with calmodulin to accelerate recovery from depression in cultured hippocampal neurons (Liu et al., 2014). Syt7 also acts as a high-affinity calcium sensor for exocytosis from non-neuronal cells (Gustavsson et al., 2008; Martinez et al., 2000; Schonn et al., 2008; Sugita et al., 2001). Thus syt7 functions as a high-affinity calcium sensor to regulate release in multiple ways in both neurons and non-neuronal cells.
There are still many unresolved questions regarding syt7. Syt7 has 3 alternative splice variants in mice and humans (Fukuda et al., 2002) and more in rats (Sugita et al., 2001). Rescue experiments have been performed using syt7α, the dominant splice variant in the brain (Bacaj et al., 2013; Jackman et al., 2016; Liu et al., 2014), and it is not known if splice variants make different contributions to facilitation and other processes. Finally, syt7 is expressed by neurons that exhibit net depression, such as Purkinje cells, and it will be interesting to determine the role of syt7 in transmission at other such synapses. At high p synapses facilitation may still be present, but obscured by prominent depression (Dittman et al., 2000). Facilitation can still make important contributions during repetitive activation at depressing synapses (Turecek et al., 2016).
How does syt7 produce facilitation?
The structural similarity between syt7 and the fast isoforms, syt1 and syt2, suggests they may have related functions in vesicle fusion. It is therefore useful to review current models of how syt1 acts as a calcium sensor for fusion, with the caveat that the mechanism by which syt1 triggers fusion has yet to be resolved (Jahn and Fasshauer, 2012). The core machinery for vesicle fusion is composed of syntaxin-1, synaptobrevin, and SNAP-25, which together form SNARE complexes that link vesicles to the plasma membrane (Sudhof and Rothman, 2009). The two C2 domains of syt1, C2A and C2B, coordinate calcium binding (Ubach et al., 1998) and confer calcium sensitivity to SNARE-mediated fusion through calcium-dependent interactions with membranes and SNAREs (Chapman, 2008; Sudhof, 2013). Calcium binding induces a charge switch in the C2 domains that leads to fusion through mechanisms that likely involve SNARE and membrane binding.
Although both C2 domains are structurally similar, mutations of the calcium-binding pocket of the C2B cause greater deficits in neurotransmitter release than similar mutations in C2A (Fernandez-Chacon et al., 2002; Gaffaney et al., 2008; Mackler et al., 2002; Nishiki and Augustine, 2004; Robinson et al., 2002; Stevens and Sullivan, 2003; Xue et al., 2008). The C2B domain also contains basic regions that support calcium-independent binding to SNAREs and the acidic phospholipid phosphatidylinositol-4,5-bisphosphate (PIP2) (Bai et al., 2004; Wang et al., 2016; Zhou et al., 2015). C2B binding to PIP2 may target vesicles to release sites that are enriched in PIP2 (Honigmann et al., 2013), coordinate SNAREs with the opposing membrane to stabilize vesicles in a pre-fusion state (Chicka et al., 2008; de Wit et al., 2009; Mohrmann et al., 2013; van den Bogaart et al., 2011; Zhou et al., 2015), and raise p by increasing the calcium affinity of syt1 (Bai et al., 2004; Li et al., 2006; Mackler and Reist, 2001; Radhakrishnan et al., 2009; van den Bogaart et al., 2012).
A prominent model posits that, upon binding calcium, the top loops of C2B insert into the plasma membrane while the basic regions remain bound to SNAREs and PIP2 (Wang et al., 2016; Zhou et al., 2015). This calcium-dependent membrane insertion appears to be essential for vesicle fusion, as neurotransmitter release is abolished by a point mutation that blocks membrane insertion (Bai et al., 2002; Herrick et al., 2006; Paddock et al., 2011). It is thought that by penetrating the plasma membrane while remaining bound to SNAREs, syt1 induces inward curvature in the plasma membrane (Bharat et al., 2014; Martens et al., 2007; Wang et al., 2016). Curvature may lower the energy barrier for fusion by placing elastic stress on the membrane and bringing the vesicle and membrane closer together (Kozlovsky and Kozlov, 2002; Kuzmin et al., 2001).
It is likely that syt7 produces facilitation by increasing the probability of syt1-mediated release, rather than by triggering fusion on its own. Although syt7 may mediate calcium-dependent release from non-neuronal cells (Martinez et al., 2000; Sugita et al., 2001), syt7 is ineffective at triggering release from neurons that lack syt1 (Bacaj et al., 2013; Xu et al., 2007). Syt7 has been shown to bind membranes and SNAREs in a calcium-dependent manner (Osborne et al., 2007; Sugita et al., 2002), but syt7 has not been biochemically characterized to the same degree as syt1 and many questions must be answered to understand how syt7 raises p during facilitation. Several membrane-binding properties of syt7 could help to explain its role in facilitation. Syt7 exhibits calcium-dependent phospholipid binding with ~10-fold higher affinity than syt1, and is activated by submicromolar calcium concentrations (Sugita et al., 2002). This makes syt7 capable of sensing the Cares signals that drive facilitation. Phospholipid binding is also much slower for syt7 than for syt1 (Hui et al., 2005), with binding and unbinding proceeding 2-fold and 60-fold slower, respectively (Brandt et al., 2012) (Figure 4C).
If phospholipid binding is a determinant for triggering fusion, syt7’s slow kinetics are optimal for a facilitation sensor. This is illustrated by simulating how syt1 and syt7 bind dynamically to membranes in response to the calcium signals produced by action potentials using the measured calcium-affinity, cooperativity, and kinetics of phospholipid binding (Brandt et al., 2012). Consider an action potential that produces Calocal of 25 μM for 0.5 ms, followed by Cares of 400 nM that decays with a time constant of 40 ms back to a resting calcium of 50 nM (Figure 4D). Calocal causes syt1 to transiently bind to membranes, but Cares is not effective at activating syt1 because of its low affinity for calcium (Figure 4E). In contrast, Calocal is so brief that it does not lead to much syt7 binding to the membrane, but the longer lasting Cares activates syt7 due to its high calcium affinity. Syt7 will continue to bind to membranes tens of milliseconds after an initial action potential. Even after Cares returns to resting levels, slow dissociation keeps syt7 bound to membranes where it could facilitate release during subsequent action potentials. The slow dissociation of syt7 could allow facilitation to outlast Cares increases (Atluri and Regehr, 1996).
Remarkably, simply multiplying the amount of membrane bound syt1 and syt7 can account for facilitation (Figure 4F-H). This is consistent with the idea that syt1 and syt7 membrane binding lowers the energy barrier for membrane fusion by Esyt1 and Esyt7, respectively. The rate of fusion (k+) predicted by the Arrhenius equation is then given by:
| (4) |
where EA is the activation energy in the absence of syt binding, k is the Boltzmann constant, and T is the temperature. This predicts that the fusion rate depends on the multiplied contributions of the two bound isoforms. Theoretical studies of membrane fusion suggest that EA ~40 kT (Kozlovsky and Kozlov, 2002; Kuzmin et al., 2001), for saturating concentrations of membrane-bound syt1 Esyt1 ~20 kT (Martens et al., 2007) and Esyt7 is not known. Using these parameters, simulated rates of fusion with realistic amounts of facilitation were obtained when membrane-bound syt7 lowered the energy barrier only modestly (Esyt7 =~2-5 kT).
The prediction that syt7 contributes far less than syt1 (or syt2) to lowering the energy barrier may help to explain why syt7 produces facilitation in the presence of syt1, and supports asynchronous release in the absence of syt1. When syt1 is present, a small contribution from syt7 could dramatically increase the probability of release for syt1-triggered fusion because the fusion rate depends exponentially on the energy barrier. In the absence of syt1, syt7 could drive fusion on its own but would do so at much lower rates, and syt7-triggered fusion would persist for tens to hundreds of milliseconds. This is consistent with the syt7-triggered asynchronous release observed in neurons lacking syt1 (Bacaj et al., 2015; Bacaj et al., 2013). Syt7-triggered asynchronous release rates are orders of magnitude lower than syt1-triggered synchronous release rates, but release continues for hundreds of milliseconds. Interestingly, asynchronous release and facilitation have been suggested to rely on the same mechanism (Rahamimoff and Yaari, 1973; Zucker, 1996). While the time-course and magnitude of facilitation predicted by this model support membrane binding as a critical determinant of syt7-mediated facilitation, more work is required to understand how syt7 and other syt isoforms interact with vesicles and fusion machinery to promote exocytosis.
Use-dependent increases in calcium entry
It has been hypothesized that use-dependent increases in presynaptic calcium entry could underlie facilitation. This could occur either as a consequence of a change in the presynaptic waveform that leads to the opening of more presynaptic calcium channels, or as a result of calcium channel modulation that enhances calcium entry. Based on the steep calcium-dependence of release, even a small change in calcium entry could produce large facilitation.
