

Abstract
Periostin is a 90-kDa extracellular matrix protein, which is secreted primarily from fibroblasts and is expressed in the lungs, kidneys and heart valves. Angiotensin II (AT-II) serves pivotal roles in the pathogenesis of several diseases with accompanying fibrosis, including chronic liver diseases. AT-II induces periostin expression by regulating transforming growth factor-β1 (TGF-β1)/Smad signaling during cardiac fibrosis. The aim of the present study was to investigate the interaction between AT-II and periostin during liver fibrosis development. Fischer 344 rats were fed a choline-deficient L-amino-acid (CDAA)-defined diet for 12 weeks to simulate the development of steatohepatitis with liver fibrosis. Losartan, an AT-II type I receptor blocker, was administered to inhibit the effect of AT-II. The therapeutic effect of losartan on hepatic fibrosis development and on periostin expression was then evaluated. Several in vitro experiments were performed to examine the mechanisms underlying the interaction between AT-II and periostin in activated hepatic stellate cells (Ac-HSCs). Treatment with losartan suppressed the development of liver fibrosis induced by the CDAA diet, and reduced hepatic periostin expression. In addition, losartan treatment suppressed hepatic Ac-HSC expansion and hepatic TGF-β1 expression. In vitro analysis using LX2 HSC cells indicated that AT-II can augment TGF-β1 and collagen type I α1 mRNA expression via periostin expression, suggesting that the interaction between AT-II and periostin may serve a role in liver fibrosis development. In conclusion, blockade of AT-II-induced periostin may suppress the progression of liver fibrosis development.
Keywords: periostin, angiotensin II, liver fibrosis, renin-angiotensin system, choline-supplemented amino acid
Introduction
Liver fibrosis is a frequent and chronic clinical insult that induces wound-healing responses in the liver. It is characterized by the excessive deposition of extracellular matrix (ECM). Fibrosis is the predominant complication of chronic liver diseases that are caused by viral infection, autoimmune hepatitis, alcohol consumption, biliary obstruction and non-alcoholic fatty liver disease (1,2). The fibrotic process is characterized by the loss of parenchymal tissue and excessive accumulation of ECM, followed by activation of hepatic stellate cells (HSCs) (1,2).
The renin-angiotensin system (RAS) is a key mediator of arterial blood pressure and body fluid homeostasis; it serves an important role in the regulation of local hemodynamics in several organs (3–6), and in the progression of chronic liver diseases (3–6). In the RAS, angiotensin II (AT-II), which is an octapeptide produced by the enzymatic cleavage of angiotensin I by angiotensin I converting enzyme (ACE), serves a central role in the activation and proliferation of HSCs into activated HSCs (Ac-HSCs) (3). Previous studies have demonstrated that pharmacological inhibition of RAS effectively attenuates the development of liver fibrosis (3–7). For instance, inhibitors of the renin-angiotensin-aldosterone system, including ACE inhibitors (ACE-I), AT-II type I receptor blockers (ARBs), or selective aldosterone blockers (SABs), which are commonly used as antihypertensive agents, reportedly suppress the progression of hepatic fibrosis (4).
A previous study demonstrated that AT-II induced the expression of tissue inhibitor of metalloproteinase 1 (TIMP-1), which is a central molecule involved in fibrosis development (7). TIMP-1 was induced in Ac-HSCs, and this occurred via the AT-II type I receptor and phosphokinase C. In addition, a number of previous studies have indicated that the levels of periostin, a novel 90-kDa ECM protein, were reported to increase in a rat model of cardiac fibrosis development induced by continuous AT-II infusion, suggesting an interaction between AT-II and periostin (8,9). Periostin is secreted primarily from osteoblasts and fibroblasts and is expressed under normal conditions in the bone and to a lesser extent in the lungs, kidneys and heart valves of adult mammals (10). The effect of AT-II on periostin expression in hepatic fibrosis is currently unclear. Therefore, the aim of the present study was to explore the role of periostin in the development of liver fibrosis.
In the current study it was observed that periostin expression, which was attenuated by ARB treatment, was significantly and exclusively increased in Ac-HSCs in the choline-deficient L-amino acid (CDAA)-defined diet-induced rat model of liver fibrosis. The in vitro experiments indicated that periostin augmented transforming growth factor (TGF) β1 and collagen type 1 α1 (Col1a1) mRNA expression in human Ac-HSCs, suggesting a potential role of an AT-II-periostin interaction in liver fibrosis development.
