

Abstract
The histone deacetylase family comprises 18 enzymes that catalyze deacetylation of acetylated lysine residues, however, the specificity and substrate profile of each enzyme remains largely unknown. Due to transient enzyme-substrate interactions, conventional co-immunoprecipitation methods frequently fail to identify enzyme-specific substrates. Additionally, compensatory mechanisms often limit the ability of knockdown or chemical inhibition studies to achieve significant fold-changes observed by acetylation proteomics methods. Furthermore, measured alterations do not guarantee a direct link between enzyme and substrate. Here we present a chemical crosslinking strategy that incorporates a photo-reactive, non-natural amino acid, p-benzoyl-L-phenylalanine, into various positions of the structurally characterized isozyme histone deacetylase 8 (HDAC8). After covalent capture, co-immunoprecipitation, and mass spectrometric analysis, we identified a subset of HDAC8 substrates from human cell lysates, which were further validated for catalytic turnover. Overall, this chemical-crosslinking approach identified novel HDAC8 specific substrates with greater catalytic efficiency, thus presenting a general strategy for unbiased deacetylase substrate discovery.
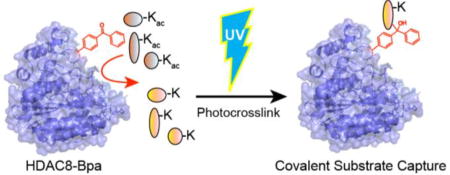
Graphical Abstract
INTRODUCTION
Lysine acetylation is an essential post-translational modification involved in chromatin remodeling1, protein-protein interactions, protein stability, and protein-DNA interactions.2 Lysine acetyltransferases (KATs) catalyze acetyl group transfer from acetyl-CoA to select lysine residues in proteins, which can then be catalytically removed by either NAD-dependent sirtuins (class III) or the metal-dependent Class I (HDACs 1, 2, 3, and 8), class II (HDACs 4, 5, 6, 7, 9, and 10) or class IV (HDAC 11) histone deacetylases (HDACs). Parsing deacetylase-substrate interactions remains a major challenge, yet is critical to understand the function and potential outcomes of isoform-selective inhibitors.
Active site mutants of HDAC1 can be used as substrate traps for affinity purification of bound substrates3. Using this approach, HDAC1 was demonstrated to deacetylate the kinesin motor protein Eg5, which is required for cell cycle progression. Other studies have attempted to indirectly map deacetylase-substrate interactions, typically by inhibitor-dependent accumulation profiled by anti-acetyl-lysine affinity purification and mass spectrometry4. Conversely, direct affinity purification of select HDAC-GFP fusions reported a number of interactions across HDAC isoforms5, yet transient HDAC-substrate interactions are likely lost during enrichment. Thus, epitope-tagged co-immunoprecipitation can report HDAC protein-protein interactions, but is less likely to identify low affinity HDAC substrates.
While resin-bound HDAC inhibitors can be used to identify native HDAC complexes, binding to the resin-linked inhibitor requires substrate displacement6. Therefore, new methods capable of capturing transient HDAC-substrate interactions are needed to accelerate substrate discovery and function of each HDAC isoform. Ideally, newly developed methods should require a small surface area to capture interactions while simultaneously being compatible with current mass spectrometry and proteomic approaches.
The Class I metal-dependent deacetylase HDAC8 has been crystallized with a wide variety of ligands and peptides, yet HDAC8 substrates remain largely unknown7. To date, only a few HDAC8 substrates have been identified and validated in vivo. The first HDAC8 substrate, ERR-α, was identified using in vitro radiolabeling and deacetylation assays8. The second validated HDAC8 substrate, SMC3, was identified through human genetics, since HDAC8 missense mutations increase acetylation of the SMC3-cohesion complex and development of Cornelia de Lange Spectrum disorders9. A recent quantitative proteomic study identified HDAC8 substrates by treating cells with an HDAC8-specific inhibitor, followed by immuno-enrichment of acetylated peptides for quantitative proteomics4. Several putative substrates were identified, including AT-rich interactive domain-containing protein 1A (ARID1A), cysteine-rich protein 2-binding protein (CSRP2BP), and nuclear receptor coactivator 3 (NCOA3)4. Furthermore, affinity-based chemoproteomic methods failed to assign HDAC8 to any specific protein complexes6. Clearly, affinity-based approaches have many challenges for identifying HDAC8 substrates, especially since many substrates are likely substrates of multiple HDACs.