Spike Broadening
Studies of peptide release from presynaptic terminals in the neurohypophysis provided the first indication that changes in presynaptic waveform in presynaptic terminals could lead to use-dependent enhancement of release. Peptide release from pituitary nerve terminals is highly frequency dependent. Optical measurements of presynaptic waveform revealed frequency-dependent spike broadening occurs and likely contributes to the frequency-dependence of peptide release (Dreifuss et al., 1971; Gainer et al., 1986; Salzberg et al., 1986). Similarly, whole-cell recordings from pituitary nerve terminals confirmed that use-dependent spike broadening occurs, likely as a consequence of potassium channel inactivation, and that spike broadening increases presynaptic calcium entry (Jackson et al., 1991).
Use-dependent spike broadening (Figure 3B) was subsequently shown to occur at some fast glutamatergic synapses in the mammalian brain. Mossy fiber synapses between hippocampal granule cells and CA3 pyramidal cells have the unusual property that they undergo profound use-dependent enhancement during sustained activation for a broad range of stimulus frequencies (Regehr et al., 1994; Salin et al., 1996). Whole-cell patch clamp recordings from MFBs revealed activity-dependent spike broadening and increases in calcium entry during high frequency stimulation as a result of inactivation of a DTX-sensitive potassium channel (Geiger and Jonas, 2000). This spike broadening is sufficient to make a major contribution to synaptic enhancement during prolonged stimulation, which could explain the remaining facilitation observed during train stimulation at mossy fiber synapses in syt7 KO animals (~30% compared to WT animals, (Jackman et al., 2016)). However, spike broadening was not prominent for pairs of closely spaced stimuli, suggesting that it is unlikely to contribute significantly to paired-pulse facilitation at this synapse. In presynaptic boutons of layer 5 pyramidal cells a slowly inactivating K channel, possibly Kv1.2, can also influence presynaptic waveform during prolonged somatic depolarization (Shu et al., 2007), but waveform changes are small and unlikely to contribute to facilitation during brief trains (Shu et al., 2006).
It appears that spike broadening does not contribute to facilitation at most synapses. The contribution of waveform changes to facilitation was first tested at the squid giant synapse where direct measurements from presynaptic terminals were possible. Facilitation is observed at the squid giant synapse in the presence of low external calcium. Use-dependent changes in the presynaptic action potentials were small and insufficient to account for this facilitation (Charlton and Bittner, 1978). At synapses made by cerebellar granule cells presynaptic waveforms were monitored by measuring the presynaptic volley, which reflects the current flow associated with a presynaptic action potential, and by using voltage sensitive dyes (Sabatini and Regehr, 1997). Granule cell to Purkinje cell synapses exhibit robust facilitation (Atluri and Regehr, 1996), but do not exhibit use-dependent spike broadening (Kreitzer and Regehr, 2000; Sabatini and Regehr, 1997). Similarly, for hippocampal CA3 to CA1 synapses, presynaptic volleys and presynaptic waveforms measured with voltage sensitive dyes do not show use-dependent waveform changes (Qian and Saggau, 1999). At the calyx of Held, whole cell recordings indicated that presynaptic waveforms are essentially unchanged for pairs of stimuli that produce almost a doubling of synaptic strength (Muller et al., 2008). High-frequency (40 Hz) stimulation does not lead to spike broadening in presynaptic boutons of cerebellar interneurons (Pouzat and Hestrin, 1997; Rowan et al., 2014). Synapses made between cultured Purkinje cells facilitate, but waveform changes are not responsible for facilitation (Diaz-Rojas et al., 2015).
Thus, use-dependent waveform changes only occur at specialized synapses, such as mossy fiber synapses, and even at those synapses spike broadening is not responsible for the bulk of synaptic facilitation. It is known that specific complements of ion channels are present in presynaptic boutons (Trimmer, 2015), and the finding that most synapses allow reliable action potential propagation and stereotyped presynaptic waveforms during repetitive activation suggests that the density and complement of ion channels in presynaptic terminals allow presynaptic terminals at most types of synapses to avoid waveform changes.
Calcium current facilitation
It has also been proposed that calcium-dependent facilitation of calcium entry (CDF) that is independent of presynaptic waveform changes could contribute to synaptic facilitation (Figure 3C). It is predicted that if CDF contributes to synaptic facilitation there should be use-dependent increases in presynaptic calcium influx that are not a consequence of changes in presynaptic waveform. This could result in an increase in presynaptic calcium current or an increase in stimulus-induced presynaptic calcium increases. At the squid giant synapse, measurements of presynaptic calcium with arsenazo III, an early calcium indicator, found that each of two closely-spaced action potentials produced the same incremental increase in Cares (Charlton et al., 1982). These studies suggested that at the squid giant synapse facilitation is not a consequence of use-dependent increases in calcium entry. Similarly Cares measurements suggest that there is no use-dependent increase in calcium entry at other facilitating synapses, including the granule cell to Purkinje cell synapse and hippocampal synapses (Atluri and Regehr, 1996; Jackman et al., 2016). It is difficult, however, to exclude this mechanism because Cares reflects influx through all calcium channels in a presynaptic bouton, and in theory CDF could selectively occur at those calcium channels near release sites. Elevation of intracellular calcium can either inhibit or facilitate calcium entry (Ben-Johny and Yue, 2014; Christel and Lee, 2012). Use-dependent increases in presynaptic calcium currents were first described at the calyx of Held (Borst and Sakmann, 1998; Cuttle et al., 1998). Calcium currents generated by a pair of 1 ms voltage steps facilitated by approximately 20% for 10 ms ISIs (Cuttle et al., 1998) (Figure 5A,C). High-frequency trains of these voltage steps transiently facilitated calcium currents by almost a factor of 2. This facilitation was strongly attenuated in low external calcium, eliminated when external calcium was replaced with either sodium or barium, and was reduced by about 50% when 10 mM EGTA was included in the recording pipette (Cuttle et al., 1998).
Figure 5. Calcium current facilitation.
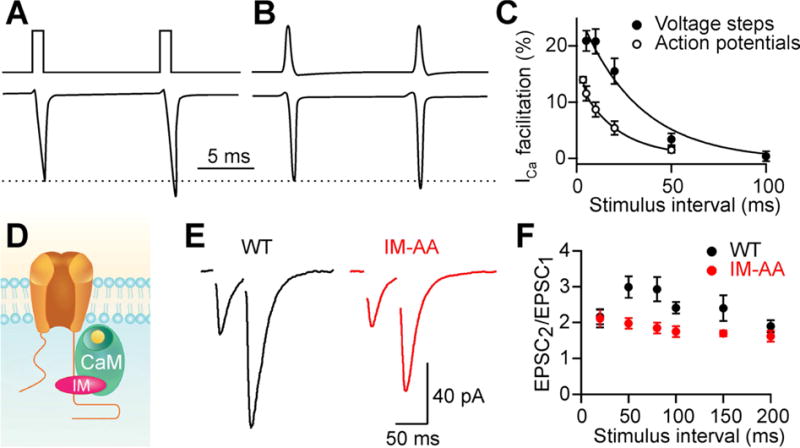
A) Ca2+ influx at the Calyx of Held evoked by a pair of 1 ms depolarizing steps (adapted from Cuttle et al. (1998)) and
B) action potentials (adapted from Di Guilmi et al. (2014)).
C) Paired-pulse facilitation of Ca2+ influx at different stimulus intervals produced by voltage steps at the Calyx of Held steps (adapted from(Cuttle et al., 1998)) and action potentials between cultured Purkinje cell synapses. Adapted from Diaz-Rojas et al. (2015).
D) The Ca2+ sensors NCS-1 and CaM bind to the IQ-like motif (IM) of P/Q-type Ca2+ channels to produce facilitation of Ca2+ entry. Substituting alanine for a pair of isoleucine and methionine residues in IM (IM-AA) disrupts this interaction.
E) EPSCs recorded from CA1 pyramidal cells in WT and IM-AA knockin mice. Recordings were made in the presence of ω-conotoxin GVIA to block N-type Ca2+ channels.
F) Paired-pulse facilitation in WT and IM-AA mice. (E) and (F) adapted from Nanou et al. (2016).
Comparing the degree of facilitation of presynaptic calcium current and synaptic responses is particularly illuminating. For action potential driven release at the calyx of Held synaptic facilitation more than doubled synaptic strength, and the facilitation of calcium channels accounted for less than half of the synaptic facilitation (Muller et al., 2008). At synapses between cultured Purkinje cells, there is a 16 % enhancement of calcium current and a doubling of synaptic strength for stimuli separated by 10 ms. Calcium current enhancement is sufficient to account for synaptic enhancement, provided release is steeply dependent on calcium influx (4.5 power, (Diaz-Rojas et al., 2015)). In cultured superior cervical ganglion neurons that express P-type calcium channels, both calcium entry and synaptic strength show facilitation, but quantitative comparisons were complicated because calcium currents were evoked by 5 ms voltage steps and synaptic responses were evoked by action potentials (Yan et al., 2014).