Materials and methods
Animals and reagents
A total of 22 male Fischer 344 rats (age, 6 weeks; weight, 100–120 g) were purchased from Japan SLC, Inc. (Hamamatsu, Japan). Rats were individually housed under the following regulated conditions: Controlled temperature (23.5±2°C) and relative humidity (50±10%), with 10–15 air changes/h and 12-h light/dark cycles. The losartan ARB was purchased from Merck Sharp and Dohme (Shanghai, China). AT-II and conventional chemical agents were purchased from Nacalai Tesque, Inc. (Kyoto, Japan). Human HSCs (LX2) were obtained from the Japanese Collection of Research Bioresources Cell Bank (JCRB, Osaka, Japan).
Animal treatment
The experimental period for all animal experiments was 12 weeks. Rats were randomly divided into the following 3 groups: G1 (n=6), the negative control group where rats received a choline-supplemented amino acid (CSAA) diet (Japan SLC, Inc., Shizuoka, Japan) and normal water; G2 (n=6), a liver fibrosis model group where rats received a CDAA-defined diet (Japan SLC, Inc.) and normal water; G3 (n=10), a liver fibrosis model group where rats received a CDAA-defined diet and were administered with 30 mg/kg/day losartan in their drinking water. The animals were allowed free access to food and water throughout the acclimation and experimental periods. All rats were sacrificed at the end of the experimental period, and blood was collected via cardiac puncture. Serum markers, including albumin, bilirubin and alanine aminotransferase (ALT) were measured by SRL, Inc. (Tokyo, Japan) following sacrifice. Pieces of rat liver were cut from each of the main 3 lobes and incubated in 10% formaldehyde for liver sectioning as described below. The remainder of the liver was snap frozen in liquid nitrogen and stored at −80°C for RNA and protein analysis. All animal procedures were performed in accordance with the recommendations for the proper care and use of laboratory animals at Nara Medical University (Kashihara, Japan). The animal experiments performed in the present study were approved by the Animal Experimentation Committee of Nara Medical University.
Immunohistochemical staining and semi-quantification
In all experimental groups, liver was fixed in 10% formaldehyde for 24 h and embedded in paraffin. Liver sections were cut at 5 µM thick from the liver block. The first liver section was routinely stained with hematoxylin and eosin for histologic examination and observed by light microscope (data not shown). A separate tissue section was stained with Sirius red at Narabyouri research Co. Ltd. (Nara, Japan) for the detection of liver fibrosis. The remaining sections were used for immunohistochemical staining with antibodies against α-smooth muscle actin (αSMA; Cat. No. ab5694; Abcam, Tokyo, Japan) and periostin (Cat. No. ab14041; Abcam). Antigen retrieval was performed with enzymatic antigen retrieval solution (Cat. No. 415281; Nichirei Biosciences, Inc., Tokyo, Japan) for 15 min at 121°C. Endogenous peroxidase was blocked using 0.3% H2O2 for 15 min at room temperature, and then the sections were incubated overnight at 4°C with each primary antibody or a rabbit IgG isotypic control (Cat. No. ab27478; Abcam), diluted 1:100 in PBS + 4% goat serum (cat. No. CL1200-100; Cosmo Bio Co., Ltd., Tokyo, Japan). The slides were then incubated with MAX-PO Multi secondary antibody (cat. No. 424151; Nichirei Biosciences, Inc.) for 60 min at 37°C, and subsequently stained with 3,3′-diaminobenzidine solution for 45 sec at 37°C. Sections were then counterstained with hematoxylin solution for 10 sec at room temperature. To quantify Sirius red, periostin and αSMA positive areas, 10 independent fields (magnification, ×100) were semi-quantified using ImageJ software ver.1.45 (National Institutes of Health, Bethesda, MD, USA) and the mean ± standard deviation (SD) were calculated as described previously (10).