Here, we report an alternative approach to identify candidate HDAC8 substrates using active-site directed chemical covalent capture. Our strategy leverages recent advances in genetically encoded unnatural amino acid mutagenesis. Epitope-tagged HDAC8 mutants were expressed in bacteria encoding an orthogonal tRNA/aminoacyl-tRNA synthetase for incorporation of the photoreactive non-natural amino acid p-benzoyl-L-phenylalanine (Bpa) at positions near the HDAC8 active site10. Similar approaches have been used to map protein binding interfaces11,12, identify stable interacting proteins, or to trap low affinity transcriptional protein-protein interactions.10b,13. Based on these studies, we now present the first application of unnatural amino acid mutagenesis to capture transient enzyme substrates. Mammalian cell lysates were then incubated with the recombinant protein and irradiated with ultraviolet (UV) light. After photocrosslinking, captured HDAC8 substrates were enriched by immunoprecipitation and identified by mass spectrometry. Identified HDAC8 substrates prefer adjacent aromatic residues, and achieve deacetylation rates equal or greater than that of SMC3 Lys106, the most validated HDAC8 substrate14.
EXPERIMENTAL METHODS
Purification of HDAC8-Bpa mutants
Site directed mutagenesis (QuikChange;Stratagene) was performed on a pHD2-TEV-His-HDAC8 plasmid to mutate Y100, I94, or F191 to an amber stop codon. The Methanocaldococcus jannaschii aminoacyl-tRNA sinthetases/suppressor tRNA pairs were purchased from AddGene.10c Recombinant HDAC8-Bpa variants were expressed in BL21(DE3) Escherichia coli. Cells were grown at 37°C until OD600 reached 0.4 – 0.6, at which point the temperature was lowered to 20°C. After 1 hour, cells were induced by the addition of 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG), 0.2% w/v L-arabinose, 200µM Bpa-OH (Chem-Impex Int’l) and 200µM ZnSO4, and grown overnight at 20°C. Cells were lysed using a microfluidizer (Microfluidics) followed by centrifugation (27,000 × g, 45 mins, 4°C). The cleared lysate was applied to an NiSO4-charged immobilized Sepharose fast flow metal affinity column and HDAC8-Bpa mutants were eluted with a gradient of increasing imidazole concentration (25 – 200 mM imidazole in 30 mM HEPES pH 8.0, 150 mM NaCl, 1 mM TCEP and 5 mM KCl). HDAC8-Bpa mutants were then dialyzed overnight at 4°C using a metal chelating buffer (25 mM MOPS pH 7.5, 1 mM EDTA, 1 mM TCEP and 5 mM KCl) to remove any trace metals and then dialyzed overnight against metal free buffer (25 mM MOPS pH 7.5, 1 mM TCEP and 5 mM KCl) to ensure complete trace metal removal. The final yields for each mutant were 0.5 – 0.7 mg / L of bacterial culture. HDAC8-Bpa mutants were concentrated to 150 – 300µM, flash frozen and stored at −80°C. Bpa incorporation for each mutant was verified by mass spectrometry (Supplementary Figure 1).