Facilitation of calcium current is short-lived. A recent study quantified the time course of calcium-dependent facilitation of P-type calcium channels in Purkinje cells using optical manipulations of intracellular calcium levels (Lee et al., 2015). They found that the onset of facilitation is rapid (τon=2 ms) and following a return to resting calcium levels enhancement is short-lived (τoff=31 ms). The time course of paired-pulse facilitation of calcium current had a similar time course at the calyx of Held (Cuttle et al., 1998) and in Purkinje cell presynaptic boutons (Diaz-Rojas et al., 2015) (Figure 5C). These experiments were performed at room temperature, so it is likely that the onset and decay of facilitation of calcium entry is even more rapid at physiological temperatures. Maximal CDF of P-type calcium channels in Purkinje cells (Lee et al., 2015) was approximately a doubling of the current and it occurred at a half maximal value of approximately 500 nM with a Hill coefficient of 1.8. These properties suggest that facilitation of calcium currents can occur in response to high Calocal increases that only last for about a millisecond, but that Cares could also be sufficiently large to enhance presynaptic calcium currents.
A number factors influence the magnitude of CDF. First, the voltage command used to induce CDF has a major influence on the extent of facilitation. At the calyx of Held, 1 ms voltage steps facilitated calcium current by 21% (Cuttle et al., 1998), but for action potentials there was only a 7% enhancement (Di Guilmi et al., 2014) (Figure 5B). 100 Hz trains of 1 ms steps enhanced calcium influx by 100%, whereas action potentials trains enhanced influx by 20% (Cuttle et al., 1998; Di Guilmi et al., 2014). This suggests that studies that used voltage steps of 5 ms and longer significantly overestimate the extent of enhancement that occurs under physiological conditions. The waveform of the presynaptic action potential, which is synapse specific, will also influence the extent of CDF. Second, alternative splicing of P-type calcium has a major influence on CDF. Multiple splice variants of P-type channels have been identified, and can be grouped into two classes, EFa, which supports CDF, and EFb, which does not (Chaudhuri et al., 2004). Alternative splicing can also regulate whether CDF is driven by high local calcium (insensitive to calcium buffers) or by global calcium (highly sensitive to calcium buffers). Splice variants are developmentally regulated. Early in development EFb is the predominantly expressed, but EFa becomes equally prevalent in the adult brain. Moreover, in the adult brain, the relative expression of each variant is regionally specific, with EFa being highly enriched in the thalamus, amygdala, and cerebellar Purkinje cells (Bourinet et al., 1999; Chaudhuri et al., 2004). The developmental and regional specificity of the different splice variants suggest they could act as a molecular switch to set the degree of CDF and determine the properties of synaptic plasticity.
Calcium sensors for CDF
In considering how calcium current facilitation could occur, and how it could contribute to synaptic facilitation, it is most relevant to consider the three types of calcium channels responsible for most synaptic transmission: P-type (CaV 2.1), N-type (CaV 2.2), and R-type (CaV 2.3) calcium channels. All of these channels can undergo calcium/calmodulin (CaM) dependent inhibition, but only P-type calcium channels exhibit calcium-dependent facilitation (Ben-Johny and Yue, 2014; Christel and Lee, 2012; Lee et al., 1999). In support of the importance of P-type calcium channels, it was found that in P-type calcium channel knockout animals in which N-type calcium channels take the place of P-type channels at the calyx of Held, presynaptic calcium current facilitation was absent and synaptic facilitation was strongly attenuated (Inchauspe et al., 2004). Calcium-dependent facilitation of P-type calcium currents is impaired by mutating CaM, or by mutating an “IQ-like” motif (IM) on the carboxy tail of P-type calcium channels (DeMaria et al., 2001; Lee et al., 1999). These observations suggest that calcium binds to CaM, which in turn binds to the IM domain of P-type calcium channels (Mori et al., 2008) to produce CDF.
Calmodulin was the first calcium sensor to be implicated in calcium channel modulation. Expression of a calcium insensitive form of CaM suppresses CDF of P-type calcium channels (DeMaria et al., 2001; Lee et al., 2003) and mutagenesis of P-type calcium channels has established the importance of the IQ domain for CaM binding and CDF (DeMaria et al., 2001; Kim et al., 2008; Lee et al., 2003; Mori et al., 2008). Remarkably, the C lobe of CaM triggers rapid CDF of P-type calcium channels, but the N-lobe triggers slow CDI (Ben-Johny and Yue, 2014; DeMaria et al., 2001; Lee et al., 2003). An important feature of CaM-mediated modulation is that CaM pre-associates with P-type calcium channels by binding to the IQ domain (Erickson et al., 2001).
The calcium-binding protein NCS-1 is also proposed to play a role in synaptic facilitation, but there is considerable controversy over the mechanism by which NCS-1 influences facilitation. NCS-1 binds calcium with high affinity and is present in presynaptic boutons (Dason et al., 2012). At the drosophila NMJ (Pongs et al., 1993), overexpression of the NCS-1 homolog frequenin enhanced frequency-dependent facilitation. However, a subsequent study at the fly NMJ found that frequenin overexpression increased basal synaptic strength, increased depression in high Cae but did not alter paired-pulse plasticity in low Cae. In frequenin null mutants basal synaptic strength decreased, depression in high Cae decreased, and paired-pulse plasticity in low Cae was unaltered (Dason et al., 2009). These findings are reminiscent of the calyx of Held, where introducing NCS-1 into the presynaptic terminal enhances presynaptic calcium currents and occludes activity dependent increases in calcium entry, and a peptide that blocks NCS-1 interactions with its targets prevents CDF of calcium current (Tsujimoto et al., 2002). NCS-1 binds to the IQ-like domain and the CaM binding domain of P-type calcium channels to reduce calcium-dependent inactivation of calcium channels, thereby allowing calcium/CaM-dependent facilitation to dominate (Yan et al., 2014). These findings suggest that calcium binding to NCS-1 leads to synaptic facilitation by contributing to CDF.
Numerous other calcium sensors could potentially detect calcium and modulate calcium channels (Burgoyne and Haynes, 2015). Visinin-like protein 2 (VILIP-2) is highly expressed in the neocortex and hippocampus, blocks inactivation of calcium currents, reduces depression and increases facilitation (Lautermilch et al., 2005; Leal et al., 2012).
The contribution of CDF to synaptic facilitation was assessed using IM-AA mice in which native P-type calcium channels were replaced by calcium channels that cannot undergo CDF because they have a mutated IQ-like (IM) domain (Nanou et al., 2016) (Figure 5D-F). They studied facilitation at hippocampal synapses that normally rely on both N-type and P-type calcium channels. To isolate the contribution of P-type calcium channels to transmission, experiments were performed in the presence of antagonists of N-type calcium channels. The IM-AA mutation did not alter the basal properties of synaptic transmission, but it eliminated the small synaptic facilitation (~20% enhancement) observed in cultured hippocampal cells. Facilitation was also assessed in hippocampal slices. Previous studies found that facilitation is still present at these synapses when P-type calcium channels are blocked and transmission is mediated by N-type calcium channels (Scheuber et al., 2004), which indicates that CDF of P-type calcium channels does not account for all facilitation at this synapse. In hippocampal slices facilitation was still prominent in IM-AA mice (PPR~2) (Nanou et al., 2016), but it was reduced in magnitude compared to wildtype mice. This suggests that CDF can contribute to facilitation at this synapse, although syt7 appears to be the dominant mechanism of facilitation (Jackman et al., 2016). It will be important to determine the contribution of CDF to facilitation in IM-AA mice in control conditions when both P-type and N-type calcium channels mediate transmission.
Buffer Saturation Model
The properties of calcium regulation in presynaptic terminals can to lead to elevated Calocal during repetitive activation, even if successive action potentials evoke the same calcium entry. This is a consequence of the properties of calcium binding, which Neher and his colleagues realized could potentially contribute to synaptic facilitation (Klingauf and Neher, 1997; Neher, 1998; Rozov et al., 2001). In order to understand this mechanism, it is necessary to appreciate several aspects of presynaptic calcium signaling. First, the vast majority of calcium inside terminals is bound to calcium-binding proteins (Matthews and Dietrich, 2015; Neher, 1995; Smith and Zucker, 1980). Second, vesicle fusion is triggered when Calocal reaches tens of micromolar, which only occurs within tens of nanometers of open calcium channels. Third, calcium-binding proteins can intercept some of the calcium that enters the presynaptic terminal before it reaches the calcium sensor for release, which can reduce p for the initial EPSC. Calcium-binding proteins decrease the initial p most effectively when: (a) they are present at high concentrations, (b) calcium sensors for release are distant from calcium channels, and (c) the buffers are fast. Fourth, if the calcium buffer has a high affinity for calcium, less free calcium buffer is available to bind calcium during repetitive activation. This could lead to progressively larger Calocal signals during high-frequency firing and could produce facilitation.
Experiments at the layer 2/3 pyramidal cell to bitufted interneurons were the first to implicate buffer saturation in facilitation (Rozov et al., 2001). At this and many other synapses paired-pulse facilitation gets smaller when external calcium (Cae) is increased. This is thought to be a secondary consequence of increased Cae elevating p, which increases depletion and reduces the extent of facilitation. However, introducing the fast calcium-chelator BAPTA into the presynaptic cell led to facilitation that increased when Cae was elevated. This is compatible with the partial buffer saturation model that predicts that as Cae increases, buffer saturation becomes more prominent, which results in more facilitation. Rozov et al concluded that introducing a fast high-affinity calcium buffer into presynaptic terminals led artificial facilitation that relied on partial buffer saturation.