Cell culture and WST-1 assay
Human LX2 HSCs (3×103 cells/200 µl) were seeded into a 96-well plate and cultured in Dulbecco's Modified Eagle's Medium (Nacalai Tesque, Inc.) supplemented with 10% fetal bovine serum (Cat. No. FB-1365/500; Biosera, Kansas, USA) and 0.01% penicillin-streptomycin-L-glutamine solution in a 5% CO2 humidified atmosphere at 37°C. The growth medium was refreshed every other day. Cell viability was investigated using a WST-1 assay (cat. No. MK400; Premix WST-1 Cell Proliferation Assay System; Takara Bio, Inc., Otsu, Japan) in the presence of periostin and/or losartan, according to the manufacturer's protocol. Briefly, cells were incubated for 24 h before the addition of media containing periostin (0.1 or 1.0 µg/ml; Cat. No. C-60045; PromoCell GmbH, Heidelberg, Germany) or Losartan (10 µM; Cat. No. 12353-04; Nacalai Tesque, Inc.). Cells were incubated for a further 48 h and then analyzed using a WST-1 assay. WST positivity was assessed using a plate reader at 450 nm.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from liver tissue samples and LX2 cells using an RNeasy RNA extraction kit (Qiagen GmbH, Hilden, Germany). Total RNA (1 µg) was reverse transcribed to cDNA using a High Capacity RNA-to-cDNA kit (Cat. No. 4387406; Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer's protocol. The mRNA levels of TGF-β1, Col1a1 and periostin in the liver and HSC-LX2 cells were measured by qPCR using a StepOnePlus™ Real-Time PCR system (Applied Biosystems; Thermo Fisher Scientific, Inc.) as described previously (11). The primer sequences were as follows: GAPDH, forward, 5′-CGACCACTTTGTCAAGCTCA-3′, and reverse, 5′-AGGGGAGATTCAGTGTGGTG-3′; TGF-β1, forward, 5′-CGGCAGCTGTACATTGACTT-3′, and reverse, 5′-AGCGCACGATCATGTTGGAC-3′; Col1a1, forward, 5′-AGCTCCTGGGCCTATCTGATGA-3′, and reverse, 5′-AATGGTGCTCTGAAACCCTGATG-3′; periostin, forward, 5′-GCCCAATTAGGCTTGGCATC-3′, and reverse, 5′-GTTTCCAGTATTTGCCCGTTGTA-3′. The thermal cycling conditions employed were as described previously (11). Semi-quantification of the mRNA expression level was performed according to the 2−ΔΔCq method as described previously (12).
Expression of periostin in the liver
The periostin protein levels in the liver tissues from rats in each experimental group were measured using an enzyme-linked immunosorbent assay kit (Cat. No. SEH339Ra; Cloud-Clone Corp., Katy, TX, USA) according to the manufacturer's protocol. Protein was extracted from frozen liver using T-PER Tissue Protein Extraction Regent with HALT™ protease inhibitor cocktail (both from Thermo Fisher Scientific, Inc.). The protein concentration in each sample were measured at 280 nm using a NanoDrop™ Lite (Thermo Fisher Scientific, Inc.) and 100 µg protein was used for the assessment of periostin protein levels.
Statistical analysis
Results were presented as the mean ± SD. To assess the statistical significance of inter-group differences, one-way analysis of variance was performed followed by Bonferroni's multiple comparison tests using software EZR version 3.1.2 (13). Bartlett's test was subsequently performed to determine the homology of variance. P<0.05 was considered to indicate a statistically significant difference.
Results
Clinical parameters
Information regarding the number of rats in each group, their final body weights and relative liver weights in all experimental groups is provided in Table I. CDAA-fed rats in groups G2 and G3 exhibited significantly higher serum ALT levels, lower body weights and higher liver/body weight rates when compared with rats in the G1 group. This is consistent with the results described previously (11). Losartan-treated G3 rats did not exhibit significantly different clinical parameters from the rats in the G2 group (Table I).
Table I.
Physical and serum parameters of rats in the three experimental groups.
| Parameter | G1 | G2 | G3 |
|---|---|---|---|
| Number of rats | 6 | 6 | 10 |
| Body weight (g) | 325.1±8.5 | 239±10.5b | 244±16.4b |
| Liver/body weight (%) | 9.9±0.05 | 11±0.09a | 11.1±0.05a |
| Alb (g/dl) | 4.4±0.1c | 4.1±0.1 | 4.3±0.1c |
| Total bilirubin (mg/dl) | 0.06±0.006 | 0.13±0.01b | 0.12±0.03b |
| ALT (IU/l) | 57±10 | 245±50b | 300±31b |
Data are presented as the mean ± standard deviation.
P<0.05
P<0.01 vs. G1
P<0.05 vs. G2.G1, choline-supplemented amino acid diet fed control rats; G2, CDAA-defined diet fed rats; G3, losartan-treated CDAA diet-fed rats; Alb, serum albumin; LDH, lactate dehydrogenase; ALT, alanine aminotransferase; CDAA, choline-deficient L-amino acid.