HDAC8 activity assay
Apo-HDAC8-Bpa (10µM) was incubated with Zn2+ at a stoichiometric ratio on ice for 1 hour prior to dilution into the assay. The catalytic activity of HDAC8-Bpa mutants was measured using the Fluor-de-Lys tetrapeptide substrate Ac-Arg-His-Lys(ac)-Lys(ac)-aminomethylcoumarin (Enzo Life Sciences). Activity assays were run at 30°C in 1x assay buffer (25 mM HEPES pH 7.8, 137 mM NaCl and 3 mM KCl) that contained 100µM peptide and 1µM HDAC8-Bpa mutant. The ratio of product fluorescence (ex, = 340 nm, em. = 450 nm) divided by the substrate fluorescence (ex. = 340 nm, em. = 380 nm) increases with product concentration. The concentration of product was determined using a standard curve and the initial rate was determined from the product formed over time. The catalytic (kcat/KM,app) was determined from dividing the initial rate by the substrate and enzyme concentrations, assuming a linear fit.
HDAC8 chemical covalent capture assay
Anti-6xHIS Epitope tag monoclonal antibodies (Thermo Scientific) (100µg/mL) were coupled to M-270 epoxy Dynabeads (Invitrogen) according to the manufacturer’s suggestion. HDAC8-Bpa mutants were diluted in PBS (pH 7.4) to 40µM and kept on ice. HEK293 cells (~10–15 million cells) were resuspended in PBS (800µL), sonicated (1 – 3 minutes, 10% duty cycle), and centrifuged (17,000×g, 25 mins, 4°C) to remove insoluble material. Each HDAC8-Bpa mutant was mixed individually with HEK293 lysate to a final volume of 140µL (HDAC8-Bpa = [5µM]). Samples were incubated at 4°C for 15 minutes and then either irradiated using an ultraviolet light lamp (Thermo Scientific) at a wavelength of 365 nm (115 volts, 60 Hz) for 20–30 minutes or kept in the dark for the same amount of time for the non-UV control samples. The length of UV light exposure was varied to optimize the gel-shift efficiency, which reached saturation after 30 minutes. The crosslinked and non-crosslinked samples were then added to the coupled beads and incubated for 1 – 2 hours with rotation at 4°C. After extensive washes, the beads were analyzed by mass spectrometry or gel electrophoresis and western blotting (anti-6xHIS monoclonal antibody (Fisher), mouse anti-HDAC8 monoclonal antibody (Santa Cruz Biotech)). Blots were imaged using a C600 Series imager (Azure Biosystems).
Mass Spectrometry and data processing
After affinity purification, washed beads were resuspended in ice-cold 6 M urea. Samples were then incubated with 10 mM dithiothreitol (DTT) for 30 minutes at 37°C, followed by alkylation using 55 mM iodoacetamide for 30 minutes at 25°C in the dark. Samples were diluted to 1 M urea using PBS and digested overnight by incubation with sequence-grade Trypsin/Lys-C (Promega). Peptide samples were desalted using Oasis HLB PrimeµElution C18 solid-phase extraction cartridges (Waters), and reconstituted in water with 3% acetonitrile, 0.1% formic acid and 10 fM/µL yeast alcohol dehydrogenase digest (Waters) as an internal standard. In triplicate, samples were analyzed in positive mode on a Synapt G2-S HDMS traveling wave ion mobility time-of-flight (TOF) mass spectrometer (Waters). Reversed-phase liquid chromatography and data-independent acquisition were performed as described.15 Peptide identification and label-free protein quantitation were performed with Progenesis QI for Proteomics 2.0.5 (Nonlinear Dynamics) against a database of the human proteome. Peptide and protein identification were made using the following criteria: Tryptic cleavage rules with one missed cleavage allowed, carbamidomethyl cysteine as a fixed modification and methionine oxidation as a variable modification, a minimum of two identified fragment ions per peptide and a minimum of five fragments per protein, and at least two identified peptides per protein. The global false discovery rate (FDR) for protein identification was set at 1% using a reversed database. Peptide identifications with a calculated mass error greater than 10 ppm were not considered.