It was subsequently found that partial buffer saturation contributed to facilitation at native synapses that express high concentrations of the endogenous calcium-binding protein calbindin-D28k (Blatow et al., 2003). The mossy fiber synapse between hippocampal dentate granule cells and CA3 pyramidal cells is unusual in that facilitation gets larger when Cae is increased. In calbindin knockout animals facilitation gets smaller as Cae is increased. Thus it was suggested that in control conditions facilitation is a consequence of saturation of the fast, high-affinity calcium buffer calbindin. Calbindin is likely effective at lowering initial p and producing facilitation at mossy fiber synapses because of its calcium-binding properties (fast on-rate of 8 × 107 M−1s−1, and high affinity of 290 nM, (Eggermann et al., 2012; Nagerl et al., 2000)), and the loose coupling between calcium channels and release sensors at this synapse (Vyleta and Jonas, 2014). However, when calbindin was eliminated a form of facilitation remained that was similar to that observed at other synapses, such as the CA3 to CA1 synapse (Blatow et al., 2003).
Simulations suggest that partial buffer saturation can be a consequence of either global saturation of a mobile buffer, or local saturation of an immobile buffer (Matveev et al., 2004). If global saturation of a mobile buffer occurs within the presynaptic bouton, optical measurements using calcium indicators should detect use-dependent increases in action potential evoked calcium signals. If, instead, local saturation of an immobile buffer occurs, then larger calcium signals would only occur very locally, which would be very difficult to detect with standard fluorescence approaches. Although it is difficult to exclude the possibility that local buffer saturation occurs, there is no evidence that it contributes to facilitation at most synapses.
Conclusion
In the 70 years since its initial description by T.P Feng and others, substantial progress has been made in clarifying the mechanism of facilitation, while modeling studies have provided insight into possible functional roles of facilitation. It appears that a primary mechanism of facilitation is that the Cares activates syt7 to increase the probability of release. Calcium modulation of P-type calcium channels can also contribute to short-lived synaptic facilitation at many synapses. Other mechanisms such as spike broadening and local calcium buffer saturation contribute to facilitation at some synapses, but they appear to be less prevalent. Further studies will determine whether other presynaptic calcium sensors or additional mechanisms also contribute to facilitation, and whether multiple mechanisms interact to produce facilitation.
A concrete understanding of how facilitation affects neural processing and behavior is lacking because no study has addressed the impact of eliminating facilitation on behavioral phenotypes or in vivo circuit performance. Discovering the underlying mechanisms is an important first step in determining the functional importance of facilitation. In the case of syt7, only global constitutive knockout lines are currently available, making it difficult to evaluate the impact of removing facilitation from specific circuits. Synapse-specific elimination of facilitation using genetic approaches will make it possible to test theories of the function of facilitation and provide insight into its role in neural processing, neuropsychiatric diseases and animal behavior.
Acknowledgments
We thank Jasmine Vazquez for illustrations, and Pascal Kaeser, Stephanie Rudolph, Josef Turecek, Chong Guo and Chris Chen for comments on the manuscript. This work was supported by grants from the National Institutes of Health (R01NS032405 and R35NS097284) to W.R. and a Nancy Lurie Marks Fellowship to S.L.J.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott LF, Regehr WG. Synaptic computation. Nature. 2004;431:796–803. doi: 10.1038/nature03010. [DOI] [PubMed] [Google Scholar]
- Abbott LF, Varela JA, Sen K, Nelson SB. Synaptic depression and cortical gain control. Science. 1997;275:220–224. doi: 10.1126/science.275.5297.221. [DOI] [PubMed] [Google Scholar]
- Alger BE, Nicoll RA. Feed-forward dendritic inhibition in rat hippocampal pyramidal cells studied in vitro. The Journal of physiology. 1982;328:105–123. doi: 10.1113/jphysiol.1982.sp014255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashmore J. Cochlear outer hair cell motility. Physiol Rev. 2008;88:173–210. doi: 10.1152/physrev.00044.2006. [DOI] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Determinants of the time course of facilitation at the granule cell to Purkinje cell synapse. J Neurosci. 1996;16:5661–5671. doi: 10.1523/JNEUROSCI.16-18-05661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci. 1998;18:8214–8227. doi: 10.1523/JNEUROSCI.18-20-08214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atwood H. Parallel ‘phasic’ and ‘tonic’ motor systems of the crayfish abdomen. J Exp Biol. 2008;211:2193–2195. doi: 10.1242/jeb.010868. [DOI] [PubMed] [Google Scholar]
- Atwood HL, Karunanithi S. Diversification of synaptic strength: presynaptic elements. Nature reviews Neuroscience. 2002;3:497–516. doi: 10.1038/nrn876. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Adler EM, Charlton MP. The calcium signal for transmitter secretion from presynaptic nerve terminals. Ann N Y Acad Sci. 1991;635:365–381. doi: 10.1111/j.1749-6632.1991.tb36505.x. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Burre J, Malenka RC, Liu X, Sudhof TC. Synaptotagmin-1 and -7 Are Redundantly Essential for Maintaining the Capacity of the Readily-Releasable Pool of Synaptic Vesicles. PLoS biology. 2015;13:e1002267. doi: 10.1371/journal.pbio.1002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, Sudhof TC. Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron. 2013;80:947–959. doi: 10.1016/j.neuron.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nature structural & molecular biology. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- Bai J, Wang P, Chapman ER. C2A activates a cryptic Ca(2+)-triggered membrane penetration activity within the C2B domain of synaptotagmin I. Proc Natl Acad Sci U S A. 2002;99:1665–1670. doi: 10.1073/pnas.032541099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestero J, Zorrilla de San Martin J, Goutman J, Elgoyhen AB, Fuchs PA, Katz E. Short-term synaptic plasticity regulates the level of olivocochlear inhibition to auditory hair cells. J Neurosci. 2011;31:14763–14774. doi: 10.1523/JNEUROSCI.6788-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J, Reim K, Sakaba T. Target-dependent feedforward inhibition mediated by short-term synaptic plasticity in the cerebellum. J Neurosci. 2010;30:8171–8179. doi: 10.1523/JNEUROSCI.0276-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak O, Tsodyks M. Persistent activity in neural networks with dynamic synapses. PLoS computational biology. 2007;3:e35. doi: 10.1371/journal.pcbi.0030035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF, Stevens CF. The kinetics of transmitter release at the frog neuromuscular junction. The Journal of physiology. 1972;227:691–708. doi: 10.1113/jphysiol.1972.sp010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartley AF, Dobrunz LE. Short-term plasticity regulates the excitation/inhibition ratio and the temporal window for spike integration in CA1 pyramidal cells. Eur J Neurosci. 2015;41:1402–1415. doi: 10.1111/ejn.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Johny M, Yue DT. Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J Gen Physiol. 2014;143:679–692. doi: 10.1085/jgp.201311153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best AR, Regehr WG. Inhibitory regulation of electrically coupled neurons in the inferior olive is mediated by asynchronous release of GABA. Neuron. 2009;62:555–565. doi: 10.1016/j.neuron.2009.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharat TA, Malsam J, Hagen WJ, Scheutzow A, Sollner TH, Briggs JA. SNARE and regulatory proteins induce local membrane protrusions to prime docked vesicles for fast calcium-triggered fusion. EMBO reports. 2014;15:308–314. doi: 10.1002/embr.201337807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blatow M, Caputi A, Burnashev N, Monyer H, Rozov A. Ca2+ buffer saturation underlies paired pulse facilitation in calbindin-D28k-containing terminals. Neuron. 2003;38:79–88. doi: 10.1016/s0896-6273(03)00196-x. [DOI] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Facilitation of presynaptic calcium currents in the rat brainstem. The Journal of physiology. 1998;513(Pt 1):149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of alpha 1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- Bradacs H, Cooper R, Msghina M, Atwood H. Differential physiology and morphology of phasic and tonic motor axons in a crayfish limb extensor muscle. J Exp Biol. 1997;200:677–691. doi: 10.1242/jeb.200.4.677. [DOI] [PubMed] [Google Scholar]