Losartan reduces liver fibrosis development
The effects of clinically comparable doses of losartan on the development of liver fibrosis were investigated (3). The CDAA diet induced extensive fibrosis and severe steatohepatitis (Fig. 1). Treatment of rats in the G3 group with losartan resulted in significant attenuation of liver fibrosis when compared with the losartan-untreated G2 group (P<0.01); however the level of steatosis was unaffected (Fig. 1). No liver fibrosis development was observed in the CSAA diet-fed control rats in the G1 group (Fig. 1). Immunohistochemical analysis of αSMA was performed to confirm the inhibitory effect of losartan on HSC activation during the development of liver fibrosis. The number of Ac-HSCs, which express αSMA (1,2), were significantly reduced in the livers of the losartan-treated rats in the G3 group when compared with untreated rats in the G2 group (P<0.01; Fig. 2).
Figure 1.

Losartan reduces liver fibrosis in rats. (A) Representative photomicrographs of fibrosis in liver sections from rats in the G1, G2 and G3 experimental groups, as determined by Sirius red staining (magnification, ×100). Fibrosis is not observed liver tissues from rats in the G1 group; however, extensive fibrosis accompanied by severe steatosis is evidence in rats from the G2 group. A reduction in liver fibrosis is observed in rats from the G3 group when compared with the G2 group. (B) Semi-quantitative analysis of the area of liver fibrosis among the different experimental groups. The results are presented as the mean ± standard deviation (G1 and G2, n=6; G3, n=10). *P<0.01, as indicated. G1, choline-supplemented amino acid diet-fed control rats; G2, CDAA diet-fed rats; G3, losartan-treated CDAA diet-fed rats; CDAA, choline-deficient L-amino acid.
Figure 2.

Effect of losartan on Ac-HSC expansion during liver fibrosis development. (A) Immunohistochemical analysis αSMA expression in the liver tissues of rats from the G1, G2 or G3 experimental groups (magnification, ×100). (B) Semi-quantification of αSMA expression in the liver. The results are presented as the mean ± standard deviation (G1 and G2, n=6; G3, n=10). *P<0.01, as indicated. Ac-HSC, activated hepatic stellate cells; αSMA, α smooth muscle actin; G1, choline-supplemented amino acid diet-fed control rats; G2, CDAA diet-fed rats; G3, losartan-treated CDAA diet-fed rats; CDAA, choline-deficient L-amino acid.
Immunohistochemistry was used to examine the levels of periostin in the experimental models, in order to investigate the association between liver fibrosis development and periostin. The CDAA diet significantly induced periostin expression along the fibrotic septa when compared with the CSAA diet (Fig. 3). Compared with rats in the G2 group, those in the G3 group exhibited a significant reduction in periostin expression in the regions of fibrosis (P<0.05; Fig. 3). No periostin expression was observed in rats from the G1 group.
Figure 3.
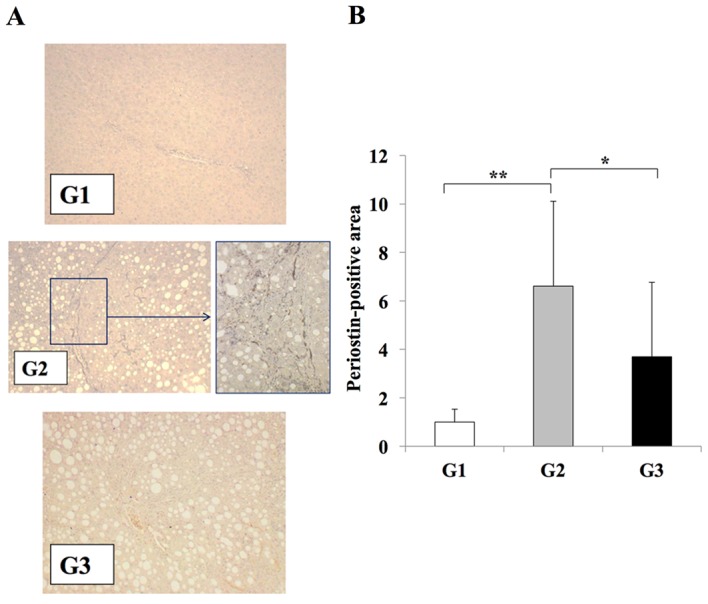
Effects of losartan on periostin expression during liver fibrosis. (A) Immunohistochemical analysis of periostin expression in the liver tissues of rats from G1, G2 and G3 experimental groups (magnification: G1, G3, ×100; G2, ×100 (left) and ×400 (right) demonstrated that periostin expression was induced in G2 group rats exclusively along the fibrotic septa. (B) Semi-quantification of the periostin-positive area using an image analyzer system. A significant reduction in periostin expression was observed in the liver tissues of rats in the G3 group when compared with the G2 group, whereas periostin expression was weak in rat liver tissues from the G1 group. The results are presented as the mean ± standard deviation (G1 and G2, n=6; G3, n=10). *P<0.05 and **P<0.01, as indicated. G1, choline-supplemented amino acid diet-fed control rats; G2, CDAA diet-fed rats; G3, losartan-treated CDAA diet-fed rats; CDAA, choline-deficient L-amino acid.