Peptide identification and kinetic assays
All specifically enriched proteins were screened for acetylated lysine positions using PhosphoSite Plus. Any annotated acetyl-lysine positions were then evaluated using the Rosetta Flex-Pep-Bind structure-based protocol tailored to HDAC8.14a Acetylated lysine residues with the highest overall algorithm score were considered as the best candidates for in vitro peptide testing. A library of 39 desalted peptides with a single acetyl-lysine were purchased (N-terminal N-acetylation, C-terminal amidation, >75% purity) (Synthetic Biomolecules). HDAC8 activity assays were performed using an enzyme-coupled assay to measure acetate production. 23 Reactions were performed using a standard HDAC reaction buffer (25 mM HEPES pH 7.8, 137 mM NaCl, 3 mM KCl at 30°C). Reactions measuring deacetylation of acetylated peptides (12.5 – 100µM) were initiated by the addition of recombinant Zn(II)-HDAC8 (1µM). The reactions were quenched with 10% HCl, and the acetate product, as reflected by an increase in NADH fluorescence, was measured at 5 time points (up to 50 mins) and used to calculated the initial rates. The steady state kinetic parameter (kcat/KM) was calculated by fitting a line to the dependence of the initial rate on the peptide concentration. For peptides that demonstrated curvature, namely ITGB1 and UPF1, a Michaelis-Menten equation was fit to the data resulting in kcat values of 0.01 ± 0.002 and 0.009 ± 0.002, respectively.
RESULTS AND DISCUSSION
Expression and catalytic activity of HDAC8 Bpa mutants
HDAC8 mutants were designed using amber stop codon nonsense suppression in E. coli10a for site-specific Bpa incorporation, recombinant expression, and purification. Three solvent exposed positions (Y100, I94 and F191) were selected in HDAC8 for Bpa mutagenesis (Figure 1A). Y100 is located in immediate proximity to the active site tunnel and has been shown to interact with a methylcoumarin-bound peptide substrate, while I94 is located on a disordered loop proposed to aid in substrate binding and release7, 14. F191 is located 24 Å away from the active site tunnel, providing an important control for non-specific or distal protein-protein interactions. Since Bpa can undergo repeated cycles of excitation before crosslinking, we hypothesized its incorporation would increase the likelihood of successful substrate capture, although aryl-azides and diazirines were not tested in this study. After expression and purification, efficient incorporation was confirmed by high resolution mass spectrometry (Supplementary Figure 1). Each enzyme retained catalytic activity with mutants showing only modest effects on catalytic turnover (Figure 1B).
Figure 1.

Bpa incorporation in HDAC8 does not dramatically affect hydrolytic activity. (A) Positions for Bpa incorporation were chosen based on proximity to a coumarin-linked peptide substrate (Ac-Arg-His-Lys(ac)-Lys(ac)-methylcoumarin) visualized in the co-crystal structure of HDAC8 (PDB ID: 2V5W). (B) Measurement of the activity of HDAC8-Bpa mutants assayed using Fluor-de-Lys assay with a coumarin-linked peptide substrate. Assays were performed using 50µM peptide and 1µM Co2+-HDAC8. The value of kcat/Km (app) was calculated assuming a linear dependence on peptide concentration.
Identification of HDAC8-Bpa active-site proximal proteins
Bacterially expressed Bpa mutants were incubated with human embryonic kidney cell (HEK293) lysates with or without ultraviolet (UV) irradiation and analyzed by western blot (Figure 2). Several high molecular weight bands formed after UV irradiation, indicating covalent capture of a number of HDAC8-Bpa interacting proteins or crosslinking between HDAC8 monomers. Based on these results, HDAC8-Bpa mutants (+/− UV) were immunoprecipitated, digested with trypsin, and analyzed by mass spectrometry. Using data-independent acquisition (DIA) methods and label-free quantitation, cutoffs were applied where proteins showed: (i) an overall fold enrichment (+UV/−UV) of ≥ 2.5-fold; (ii) a p-value of ≤ 0.05 (+UV/−UV); and (iii) at least two peptide annotations per protein (Figure 3A and Supplementary Tables 1–3). Analogous to SWATH methods16, our ion mobility-resolved DIA approach ensures reproducible acquisition across replicate datasets, and provides cross extraction of missed features for enhanced statistical analysis15, 17. In addition, data was normalized to the total level of HDAC9-Bpa, providing a second filter to reduce run to run variance and increase statistical confidence. Importantly, since HDAC8 may catalyze deacylation of a variety of short or long chain fatty acyl groups18, this method is agnostic to the specific modification.