- Brandt DS, Coffman MD, Falke JJ, Knight JD. Hydrophobic contributions to the membrane docking of synaptotagmin 7 C2A domain: mechanistic contrast between isoforms 1 and 7. Biochemistry (Mosc) 2012;51:7654–7664. doi: 10.1021/bi3007115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG. Reliability and heterogeneity of calcium signaling at single presynaptic boutons of cerebellar granule cells. J Neurosci. 2007;27:7888–7898. doi: 10.1523/JNEUROSCI.1064-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Kujawa SG, Duca ML. Single olivocochlear neurons in the guinea pig. I. Binaural facilitation of responses to high-level noise. J Neurophysiol. 1998;79:3077–3087. doi: 10.1152/jn.1998.79.6.3077. [DOI] [PubMed] [Google Scholar]
- Brownell WE, Bader CR, Bertrand D, de Ribaupierre Y. Evoked mechanical responses of isolated cochlear outer hair cells. Science. 1985;227:194–196. doi: 10.1126/science.3966153. [DOI] [PubMed] [Google Scholar]
- Bruner J, Kennedy D. Habituation: occurrence at a neuromuscular junction. Science. 1970;169:92–94. doi: 10.1126/science.169.3940.92. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD. Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signalling. Nature reviews Neuroscience. 2007;8:182–193. doi: 10.1038/nrn2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne RD, Haynes LP. Sense and specificity in neuronal calcium signalling. Biochim Biophys Acta. 2015;1853:1921–1932. doi: 10.1016/j.bbamcr.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti S, Kobayashi KS, Flavell RA, Marks CB, Miyake K, Liston DR, Fowler KT, Gorelick FS, Andrews NW. Impaired membrane resealing and autoimmune myositis in synaptotagmin VII-deficient mice. J Cell Biol. 2003;162:543–549. doi: 10.1083/jcb.200305131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER. How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- Charlton MP, Bittner GD. Presynaptic potentials and facilitation of transmitter release in the squid giant synapse. J Gen Physiol. 1978;72:487–511. doi: 10.1085/jgp.72.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton MP, Smith SJ, Zucker RS. Role of presynaptic calcium ions and channels in synaptic facilitation and depression at the squid giant synapse. The Journal of physiology. 1982;323:173–193. doi: 10.1113/jphysiol.1982.sp014067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Chang SY, DeMaria CD, Alvania RS, Soong TW, Yue DT. Alternative splicing as a molecular switch for Ca2+/calmodulin-dependent facilitation of P/Q-type Ca2+ channels. J Neurosci. 2004;24:6334–6342. doi: 10.1523/JNEUROSCI.1712-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Das B, Nakamura Y, DiGregorio DA, Young SM., Jr Ca2+ channel to synaptic vesicle distance accounts for the readily releasable pool kinetics at a functionally mature auditory synapse. J Neurosci. 2015;35:2083–2100. doi: 10.1523/JNEUROSCI.2753-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicka MC, Hui E, Liu H, Chapman ER. Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+ Nature structural & molecular biology. 2008;15:827–835. doi: 10.1038/nsmb.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christel C, Lee A. Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim Biophys Acta. 2012;1820:1243–1252. doi: 10.1016/j.bbagen.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie JM, Jahr CE. Multivesicular release at Schaffer collateral-CA1 hippocampal synapses. J Neurosci. 2006;26:210–216. doi: 10.1523/JNEUROSCI.4307-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DL, Schwindt PC, Grande LA, Spain WJ. Synaptic depression in the localization of sound. Nature. 2003;421:66–70. doi: 10.1038/nature01248. [DOI] [PubMed] [Google Scholar]
- Corbalan-Garcia S, Gomez-Fernandez JC. Signaling through C2 domains: more than one lipid target. Biochim Biophys Acta. 2014;1838:1536–1547. doi: 10.1016/j.bbamem.2014.01.008. [DOI] [PubMed] [Google Scholar]
- Craxton M. A manual collection of Syt, Esyt, Rph3a, Rph3al, Doc2, and Dblc2 genes from 46 metazoan genomes–an open access resource for neuroscience and evolutionary biology. BMC genomics. 2010;11:37. doi: 10.1186/1471-2164-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. The Journal of physiology. 1998;512(Pt 3):723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dason JS, Romero-Pozuelo J, Atwood HL, Ferrus A. Multiple roles for frequenin/NCS-1 in synaptic function and development. Mol Neurobiol. 2012;45:388–402. doi: 10.1007/s12035-012-8250-4. [DOI] [PubMed] [Google Scholar]
- Dason JS, Romero-Pozuelo J, Marin L, Iyengar BG, Klose MK, Ferrus A, Atwood HL. Frequenin/NCS-1 and the Ca2+-channel alpha1-subunit co-regulate synaptic transmission and nerve-terminal growth. J Cell Sci. 2009;122:4109–4121. doi: 10.1242/jcs.055095. [DOI] [PubMed] [Google Scholar]
- Davis AF, Bai J, Fasshauer D, Wolowick MJ, Lewis JL, Chapman ER. Kinetics of synaptotagmin responses to Ca2+ and assembly with the core SNARE complex onto membranes. Neuron. 1999;24:363–376. doi: 10.1016/s0896-6273(00)80850-8. [DOI] [PubMed] [Google Scholar]
- de Wit H, Walter AM, Milosevic I, Gulyas-Kovacs A, Riedel D, Sorensen JB, Verhage M. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Deco G, Rolls ET, Romo R. Synaptic dynamics and decision making. Proc Natl Acad Sci U S A. 2010;107:7545–7549. doi: 10.1073/pnas.1002333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J, Katz B. Statistical factors involved in neuromuscular facilitation and depression. The Journal of physiology. 1954;124:574–585. doi: 10.1113/jphysiol.1954.sp005130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, Yue DT. Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature. 2001;411:484–489. doi: 10.1038/35078091. [DOI] [PubMed] [Google Scholar]
- Deng PY, Klyachko VA. The diverse functions of short-term plasticity components in synaptic computations. Communicative & integrative biology. 2011;4:543–548. doi: 10.4161/cib.4.5.15870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Guilmi MN, Wang T, Inchauspe CG, Forsythe ID, Ferrari MD, van den Maagdenberg AM, Borst JG, Uchitel OD. Synaptic gain-of-function effects of mutant Cav2.1 channels in a mouse model of familial hemiplegic migraine are due to increased basal [Ca2+]i. J Neurosci. 2014;34:7047–7058. doi: 10.1523/JNEUROSCI.2526-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Rojas F, Sakaba T, Kawaguchi SY. Ca(2+) current facilitation determines short-term facilitation at inhibitory synapses between cerebellar Purkinje cells. The Journal of physiology. 2015;593:4889–4904. doi: 10.1113/JP270704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Kreitzer AC, Regehr WG. Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J Neurosci. 2000;20:1374–1385. doi: 10.1523/JNEUROSCI.20-04-01374.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. The Journal of physiology. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreifuss JJ, Kalnins I, Kelly JS, Ruf KB. Action potentials and release of neurohypophysial hormones in vitro. The Journal of physiology. 1971;215:805–817. doi: 10.1113/jphysiol.1971.sp009499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie T, Lynch G. Long-term potentiation and depression of synaptic responses in the rat hippocampus: localization and frequency dependency. The Journal of physiology. 1978;276:353–367. doi: 10.1113/jphysiol.1978.sp012239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Katz B, Kuffler SW. Nature of the “endplate potential” in curarized muscle. J Neurophysiol. 1941;4:362–387. [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P. Nanodomain coupling between Ca(2)(+) channels and sensors of exocytosis at fast mammalian synapses. Nature reviews Neuroscience. 2012;13:7–21. doi: 10.1038/nrn3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca(2+) channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- Feng TP. Studies on the neuromuscular junction. XVIII. The local potentials around n-m junctions induced by single and multiple volleys. Chin J Physiol. 1940;15:367–404. [Google Scholar]
- Fernandez-Chacon R, Shin OH, Konigstorfer A, Matos MF, Meyer AC, Garcia J, Gerber SH, Rizo J, Sudhof TC, Rosenmund C. Structure/function analysis of Ca2+ binding to the C2A domain of synaptotagmin 1. J Neurosci. 2002;22:8438–8446. doi: 10.1523/JNEUROSCI.22-19-08438.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogelson AL, Zucker RS. Presynaptic calcium diffusion from various arrays of single channels. Implications for transmitter release and synaptic facilitation. Biophys J. 1985;48:1003–1017. doi: 10.1016/S0006-3495(85)83863-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KA, Crowley JJ, Regehr WG. The influence of multivesicular release and postsynaptic receptor saturation on transmission at granule cell to Purkinje cell synapses. J Neurosci. 2005;25:11655–11665. doi: 10.1523/JNEUROSCI.4029-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann G, Segev I, Markram H, Tsodyks M. Coding of temporal information by activity-dependent synapses. J Neurophysiol. 2002;87:140–148. doi: 10.1152/jn.00258.2001. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Ogata Y, Saegusa C, Kanno E, Mikoshiba K. Alternative splicing isoforms of synaptotagmin VII in the mouse, rat and human. Biochem J. 2002;365:173–180. doi: 10.1042/BJ20011877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JM, Alexander GE. Neuron activity related to short-term memory. Science. 1971;173:652–654. doi: 10.1126/science.173.3997.652. [DOI] [PubMed] [Google Scholar]