RT-qPCR analysis indicated an inhibitory effect of losartan on the expansion of Ac-HSCs. This inhibitory effect was demonstrated by a decrease in the mRNA expression levels of Tgfb1 and Col1a1, which are representative markers of Ac-HSC expansion and directly correlate with liver fibrosis development (3) (Fig. 4). In addition, it was revealed that hepatic periostin RNA and protein levels were significantly increased in rats from the G2 group when compared with the G1 group (Fig. 5). By contrast, rats in the G3 group exhibited a significant reduction in periostin RNA and protein levels when compared with rats in the G2 group (Fig. 5). These results indicated that periostin expression may be induced during liver fibrosis, and this effect is inhibited by treatment with the losartan ARB.
Figure 4.

Effect of losartan on the hepatic expression of profibrotic genes. The expression levels of (A) TGF-β and (B) Col1a1 mRNA in the livers of rats in the G2 group were significantly increased when compared with those in the G1 group. Treatment with 30 mg/kg/day losartan significantly suppressed the expression of TGF-β1 and Col1a1 in the liver. The results are presented as the mean ± standard deviation (G1 and G2, n=6; G3, n=10). *P<0.05 and **P<0.01, as indicated. G1, choline-supplemented amino acid diet-fed control rats; G2, CDAA diet-fed rats; G3, losartan-treated CDAA diet-fed rats; CDAA, choline-deficient L-amino acid; CDAA, choline-deficient L-amino acid; TGF, transforming growth factor; Col1a1, collagen type 1 α1.
Figure 5.

Effect of losartan on hepatic periostin expression. The expression of periostin (A) mRNA (B) protein in the livers of rats in the G2 group was significantly increased when compared with rats in the G1 group. Treatment with losartan significantly suppressed the expression of periostin in the liver of rats from the G3 group. The results are presented as the mean ± standard deviation (G1 and G2, n=6; G3, n=10). *P<0.05 and **P<0.01, as indicated. G1, choline-supplemented amino acid diet-fed control rats; G2, CDAA diet-fed rats; G3, losartan-treated CDAA diet-fed rats; CDAA, choline-deficient L-amino acid.
In vitro analysis of the effects of periostin on Ac-HSCs
To further examine the possible involvement of periostin in liver fibrosis, in vitro assays were performed using the human LX2 Ac-HSC line. Periostin mRNA expression was confirmed in LX2 cells (data not shown). The mRNA expression levels of periostin in LX2 cells was significantly enhanced following treatment with 1 µM AT-II; however, this effect was significantly attenuated by supplementation with losartan (Fig. 6). This suggests that the RAS signaling pathway may positively regulate periostin expression. The direct effect of periostin on the viability of HSCs was then investigated. Exposure to 1 µg/ml periostin significantly induced the viability of LX2 cells (Fig. 6). Notably, this was not inhibited by treatment with losartan (Fig. 7). In addition, periostin significantly promoted the expression of TGF-β1 and Col1a1 in LX2 cells when compared with controls; however, treatment with losartan demonstrated no significant effect on the expression levels of these genes when compared with periostin-only treated cells (Fig. 8). These results suggest that AT-II may positively regulate periostin expression in HSCs, and periostin may serve an important autocrine role in the viability and activation of HSCs.
Figure 6.
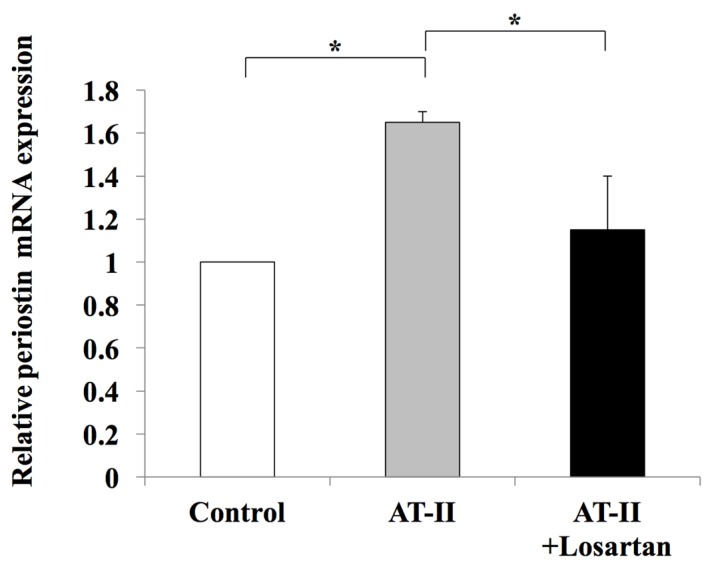
Effect of losartan on periostin mRNA expression levels. AT-II significantly induced periostin mRNA expression in hepatic LX2 stellate cells, which was abolished by losartan treatment in vitro. The results are presented as the mean ± standard deviation (n=4). *P<0.05, as indicated. AT-II, angiotensin II.