Figure 2.

Anti-HDAC8 western blot of wild type and HDAC8-Y100Bpa incubated in HEK293 cell lysates with or without exposure to UV.
Figure 3.

Mass spectrometry analysis of UV-dependent HDAC8-interacting proteins. (A) Analysis of –UV and + UV enrichment for each HDAC8 Bpa mutant. Colored data points in the greyed quadrant were specifically enriched (p ≤ 0.05, ratio ≥ 2.5, peptides / protein ≥ 2) and assigned as putative UV-dependent interaction partners. (B) Venn diagram comparing the enrichment of candidate crosslinked proteins shows little overlap between different sites of incorporation. (C) Sequence alignment of predicted acetylated sites enriched by HDAC8 covalent substrate capture reveals preference for aromatic residues.
Overall, Y100Bpa captured 119 proteins, while I94Bpa and F191Bpa identified a small (<10) non-overlapping subset (Figure 3B). Furthermore, ≥ 90% of proteins crosslinked with Y100Bpa have one or more acetylation sites listed in the Phosphosite Plus database, presenting a shortlist of candidate substrates. Based on these findings, HDAC-Bpa enriched a number of previously identified HDAC8 interacting proteins such as SMC1A5, SMC2/35, CPNE35, TPM35, TPM45 and SRSF54, as well as a large subset of putative substrates. These previously identified substrates did not meet either our established cutoffs for the +UV/−UV ratio, p-value < 0.05 or both. Since our immunoenrichment was performed under native conditions, substrates with long-lived, stable interactions are likely triaged based on the cutoff thresholds. For example, SMC3 was identified with +UV/−UV ratios < 2.5, demonstrating its affinity enrichment proceeds without covalent capture due to stable HDAC8 interactions (Supplementary Table 6). Although ARID1A and CSRP2BP identified as HDAC8 substrates in MCF7 cells, they were not observed in this dataset, which could reflect lower expression levels in HEK 293 cells, differential HDAC regulation, or poor crosslinking efficiency.4
Validation of putative HDAC8 substrate peptides
In order to validate whether the Y100Bpa-enriched proteins are HDAC8 substrates, the 119 candidate proteins were evaluated using the Rosetta Flex-Pep-Bind algorithm14a. This computational docking algorithm has been tailored for Zn2+-bound HDAC8 using inactive crystal structures. The algorithm defines the interaction strength of 6-mer peptides on the surface binding area of HDAC8 through a fixed-backbone design and minimization of the peptide structure to its lowest energy conformation, returning a reactivity score for each annotated acetylated lysine from Phosphosite (Supplementary Table 4). This algorithm has been optimized and validated through multiple iterations of experimental peptide validation and it has computationally predicted HDAC8 non-histone substrates14a. Based on these results, we purchased and assayed 39 water soluble synthetic peptides corresponding to the acetylated lysine sites with the largest fold change (+UV/−UV ratio) and the highest Flex-Pep-Bind reactivity score. HDAC8-catalyzed deacetylation was measured using an acetate-coupled assay19 with zinc-bound HDAC8 at a single substrate concentration (100µM) (Supplementary Table 4). Even as small synthetic peptides, ~30% of the peptides reported estimated kcat/KM (app) values greater than 30 M−1s−1 (Supplementary Table 5).
Peptides with kcat/KM (app) values higher than 30 M−1s−1 were then assayed as a function of peptide concentration. Importantly, nearly all (8 of 9) of the reported kcat/KM values are comparable to or faster than a peptide from SMC3, a validated, in vivo HDAC8 substrate4 (Table 1). This set includes peptides derived from previously reported HDAC8 interacting proteins, including heat shock protein 90 (HSP90AB1)20, tubulin 1-alpha (TUBA1A)21 and transcription intermediary factor beta (TRIM28)22. This analysis also validated new putative HDAC8 protein substrates peptides, including peptides from ATP-citrate lyase (ACLY) (240 ± 55 M−1s−1), integrin beta-1 (ITGB1) (435 ± 150 M−1s−1), 6-phosphofructokinase (PFKP) (128 ± 110 M−1s−1), PDZ and LIM domain protein 1 (PDLIM1) (106 ± 48 M−1s−1), and the regulator of nonsense transcripts (UPF1) (350 ± 130 M−1s−1).