- Gaffaney JD, Dunning FM, Wang Z, Hui E, Chapman ER. Synaptotagmin C2B domain regulates Ca2+-triggered fusion in vitro: critical residues revealed by scanning alanine mutagenesis. J Biol Chem. 2008;283:31763–31775. doi: 10.1074/jbc.M803355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainer H, Wolfe SA, Jr, Obaid AL, Salzberg BM. Action potentials and frequency-dependent secretion in the mouse neurohypophysis. Neuroendocrinology. 1986;43:557–563. doi: 10.1159/000124582. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Jonas P. Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron. 2000;28:927–939. doi: 10.1016/s0896-6273(00)00164-1. [DOI] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A. 1994;91:12942–12946. doi: 10.1073/pnas.91.26.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- Goutman JD, Fuchs PA, Glowatzki E. Facilitating efferent inhibition of inner hair cells in the cochlea of the neonatal rat. The Journal of physiology. 2005;566:49–59. doi: 10.1113/jphysiol.2005.087460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavsson N, Lao Y, Maximov A, Chuang JC, Kostromina E, Repa JJ, Li C, Radda GK, Sudhof TC, Han W. Impaired insulin secretion and glucose intolerance in synaptotagmin-7 null mutant mice. Proc Natl Acad Sci U S A. 2008;105:3992–3997. doi: 10.1073/pnas.0711700105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck DH, De Zeeuw CI, Jaeger D, Khodakhah K, Person AL. The neuronal code(s) of the cerebellum. J Neurosci. 2013;33:17603–17609. doi: 10.1523/JNEUROSCI.2759-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- Hennig R, Lomo T. Firing patterns of motor units in normal rats. Nature. 1985;314:164–166. doi: 10.1038/314164a0. [DOI] [PubMed] [Google Scholar]
- Herrick DZ, Sterbling S, Rasch KA, Hinderliter A, Cafiso DS. Position of synaptotagmin I at the membrane interface: cooperative interactions of tandem C2 domains. Biochemistry (Mosc) 2006;45:9668–9674. doi: 10.1021/bi060874j. [DOI] [PubMed] [Google Scholar]
- Honigmann A, van den Bogaart G, Iraheta E, Risselada HJ, Milovanovic D, Mueller V, Mullar S, Diederichsen U, Fasshauer D, Grubmuller H, et al. Phosphatidylinositol 4,5-bisphosphate clusters act as molecular beacons for vesicle recruitment. Nature structural & molecular biology. 2013;20:679–686. doi: 10.1038/nsmb.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta PT, Lisman JE. Bidirectional synaptic plasticity induced by a single burst during cholinergic theta oscillation in CA1 in vitro. Neuron. 1995;15:1053–1063. doi: 10.1016/0896-6273(95)90094-2. [DOI] [PubMed] [Google Scholar]
- Hui E, Bai J, Wang P, Sugimori M, Llinas RR, Chapman ER. Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc Natl Acad Sci U S A. 2005;102:5210–5214. doi: 10.1073/pnas.0500941102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inchauspe CG, Martini FJ, Forsythe ID, Uchitel OD. Functional compensation of P/Q by N-type channels blocks short-term plasticity at the calyx of Held presynaptic terminal. J Neurosci. 2004;24:10379–10383. doi: 10.1523/JNEUROSCI.2104-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itskov V, Hansel D, Tsodyks M. Short-Term Facilitation may Stabilize Parametric Working Memory Trace. Frontiers in computational neuroscience. 2011;5:40. doi: 10.3389/fncom.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman SL, Turecek J, Belinsky JE, Regehr WG. The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature. 2016;529:88–91. doi: 10.1038/nature16507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB, Konnerth A, Augustine GJ. Action potential broadening and frequency-dependent facilitation of calcium signals in pituitary nerve terminals. Proc Natl Acad Sci U S A. 1991;88:380–384. doi: 10.1073/pnas.88.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Zucker RS. Residual Ca2+ and short-term synaptic plasticity. Nature. 1994;371:603–606. doi: 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. The Journal of physiology. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy D, Takeda K. Reflex control of abdominal flexor muscles in the crayfish. I. The twitch system. J Exp Biol. 1965a;43:211–217. [Google Scholar]
- Kennedy D, Takeda K. Reflex control of abdominal flexor muscles in the crayfish. II. The tonic system. J Exp Biol. 1965b;43:229–246. [Google Scholar]
- Kim EY, Rumpf CH, Fujiwara Y, Cooley ES, Van Petegem F, Minor DL., Jr Structures of CaV2 Ca2+/CaM-IQ domain complexes reveal binding modes that underlie calcium-dependent inactivation and facilitation. Structure. 2008;16:1455–1467. doi: 10.1016/j.str.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingauf J, Neher E. Modeling buffered Ca2+ diffusion near the membrane: implications for secretion in neuroendocrine cells. Biophys J. 1997;72:674–690. doi: 10.1016/s0006-3495(97)78704-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klyachko VA, Stevens CF. Excitatory and feed-forward inhibitory hippocampal synapses work synergistically as an adaptive filter of natural spike trains. PLoS biology. 2006;4:e207. doi: 10.1371/journal.pbio.0040207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlovsky Y, Kozlov MM. Stalk model of membrane fusion: solution of energy crisis. Biophys J. 2002;82:882–895. doi: 10.1016/S0006-3495(02)75450-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Modulation of transmission during trains at a cerebellar synapse. J Neurosci. 2000;20:1348–1357. doi: 10.1523/JNEUROSCI.20-04-01348.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin PI, Zimmerberg J, Chizmadzhev YA, Cohen FS. A quantitative model for membrane fusion based on low-energy intermediates. Proc Natl Acad Sci U S A. 2001;98:7235–7240. doi: 10.1073/pnas.121191898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautermilch NJ, Few AP, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by the neuronal calcium-binding protein visinin-like protein-2. J Neurosci. 2005;25:7062–7070. doi: 10.1523/JNEUROSCI.0447-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal K, Mochida S, Scheuer T, Catterall WA. Fine-tuning synaptic plasticity by modulation of Ca(V)2.1 channels with Ca2+ sensor proteins. Proc Natl Acad Sci U S A. 2012;109:17069–17074. doi: 10.1073/pnas.1215172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA. Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature. 1999;399:155–159. doi: 10.1038/20194. [DOI] [PubMed] [Google Scholar]
- Lee A, Zhou H, Scheuer T, Catterall WA. Molecular determinants of Ca(2+)/calmodulin-dependent regulation of Ca(v)2.1 channels. Proc Natl Acad Sci U S A. 2003;100:16059–16064. doi: 10.1073/pnas.2237000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Adams PJ, Yue DT. Large Ca(2)(+)-dependent facilitation of Ca(V)2.1 channels revealed by Ca(2)(+) photo-uncaging. The Journal of physiology. 2015;593:2753–2778. doi: 10.1113/JP270091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Davletov BA, Sudhof TC. Distinct Ca2+ and Sr2+ binding properties of synaptotagmins. Definition of candidate Ca2+ sensors for the fast and slow components of neurotransmitter release. J Biol Chem. 1995;270:24898–24902. doi: 10.1074/jbc.270.42.24898. [DOI] [PubMed] [Google Scholar]
- Li L, Shin OH, Rhee JS, Arac D, Rah JC, Rizo J, Sudhof T, Rosenmund C. Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J Biol Chem. 2006;281:15845–15852. doi: 10.1074/jbc.M600888200. [DOI] [PubMed] [Google Scholar]
- Li YC, Chanaday NL, Xu W, Kavalali ET. Synaptotagmin-1- and Synaptotagmin-7-Dependent Fusion Mechanisms Target Synaptic Vesicles to Kinetically Distinct Endocytic Pathways. Neuron. 2017;93:616–631 e613. doi: 10.1016/j.neuron.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- Liu H, Bai H, Hui E, Yang L, Evans CS, Wang Z, Kwon SE, Chapman ER. Synaptotagmin 7 functions as a Ca2+-sensor for synaptic vesicle replenishment. eLife. 2014;3:e01524. doi: 10.7554/eLife.01524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losonczy A, Zhang L, Shigemoto R, Somogyi P, Nusser Z. Cell type dependence and variability in the short-term plasticity of EPSCs in identified mouse hippocampal interneurones. The Journal of physiology. 2002;542:193–210. doi: 10.1113/jphysiol.2002.020024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, Nicoud JF, Zemelman BV, Sternson SM, Losonczy A. Regulation of neuronal input transformations by tunable dendritic inhibition. Nat Neurosci. 2012;15:423–430. S421–423. doi: 10.1038/nn.3024. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, Toth K, McBain CJ. Target-specific expression of presynaptic mossy fiber plasticity. Science. 1998;279:1368–1370. doi: 10.1126/science.279.5355.1368. [DOI] [PubMed] [Google Scholar]
- Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE. The C(2)B Ca(2+)-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature. 2002;418:340–344. doi: 10.1038/nature00846. [DOI] [PubMed] [Google Scholar]
- Mackler JM, Reist NE. Mutations in the second C2 domain of synaptotagmin disrupt synaptic transmission at Drosophila neuromuscular junctions. J Comp Neurol. 2001;436:4–16. [PubMed] [Google Scholar]