Figure 7.
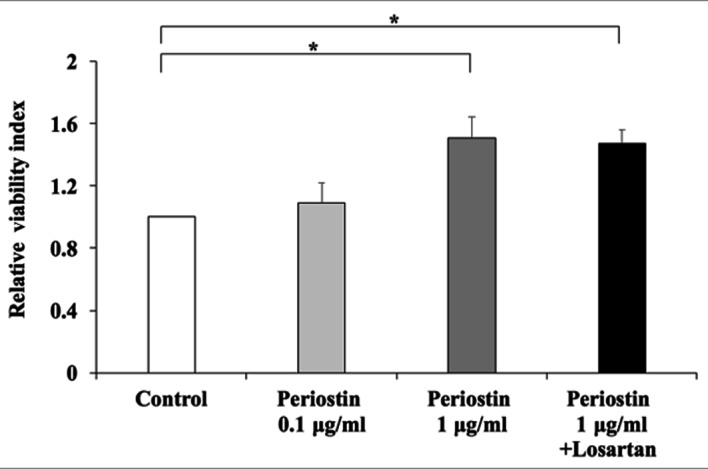
Effect of periostin on HSC viability. Periostin promoted the viability of Ac-HSCs in a concentration-dependent manner; however, this was not abolished by losartan treatment. The results are presented as the mean ± standard deviation (n=4). *P<0.05, as indicated. Ac-HSCs, activated hepatic stellate cells.
Figure 8.

Periostin induced in vitro TGF-β and Col1a1 mRNA expression in HSCs; however, losartan treatment did not reverse these increases in gene expression. The results are presented as the mean ± standard deviation (n=4). *P<0.05, as indicated. TGF-β, transforming growth factor β; Col1a1, collagen type 1 α1; HSCs, hepatic stellate cells.
Discussion
Periostin, also known as osteoblast-specific factor 2, is a recently characterized matricellular protein that binds to ECM components, such as type I collagen and fibronectin, and has been implicated in collagen fibrosis (14). This protein transmits signals from the ECM to cellular receptors to regulate a number of cellular functions, including cell adhesion, proliferation migration and tissue angiogenesis (8).
The potential involvement of AT-II-induced periostin in fibrosis development was first demonstrated in the heart (15). Cardiac fibroblasts, the predominant secretory cells that produce ECM, are important targets of AT-II and are key mediators of cardiac fibrosis (16). Continuous AT-II infusion in a rat cardiac fibrosis model demonstrated extensive interstitial fibrosis and the abundant accumulation of periostin within the interstitial space, as determined by immunofluorescence analysis (9). In cultured adult rat cardiac fibroblasts, AT-II induced the expression of periostin mRNA and protein via the Ras/p38-mitogen activated protein kinase/cyclic adenosine monophosphate responsive element binding protein 1 and extracellular signal-related kinase 1/2-TGF-β1 signaling pathways (9,17). Taken together, these results suggest the involvement of an AT-II/periostin signaling pathway in cardiac fibrosis.
A recent report demonstrated an upregulation of periostin in cirrhotic liver tissues and activated HSCs, which suggested that periostin may be a potential biomarker for hepatic fibrosis (18). In addition, previous studies have demonstrated a possible involvement of AT-II in HSC-mediated liver fibrosis development (3–6,19). A previous report demonstrated that AT-II directly stimulated HSC proliferation in vitro, and RAS inhibitors, including ACE-I, ARB and SAB, attenuated hepatic fibrosis and suppressed the proliferation of Ac-HSCs (3–6,19). These medications are commonly used antihypertensive agents that reportedly suppress the progression of hepatic fibrosis. Bataller et al (20) demonstrated that activated but not quiescent HSCs produce AT-II, which stimulate HSC proliferation in an autocrine manner. However, the association between RAS and periostin in liver fibrosis development has not yet been elucidated. The current study demonstrated that inhibition of periostin with losartan markedly inhibited the development of liver fibrosis, as evidenced by a reduced number of Ac-HSCs in the liver. These results suggested a possible interaction between the RAS and periostin in liver fibrosis development.