Table 1.
HDAC8 deacetylation of candidate substrate peptides. Catalytic efficiency for the deacetylation of synthetic acetylated peptides corresponding to a subset of identified HDAC8 substrates. These peptides demonstrated kcat/KM values equal to or faster than a validated SMC3 synthetic peptide (63 ± 11 M−1 s−1).14
| Protein | Ac-Sequence-NH2 |
kcat/KM (M−1 s−1) |
|---|---|---|
| HSP90AB1 | YKKacFYE | 120 ± 40 |
| ACLY | DHKQKacFYWGHK | 240 ± 55 |
| TUBA1A | DHKacFDL | 60 ± 13 |
| ITGB1 | TLKacFKR | 435 ± 150 |
| PFKP | HRIPKacEQW | 128 ± 110 |
| UPF1 | RYKGDLAPLWKacGIGVIKVPD | 350 ± 130 |
| TRIM28 | RMFKacQFNK | 70 ± 3 |
| PDLIM1 | GGKacDFE | 106 ± 48 |
| SMC3 | RVIGAKKacDQY | 63 ± 11 |
Consistent with previous in vitro peptide studies, HDAC8 prefers peptide substrates with aromatic phenylalanine and tyrosine near the acetyl-lysine14a, 23 (Figure 3C). The excited benzophenone diradical species can preferentially abstract the hydrogen atom adjacent to a heteroatom, as observed with methionine residues24. Since there is no specific enrichment of adjacent methionine residues, we hypothesize that substrate recognition drives association and subsequent photocrosslinking. Nonetheless, the homolytic bond dissociation energy for the benzylic C–H bonds present in the aromatic amino acids are lower than the typical C–H bonds. Accordingly, the photoexcited benzophenone could show preferential hydrogen abstraction from the weakly activated benzylic position to enhance covalent capture. Since the FlexPepBind analysis and peptide studies do not incorporate the Y100Bpa mutation, non-native Bpa-specific aromatic interactions are unlikely to impart additional substrate affinity, although aromatic stacking may enhance substrate recognition. Of eight high activity peptide substrates, six contain a phenylalanine at the +1 or +2 position. This emergent motif likely increases binding affinity similar to the observed interactions with the +1 coumarin moiety visualized in the HDAC8-peptide crystal structures14b, 25. Based on these findings, we hypothesize that the presence of an aromatic residue downstream of the acetyl-lysine further helps define HDAC8 substrate preference.14a
CONCLUSIONS
Here we present a chemical strategy to identify enzyme-substrate pairs in complex lysates. This approach is validated across a subset of HDAC8-Bpa mutants, capturing more than 100 candidate active site-engaged proteins. Acetylated peptides identified in this study possess deacetylation rates comparable to an SMC3 peptide mimic, an in vivo validated HDAC8 protein substrate. These newly identified proteins possess a diverse array of both nuclear and cytosolic functions, including cell regulation and homeostasis, chromatin and DNA remodeling and cell cytoskeleton regulation, suggesting that HDAC8 might have a role in various cellular processes not investigated previously. While this in vitro covalent capture strategy is biased towards highly abundant proteins in cell lysates out of their cellular context, future studies in live mammalian cells may provide a more representative profile of HDAC8 substrates. HDAC8 is genetically linked to the SMC3-cohesion complex, yet HDAC8 is predicted to contribute in other biological contexts, which may or may not be redundant with other enzymes. While our approach captures active-site engaged substrates, it does not distinguish selective from promiscuous substrates. Due to predicted redundancy, genetic or pharmacological inhibition of specific HDAC enzymes will not necessarily lead to substrate accumulation. Future studies will require incorporation of acetyl-lysine within select captured proteins, followed by in vitro validation with purified HDAC8 enzyme.