- MacLeod KM, Horiuchi TK, Carr CE. A role for short-term synaptic facilitation and depression in the processing of intensity information in the auditory brain stem. J Neurophysiol. 2007;97:2863–2874. doi: 10.1152/jn.01030.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magleby KL. The effect of repetitive stimulation on facilitation of transmitter release at the frog neuromuscular junction. The Journal of physiology. 1973;234:327–352. doi: 10.1113/jphysiol.1973.sp010348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Wang Y, Tsodyks M. Differential signaling via the same axon of neocortical pyramidal neurons. Proc Natl Acad Sci U S A. 1998;95:5323–5328. doi: 10.1073/pnas.95.9.5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- Martinez I, Chakrabarti S, Hellevik T, Morehead J, Fowler K, Andrews NW. Synaptotagmin VII regulates Ca(2+)-dependent exocytosis of lysosomes in fibroblasts. J Cell Biol. 2000;148:1141–1149. doi: 10.1083/jcb.148.6.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews EA, Dietrich D. Buffer mobility and the regulation of neuronal calcium domains. Frontiers in cellular neuroscience. 2015;9:48. doi: 10.3389/fncel.2015.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveev V, Zucker RS, Sherman A. Facilitation through buffer saturation: Constraints on endogenous buffering properties. Biophys J. 2004;86:2691–2709. doi: 10.1016/S0006-3495(04)74324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Lao Y, Li H, Chen X, Rizo J, Sorensen JB, Sudhof TC. Genetic analysis of synaptotagmin-7 function in synaptic vesicle exocytosis. Proc Natl Acad Sci U S A. 2008;105:3986–3991. doi: 10.1073/pnas.0712372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelsteadt T, Seifert G, Alvarez-Baron E, Steinhauser C, Becker AJ, Schoch S. Differential mRNA expression patterns of the synaptotagmin gene family in the rodent brain. J Comp Neurol. 2009;512:514–528. doi: 10.1002/cne.21908. [DOI] [PubMed] [Google Scholar]
- Mohrmann R, de Wit H, Connell E, Pinheiro PS, Leese C, Bruns D, Davletov B, Verhage M, Sorensen JB. Synaptotagmin interaction with SNAP-25 governs vesicle docking, priming, and fusion triggering. J Neurosci. 2013;33:14417–14430. doi: 10.1523/JNEUROSCI.1236-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mongillo G, Barak O, Tsodyks M. Synaptic theory of working memory. Science. 2008;319:1543–1546. doi: 10.1126/science.1150769. [DOI] [PubMed] [Google Scholar]
- Moreno A, Morris RG, Canals S. Frequency-Dependent Gating of Hippocampal-Neocortical Interactions. Cereb Cortex. 2016;26:2105–2114. doi: 10.1093/cercor/bhv033. [DOI] [PubMed] [Google Scholar]
- Mori MX, Vander Kooi CW, Leahy DJ, Yue DT. Crystal structure of the CaV2 IQ domain in complex with Ca2+/calmodulin: high-resolution mechanistic implications for channel regulation by Ca2+ Structure. 2008;16:607–620. doi: 10.1016/j.str.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Felmy F, Schneggenburger R. A limited contribution of Ca2+ current facilitation to paired-pulse facilitation of transmitter release at the rat calyx of Held. The Journal of physiology. 2008;586:5503–5520. doi: 10.1113/jphysiol.2008.155838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagerl UV, Novo D, Mody I, Vergara JL. Binding kinetics of calbindin-D(28k) determined by flash photolysis of caged Ca(2+) Biophys J. 2000;79:3009–3018. doi: 10.1016/S0006-3495(00)76537-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanou E, Sullivan JM, Scheuer T, Catterall WA. Calcium sensor regulation of the CaV2.1 Ca2+ channel contributes to short-term synaptic plasticity in hippocampal neurons. Proc Natl Acad Sci U S A. 2016;113:1062–1067. doi: 10.1073/pnas.1524636113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. The use of fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology. 1995;34:1423–1442. doi: 10.1016/0028-3908(95)00144-u. [DOI] [PubMed] [Google Scholar]
- Neher E. Usefulness and limitations of linear approximations to the understanding of Ca++ signals. Cell Calcium. 1998;24:345–357. doi: 10.1016/s0143-4160(98)90058-6. [DOI] [PubMed] [Google Scholar]
- Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. The Journal of physiology. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA. A Brief History of Long-Term Potentiation. Neuron. 2017;93:281–290. doi: 10.1016/j.neuron.2016.12.015. [DOI] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J Neurosci. 2004;24:8542–8550. doi: 10.1523/JNEUROSCI.2545-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe J, Dostrovsky J. The hippocampus as a spatial map. Preliminary evidence from unit activity in the freely-moving rat. Brain Res. 1971;34:171–175. doi: 10.1016/0006-8993(71)90358-1. [DOI] [PubMed] [Google Scholar]
- Osborne SL, Wallis TP, Jimenez JL, Gorman JJ, Meunier FA. Identification of secretory granule phosphatidylinositol 4,5-bisphosphate-interacting proteins using an affinity pulldown strategy. Molecular & cellular proteomics : MCP. 2007;6:1158–1169. doi: 10.1074/mcp.M600430-MCP200. [DOI] [PubMed] [Google Scholar]
- Paddock BE, Wang Z, Biela LM, Chen K, Getzy MD, Striegel A, Richmond JE, Chapman ER, Featherstone DE, Reist NE. Membrane penetration by synaptotagmin is required for coupling calcium binding to vesicle fusion in vivo. J Neurosci. 2011;31:2248–2257. doi: 10.1523/JNEUROSCI.3153-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongs O, Lindemeier J, Zhu XR, Theil T, Engelkamp D, Krah-Jentgens I, Lambrecht HG, Koch KW, Schwemer J, Rivosecchi R, et al. Frequenin–a novel calcium-binding protein that modulates synaptic efficacy in the Drosophila nervous system. Neuron. 1993;11:15–28. doi: 10.1016/0896-6273(93)90267-u. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Pouille F, Scanziani M. Routing of spike series by dynamic circuits in the hippocampus. Nature. 2004;429:717–723. doi: 10.1038/nature02615. [DOI] [PubMed] [Google Scholar]
- Pouzat C, Hestrin S. Developmental regulation of basket/stellate cell–>Purkinje cell synapses in the cerebellum. J Neurosci. 1997;17:9104–9112. doi: 10.1523/JNEUROSCI.17-23-09104.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Saggau P. Activity-dependent modulation of K+ currents at presynaptic terminals of mammalian central synapses. The Journal of physiology. 1999;519(Pt 2):427–437. doi: 10.1111/j.1469-7793.1999.0427m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Stein A, Jahn R, Fasshauer D. The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2009;284:25749–25760. doi: 10.1074/jbc.M109.042499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahamimoff R, Yaari Y. Delayed release of transmitter at the frog neuromuscular junction. The Journal of physiology. 1973;228:241–257. doi: 10.1113/jphysiol.1973.sp010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Delaney KR, Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid B, Slater CR, Bewick GS. Synaptic vesicle dynamics in rat fast and slow motor nerve terminals. J Neurosci. 1999;19:2511–2521. doi: 10.1523/JNEUROSCI.19-07-02511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B. Target-cell-specific facilitation and depression in neocortical circuits. Nat Neurosci. 1998;1:279–285. doi: 10.1038/1092. [DOI] [PubMed] [Google Scholar]
- Robinson IM, Ranjan R, Schwarz TL. Synaptotagmins I and IV promote transmitter release independently of Ca(2+) binding in the C(2)A domain. Nature. 2002;418:336–340. doi: 10.1038/nature00915. [DOI] [PubMed] [Google Scholar]
- Rose GJ, Fortune ES. Frequency-dependent PSP depression contributes to low-pass temporal filtering in Eigenmannia. J Neurosci. 1999;19:7629–7639. doi: 10.1523/JNEUROSCI.19-17-07629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotman Z, Deng PY, Klyachko VA. Short-term plasticity optimizes synaptic information transmission. J Neurosci. 2011;31:14800–14809. doi: 10.1523/JNEUROSCI.3231-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan MJ, Tranquil E, Christie JM. Distinct Kv channel subtypes contribute to differences in spike signaling properties in the axon initial segment and presynaptic boutons of cerebellar interneurons. J Neurosci. 2014;34:6611–6623. doi: 10.1523/JNEUROSCI.4208-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Zemelman BV, Losonczy A, Kim J, Chance F, Magee JC, Buzsaki G. Control of timing, rate and bursts of hippocampal place cells by dendritic and somatic inhibition. Nat Neurosci. 2012;15:769–775. doi: 10.1038/nn.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Burnashev N. Polyamine-dependent facilitation of postsynaptic AMPA receptors counteracts paired-pulse depression. Nature. 1999;401:594–598. doi: 10.1038/44151. [DOI] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. Journal of Physiology-London. 2001;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci. 1997;17:3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Optical measurement of presynaptic calcium currents. Biophys J. 1998;74:1549–1563. doi: 10.1016/S0006-3495(98)77867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg BM, Obaid AL, Gainer H. Optical studies of excitation and secretion at vertebrate nerve terminals. Soc Gen Physiol Ser. 1986;40:133–164. [PubMed] [Google Scholar]