In the current study, in vitro analysis of LX2 cells demonstrated that AT-II was able to induce periostin expression in Ac-HSCs, and this may serve a central role in liver fibrosis. In addition, these results suggested that Ac-HSCs may be a source of periostin. In addition, periostin treatment improved viability and induced the expression of profibrotic genes TGF-β1 and Col1a1 in Ac-HSCs, which supports the hypothesis that periostin directly affects Ac-HSCs by augmenting liver fibrosis. Notably, this direct effect of periostin was not reversed by inhibition of the RAS with losartan. The authors hypothesize that the RAS may partially induce periostin expression in Ac-HSCs in an autocrine manner. Therefore, antifibrotic treatments that target periostin directly may provide an additional anti-fibrotic effect on ARB treatment. Notably, periostin has recently been investigated as a novel target for anti-fibrotic therapy in the liver (21,22). Previous reports have demonstrated that loss of periostin in mice reduced the development of experimental liver fibrosis induced by a methionine-choline-deficient diet or treatment with carbon tetrachloride (21,22). In addition, small interfering RNA-induced loss of periostin reduced the proliferation and expression of Col1a1 and αSMA mRNA in HSCs following treatment with TGF-β (18). These observations indicate the positive involvement of periostin on HSC activation and proliferation, which serves a central role in liver fibrosis. In addition, a successful anti-fibrotic treatment targeting a receptor of periostin (αV integrin), has been reported to be a potential alternative to anti-periostin treatment (23). Further examinations are required to discover novel anti-fibrotic treatment modalities targeting periostin.
In conclusion, the present study demonstrated that AT-II-induced periostin serves an important role in liver fibrosis development. Hepatic periostin expression was significantly attenuated in parallel with hepatic fibrosis development by a pharmacological blockade of the AT-II signal. These experiments indicated that AT-II can augment the activity of HSCs by the expression of TGF-β1 and collagen type I α1 via periostin expression, which may be crucial for liver fibrosis. The results of the present study suggest that periostin may function as a novel regulator of HSC activation, and targeting periostin may therefore present a useful strategy for the treatment of liver fibrosis. Further studies, which may employ a genetically altered periostin in a rodent liver fibrosis model, would allow further investigation into the role of periostin in the development of liver fibrosis.
Glossary
Abbreviations
- AT-II
angiotensin II
- TGF-β
transforming growth factor-β
- CDAA
choline-deficient L-amino-acid
- ARB
angiotensin II type 1 receptor blocker
- Col1A1
collagen type I α1
- ECM
extracellular matrix
- HSC
hepatic stellate cells
- ACE
angiotensin I converting enzyme
- Ac-HSC
activated hepatic stellate cell
- CSAA
choline-supplemented amino acid
- ALT
alanine aminotransferase
- RT-qPCR
reverse transcription quantitative polymerase chain reaction
References
- 1.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI200524282C1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, Tsujinoue H, Fukui H. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology. 2001;34:745–750. doi: 10.1053/jhep.2001.28231. [DOI] [PubMed] [Google Scholar]
- 4.Debernardi-Venon W, Martini S, Biasi F, Vizio B, Termine A, Poli G, Brunello F, Alessandria C, Bonardi R, Saracco G, et al. AT1 receptor antagonist Candesartan in selected cirrhotic patients: Effect on portal pressure and liver fibrosis markers. J Hepatol. 2007;46:1026–1033. doi: 10.1016/j.jhep.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 5.Matono T, Koda M, Tokunaga S, Sugihara T, Ueki M, Murawaki Y. The effects of the selective mineralocorticoid receptor antagonist eplerenone on hepatic fibrosis induced by bile duct ligation in rat. Int J Mol Med. 2010;25:875–882. doi: 10.3892/ijmm_00000417. [DOI] [PubMed] [Google Scholar]
- 6.Noguchi R, Yoshiji H, Ikenaka Y, Kaji K, Shirai Y, Aihara Y, Yamazaki M, Namisaki T, Kitade M, Yoshii J, et al. Selective aldosterone blocker ameliorates the progression of non-alcoholic steatohepatitis in rats. Int J Mol Med. 2010;26:407–413. [PubMed] [Google Scholar]