In summary, this strategy adds to the growing list of alternative methods for identification of low affinity protein-substrate and protein-protein interactions such as catalytic mutant affinity enrichment, acetyl-lysine quantitative proteomics, and other to build a more comprehensive mapping network of the acetylome. Finally, since Y100 and/or an aromatic residue is conserved in the primary catalytic domain across multiple metal-dependent deacetylases, (i.e. HDAC6, HDAC10, HDAC11), the same experiment could be translated to identify substrates for other HDAC isozymes. (Supplemental Figure 2) We anticipate that this approach will provide a better understanding of how substrates form transient and long-lived interactions with HDAC enzymes, and provide a new method to correlate active-site proximity with in vitro substrate profiling.
Supplementary Material
Acknowledgments
We would like to thank Anna Mapp (Michigan) for assistance with unnatural amino acid incorporation, Rachel Pricer (Michigan) for assistance with graphical design, and Hashim Motiwala (Michigan) for helpful discussions. Financial support for these studies was provided by the National Institutes of Health F31 GM116619 (J.E.L.), T32 GM008597 (S.E.H.), R01 GM040602 (C.A.F.), DP2 GM114848 (B.R.M.), the American Heart Association 14POST20420040 (J.D.M.), and the University of Michigan.
Footnotes
ASSOCIATED CONTENT
- Supplementary Tables 1–3 (Excel)
References
- 1.Parbin S, Kar S, Shilpi A, Sengupta D, Deb M, Rath SK, Patra SK. J. Histochem. Cytochem. 2014;62(1):11–33. doi: 10.1369/0022155413506582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Glozak MA, Sengupta N, Zhang X, Seto E. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 3.Nalawansha DA, Gomes ID, Wambua MK, Pflum MKH. Cell Chem. Biol. 2017;24(4):481–492. e5. doi: 10.1016/j.chembiol.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olson DE, Udeshi ND, Wolfson NA, Pitcairn CA, Sullivan ED, Jaffe JD, Svinkina T, Natoli T, Lu X, Paulk J, McCarren P, Wagner FF, Barker D, Howe E, Lazzaro F, Gale JP, Zhang YL, Subramanian A, Fierke CA, Carr SA, Holson EB. ACS Chem. Biol. 2014;9(10):2210–6. doi: 10.1021/cb500492r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joshi P, Greco TM, Guise AJ, Luo Y, Yu F, Nesvizhskii AI, Cristea IM. Mol. Syst. Biol. 2013;9:672. doi: 10.1038/msb.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dumpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G. Nat. Biotechnol. 2011;29(3):255–65. doi: 10.1038/nbt.1759. [DOI] [PubMed] [Google Scholar]
- 7.Wolfson NA, Pitcairn CA, Fierke CA. Biopolymers. 2013;99(2):112–26. doi: 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson BJ, Tremblay AM, Deblois G, Sylvain-Drolet G, Giguere V. Mol. Endocrinol. 2010;24(7):1349–58. doi: 10.1210/me.2009-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Deardorff MA, Bando M, Nakato R, Watrin E, Itoh T, Minamino M, Saitoh K, Komata M, Katou Y, Clark D, Cole KE, De Baere E, Decroos C, Di Donato N, Ernst S, Francey LJ, Gyftodimou Y, Hirashima K, Hullings M, Ishikawa Y, Jaulin C, Kaur M, Kiyono T, Lombardi PM, Magnaghi-Jaulin L, Mortier GR, Nozaki N, Petersen MB, Seimiya H, Siu VM, Suzuki Y, Takagaki K, Wilde JJ, Willems PJ, Prigent C, Gillessen-Kaesbach G, Christianson DW, Kaiser FJ, Jackson LG, Hirota T, Krantz ID, Shirahige K. Nature. 2012;489(7415):313–7. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Decroos C, Bowman CM, Moser JA, Christianson KE, Deardorff MA, Christianson DW. ACS Chem. Biol. 2014;9(9):2157–64. doi: 10.1021/cb5003762. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Decroos C, Christianson NH, Gullett LE, Bowman CM, Christianson KE, Deardorff MA, Christianson DW. Biochemistry. 2015;54(42):6501–13. doi: 10.1021/acs.biochem.5b00881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Chin JW, Martin AB, King DS, Wang L, Schultz PG. Proc Natl Acad Sci U S A. 2002;99(17):11020–4. doi: 10.1073/pnas.172226299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Majmudar CY, Lee LW, Lancia JK, Nwokoye A, Wang Q, Wands AM, Wang L, Mapp AK. J. Am.Chem. Soc. 2009;131(40):14240–2. doi: 10.1021/ja904378z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Young T, Ahmad I, Yin J, Schultz PG. J. Mol. Biol. 2010;395(2):361–374. doi: 10.1016/j.jmb.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 11.Das D, Oliver DB. J. Biol. Chem. 2011;286(14):12371–12380. doi: 10.1074/jbc.M110.182931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohibullah N, Hahn S. Genes Dev. 2008;22:2994–3006. doi: 10.1101/gad.1724408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dugan A, Majmudar CY, Pricer R, Niessen S, Lancia JK, Fung HY, Cravatt BF, Mapp AK. J. Am.Chem. Soc. 2016;138(38):12629–35. doi: 10.1021/jacs.6b07680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Alam N, Zimmerman L, Wolfson NA, Joseph CG, Fierke CA, Schueler-Furman O. Structure. 2016;24(3):458–68. doi: 10.1016/j.str.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dowling DP, Gattis SG, Fierke CA, Christianson DW. Biochemistry. 2010;49(24):5048–56. doi: 10.1021/bi1005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernandez JL, Davda D, Majmudar JD, Won SJ, Prakash A, Choi AI, Martin BR. Mol. BioSyst. 2016;12(6):1799–1808. doi: 10.1039/c6mb00019c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Mol. Cell. Proteomics. 2012;11(6) doi: 10.1074/mcp.O111.016717. O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Distler U, Kuharev J, Navarro P, Levin Y, Schild H, Tenzer S. Nat. Methods. 2014;11(2):167–70. doi: 10.1038/nmeth.2767. [DOI] [PubMed] [Google Scholar]
- 18.Aramsangtienchai P, Spiegelman NA, He B, Miller SP, Dai L, Zhao Y, Lin H. ACS Chem. Biol. 2016;11(10):2685–2692. doi: 10.1021/acschembio.6b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolfson NA, Pitcairn CA, Sullivan ED, Joseph CG, Fierke CA. Anal. Biochem. 2014;456(1):61–9. doi: 10.1016/j.ab.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taiyab A, Rao Ch M. Biochim. Biophys. Acta. 2011;1813(1):213–21. doi: 10.1016/j.bbamcr.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 21.(a) Waltregny D, de Leval L, Glénisson W, Ly Tran S, North BJ, Bellahcène A, Weidle U, Verdin E, Castronovo V. Am. J. Pathol. 2004;165(2):553–564. doi: 10.1016/S0002-9440(10)63320-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Waltregny D, Glenisson W, Tran SL, North BJ, Verdin E, Colige A, Castronovo V. FASEB J. 2005;19(8):966–8. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- 22.Schultz DC, Friedman JR, Rauscher FJ., 3rd Genes Dev. 2001;15(4):428–43. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurard-Levin ZA, Mrksich M. Biochemistry. 2008;47(23):6242–50. doi: 10.1021/bi800053v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wittelsberger A, Thomas BE, Mierke DF, Rosenblatt M. FEBS Letters. 2006;580(7):1872–1876. doi: 10.1016/j.febslet.2006.02.050. [DOI] [PubMed] [Google Scholar]
- 25.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, Di Marco S. Proc. Natl. Acad. Sci. U. S. A. 2004;101(42):15064–9. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.