- Saraswati S, Adolfsen B, Littleton JT. Characterization of the role of the Synaptotagmin family as calcium sensors in facilitation and asynchronous neurotransmitter release. Proc Natl Acad Sci U S A. 2007;104:14122–14127. doi: 10.1073/pnas.0706711104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler BH, Charpak S. Target cell-specific modulation of transmitter release at terminals from a single axon. Proc Natl Acad Sci U S A. 1998;95:12004–12009. doi: 10.1073/pnas.95.20.12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuber A, Miles R, Poncer JC. Presynaptic Cav2.1 and Cav2.2 differentially influence release dynamics at hippocampal excitatory synapses. J Neurosci. 2004;24:10402–10409. doi: 10.1523/JNEUROSCI.1664-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Schonn JS, Maximov A, Lao Y, Sudhof TC, Sorensen JB. Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci U S A. 2008;105:3998–4003. doi: 10.1073/pnas.0712373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott P, Cowan AI, Stricker C. Quantifying impacts of short-term plasticity on neuronal information transfer. Physical review E, Statistical, nonlinear, and soft matter physics. 2012;85:041921. doi: 10.1103/PhysRevE.85.041921. [DOI] [PubMed] [Google Scholar]
- Scott R, Rusakov DA. Main determinants of presynaptic Ca2+ dynamics at individual mossy fiber-CA3 pyramidal cell synapses. J Neurosci. 2006;26:7071–7081. doi: 10.1523/JNEUROSCI.0946-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman RG, Atwood HL. Correlated electrophysiological and ultrastructural studies of a crustacean motor unit. J Gen Physiol. 1972;59:586–615. doi: 10.1085/jgp.59.5.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y, Duque A, Yu Y, Haider B, McCormick DA. Properties of action-potential initiation in neocortical pyramidal cells: evidence from whole cell axon recordings. J Neurophysiol. 2007;97:746–760. doi: 10.1152/jn.00922.2006. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, Duque A, Yu Y, McCormick DA. Modulation of intracortical synaptic potentials by presynaptic somatic membrane potential. Nature. 2006;441:761–765. doi: 10.1038/nature04720. [DOI] [PubMed] [Google Scholar]
- Simon SM, Llinas RR. Compartmentalization of the submembrane calcium activity during calcium influx and its significance in transmitter release. Biophys J. 1985;48:485–498. doi: 10.1016/S0006-3495(85)83804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippy T, Cruz-Martin A, Jeromin A, Schweizer FE. Acute changes in short-term plasticity at synapses with elevated levels of neuronal calcium sensor-1. Nat Neurosci. 2003;6:1031–1038. doi: 10.1038/nn1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SJ, Zucker RS. Aequorin response facilitation and intracellular calcium accumulation in molluscan neurones. The Journal of physiology. 1980;300:167–196. doi: 10.1113/jphysiol.1980.sp013157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo C. Viewing the brain through the master hand of Ramon y Cajal. Nature reviews Neuroscience. 2003;4:71–77. doi: 10.1038/nrn1010. [DOI] [PubMed] [Google Scholar]
- Sreenivasan KK, Curtis CE, D’Esposito M. Revisiting the role of persistent neural activity during working memory. Trends in cognitive sciences. 2014;18:82–89. doi: 10.1016/j.tics.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron. 2003;39:299–308. doi: 10.1016/s0896-6273(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Stokes MG. ‘Activity-silent’ working memory in prefrontal cortex: a dynamic coding framework. Trends in cognitive sciences. 2015;19:394–405. doi: 10.1016/j.tics.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes MG, Kusunoki M, Sigala N, Nili H, Gaffan D, Duncan J. Dynamic coding for cognitive control in prefrontal cortex. Neuron. 2013;78:364–375. doi: 10.1016/j.neuron.2013.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Synaptotagmins: why so many? J Biol Chem. 2002;277:7629–7632. doi: 10.1074/jbc.R100052200. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. A molecular machine for neurotransmitter release: synaptotagmin and beyond. Nat Med. 2013;19:1227–1231. doi: 10.1038/nm.3338. [DOI] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugase-Miyamoto Y, Liu Z, Wiener MC, Optican LM, Richmond BJ. Short-term memory trace in rapidly adapting synapses of inferior temporal cortex. PLoS computational biology. 2008;4:e1000073. doi: 10.1371/journal.pcbi.1000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Han W, Butz S, Liu X, Fernandez-Chacon R, Lao Y, Sudhof TC. Synaptotagmin VII as a plasma membrane Ca(2+) sensor in exocytosis. Neuron. 2001;30:459–473. doi: 10.1016/s0896-6273(01)00290-2. [DOI] [PubMed] [Google Scholar]
- Sugita S, Shin OH, Han W, Lao Y, Sudhof TC. Synaptotagmins form a hierarchy of exocytotic Ca(2+) sensors with distinct Ca(2+) affinities. EMBO J. 2002;21:270–280. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan WE, Konishi M. Segregation of stimulus phase and intensity coding in the cochlear nucleus of the barn owl. J Neurosci. 1984;4:1787–1799. doi: 10.1523/JNEUROSCI.04-07-01787.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Moiseff A, Konishi M. Time and intensity cues are processed independently in the auditory system of the owl. J Neurosci. 1984;4:1781–1786. doi: 10.1523/JNEUROSCI.04-07-01781.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tank DW, Regehr WG, Delaney KR. A quantitative analysis of presynaptic calcium dynamics that contribute to short-term enhancement. J Neurosci. 1995;15:7940–7952. doi: 10.1523/JNEUROSCI.15-12-07940.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer JS. Subcellular localization of K+ channels in mammalian brain neurons: remarkable precision in the midst of extraordinary complexity. Neuron. 2015;85:238–256. doi: 10.1016/j.neuron.2014.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto T, Jeromin A, Saitoh N, Roder JC, Takahashi T. Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science. 2002;295:2276–2279. doi: 10.1126/science.1068278. [DOI] [PubMed] [Google Scholar]
- Tucker WC, Edwardson JM, Bai J, Kim HJ, Martin TF, Chapman ER. Identification of synaptotagmin effectors via acute inhibition of secretion from cracked PC12 cells. J Cell Biol. 2003;162:199–209. doi: 10.1083/jcb.200302060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek J, Jackman SL, Regehr WG. Synaptic Specializations Support Frequency-Independent Purkinje Cell Output from the Cerebellar Cortex. Cell reports. 2016;17:3256–3268. doi: 10.1016/j.celrep.2016.11.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubach J, Zhang X, Shao X, Sudhof TC, Rizo J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J. 1998;17:3921–3930. doi: 10.1093/emboj/17.14.3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R. Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem. 2012;287:16447–16453. doi: 10.1074/jbc.M112.343418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G, Thutupalli S, Risselada JH, Meyenberg K, Holt M, Riedel D, Diederichsen U, Herminghaus S, Grubmuller H, Jahn R. Synaptotagmin-1 may be a distance regulator acting upstream of SNARE nucleation. Nature structural & molecular biology. 2011;18:805–812. doi: 10.1038/nsmb.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyleta NP, Jonas P. Loose coupling between Ca2+ channels and release sensors at a plastic hippocampal synapse. Science. 2014;343:665–670. doi: 10.1126/science.1244811. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- Wang S, Li Y, Ma C. Synaptotagmin-1 C2B domain interacts simultaneously with SNAREs and membranes to promote membrane fusion. eLife. 2016;5 doi: 10.7554/eLife.14211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Linhoff MW, McGinley MJ, Li GL, Corson GM, Mandel G, Brehm P. Distinct roles for two synaptotagmin isoforms in synchronous and asynchronous transmitter release at zebrafish neuromuscular junction. Proc Natl Acad Sci U S A. 2010;107:13906–13911. doi: 10.1073/pnas.1008598107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Sudhof TC. Synaptotagmin-1, -2, and -9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–581. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, Sudhof TC. Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron. 2012;73:990–1001. doi: 10.1016/j.neuron.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Ma C, Craig TK, Rosenmund C, Rizo J. The Janus-faced nature of the C(2)B domain is fundamental for synaptotagmin-1 function. Nature structural & molecular biology. 2008;15:1160–1168. doi: 10.1038/nsmb.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada WM, Zucker RS. Time course of transmitter release calculated from simulations of a calcium diffusion model. Biophys J. 1992;61:671–682. doi: 10.1016/S0006-3495(92)81872-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Leal K, Magupalli VG, Nanou E, Martinez GQ, Scheuer T, Catterall WA. Modulation of CaV2.1 channels by neuronal calcium sensor-1 induces short-term synaptic facilitation. Mol Cell Neurosci. 2014;63:124–131. doi: 10.1016/j.mcn.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zador A. Impact of synaptic unreliability on the information transmitted by spiking neurons. J Neurophysiol. 1998;79:1219–1229. doi: 10.1152/jn.1998.79.3.1219. [DOI] [PubMed] [Google Scholar]
- Zengel JE, Magleby KL. Augmentation and facilitation of transmitter release. A quantitative description at the frog neuromuscular junction. J Gen Physiol. 1982;80:583–611. doi: 10.1085/jgp.80.4.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE, et al. Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature. 2015;525:62–67. doi: 10.1038/nature14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Exocytosis: a molecular and physiological perspective. Neuron. 1996;17:1049–1055. doi: 10.1016/s0896-6273(00)80238-x. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]