- 7.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Yanase K, Namisaki T, Yamazaki M, Tsujinoue H, Imazu H, Fukui H. Angiotensin-II induces the tissue inhibitor of metalloproteinases-1 through the protein kinase-C signaling pathway in rat liver fibrosis development. Hepatol Res. 2003;27:51–56. doi: 10.1016/S1386-6346(03)00160-8. [DOI] [PubMed] [Google Scholar]
- 8.Horiuchi K, Amizuka N, Takeshita S, Takamatsu H, Katsuura M, Ozawa H, Toyama Y, Bonewald LF, Kudo A. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J Bone Miner Res. 1999;14:1239–1249. doi: 10.1359/jbmr.1999.14.7.1239. [DOI] [PubMed] [Google Scholar]
- 9.Li L, Fan D, Wang C, Wang JY, Cui XB, Wu D, Zhou Y, Wu LL. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-β1 pathways in cardiac fibroblasts. Cardiovasc Res. 2011;91:80–89. doi: 10.1093/cvr/cvr067. [DOI] [PubMed] [Google Scholar]
- 10.Yoshiji H, Kuriyama S, Kawata M, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, Tsujinoue H, Fukui H. The angiotensin-I-converting enzyme inhibitor perindopril suppresses tumor growth and angiogenesis: Possible role of the vascular endothelial growth factor. Clin Cancer Res. 2001;7:1073–1078. [PubMed] [Google Scholar]
- 11.Kaji K, Yoshiji H, Ikenaka Y, Noguchi R, Aihara Y, Douhara A, Moriya K, Kawaratani H, Shirai Y, Yoshii J, et al. Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats. J Gastroenterol. 2014;49:481–491. doi: 10.1007/s00535-013-0783-4. [DOI] [PubMed] [Google Scholar]
- 12.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 13.Kanda Y. Investigation of the freely available easy-to use software ‘EZR’ for medical statics. Bone Marrow Transplant. 2013;48:452–458. doi: 10.1038/bmt.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kudo A. Periostin in fibrillogenesis for tissue regeneration: Periostin actions inside and outside the cell. Cell Mol Life Sci. 2011;68:3201–3207. doi: 10.1007/s00018-011-0784-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snider P, Hinton RB, Moreno-Rodriguez RA, Wang J, Rogers R, Lindsley A, Li F, Ingram DA, Menick D, Field L, et al. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ Res. 2008;102:752–760. doi: 10.1161/CIRCRESAHA.107.159517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown RD, Ambler SK, Mitchell MD, Long CS. The cardiac fibroblast: Therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol. 2005;45:657–687. doi: 10.1146/annurev.pharmtox.45.120403.095802. [DOI] [PubMed] [Google Scholar]
- 17.Iekushi K, Taniyama Y, Azuma J, Katsuragi N, Dosaka N, Sanada F, Koibuchi N, Nagao K, Ogihara T, Morishita R. Novel mechanisms of valsartan on the treatment of acute myocardial infarction through inhibition of the antiadhesion molecule periostin. Hypertension. 2007;49:1409–1414. doi: 10.1161/HYPERTENSIONAHA.106.080994. [DOI] [PubMed] [Google Scholar]
- 18.Hong L, Shejiao D, Fenrong C, Gang Z, Lei D. Periostin down-regulation attenuates the pro-fibrogenic response of hepatic stellate cells induced by TGF-β1. J Cell Mol Med. 2015;19:2462–2468. doi: 10.1111/jcmm.12636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Noguchi R, Yoshiji H, Ikenaka Y, Kaji K, Aihara Y, Shirai Y, Namisaki T, Kitade M, Douhara A, Moriya K, Fukui H. Dual blockade of angiotensin-II and aldosterone suppresses the progression of a non-diabetic rat model of steatohepatitis. Hepatol Res. 2013;43:765–774. doi: 10.1111/hepr.12008. [DOI] [PubMed] [Google Scholar]
- 20.Bataller R, Sancho-Bru P, Gines P, Lora JM, Al-Garawi A, Solé M, Colmenero J, Nicolás JM, Jiménez W, Weich N, et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology. 2003;125:117–125. doi: 10.1016/S0016-5085(03)00695-4. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Wu S, Xiong S, Ouyang G. Deficiency of periostin protects mice against methionine-choline-deficient diet-induced non-alcoholic steatohepatitis. J Hepatol. 2015;62:495–497. doi: 10.1016/j.jhep.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Huang Y, Liu W, Xiao H, Maitikabili A, Lin Q, Wu T, Huang Z, Nicolás JM, Jiménez W, Weich N. Matricellular protein periostin contributes to hepatic inflammation and fibrosis. Am J Pathol. 2015;185:786–797. doi: 10.1016/j.ajpath.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]