

Abstract
As the predominant thyroid cancer, papillary thyroid cancer (PTC) accounts for 75–85% of thyroid cancer cases. This research aimed to investigate transcriptomic changes and key genes in PTC. Using RNA-sequencing technology, the transcriptional profiles of 5 thyroid tumor tissues and 5 adjacent normal tissues were obtained. The single nucleotide polymorphisms (SNPs) were identified by SAMtools software and then annotated by ANNOVAR software. After differentially expressed genes (DEGs) were selected by edgR software, they were further investigated by enrichment analysis, protein domain analysis, and protein-protein interaction (PPI) network analysis. Additionally, the potential gene fusion events were predicted using FusionMap software. A total of 70,172 SNPs and 2,686 DEGs in the tumor tissues, as well as 83,869 SNPs in the normal tissues were identified. In the PPI network, fibronectin 1 (FN1; degree=31) and transforming growth factor β receptor 1 (TGFβR1; degree=22) had higher degrees. A total of 7 PPI pairs containing the non-synonymous risk SNP loci in the interaction domains were identified. Particularly, the interaction domains involved in the interactions of FN1 and 5 other proteins (such as FN1-tenascin C, TNC) had non-synonymous risk SNP loci. Furthermore, 11 and 4 gene fusion events were identified in all of the tumor tissues and normal tissues, respectively. Additionally, the NK2 homeobox 1-surfactant associated 3 (NKX2-1-SFTA3) gene fusion was identified in both tumor and normal tissues. These results indicated that TGFβR1 and the NKX2-1-SFTA3 gene fusion may be involved in PTC. Furthermore, FN1 and TNC containing the non-synonymous risk SNP loci might serve a role in PTC by interacting with each other.
Keywords: papillary thyroid cancer, transcriptome analysis, differentially expressed gene, gene fusion, single nucleotide polymorphism
Introduction
Thyroid cancer originates from parafollicular or follicular thyroid cells, and includes papillary thyroid cancer (PTC), anaplastic thyroid cancer, medullary thyroid cancer, poorly differentiated thyroid cancer and follicular thyroid cancer (1). PTC accounts for 75–85% of all thyroid cancer cases and thus is the predominant thyroid cancer (2). It often occurs in young females, and is the most common thyroid cancer in children and patients who have undergone radiation therapy to the head and neck (3).
Research has focused on the pathogenesis of PTC. For instance, point mutation of serine-threonine protein kinase B-RAF (BRAF) occurs in approximately one-third to one-half of PTC cases, and BRAF can result in the activation of the carcinogenic mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase signaling pathway (4). The BRAF mutation in PTC patients is associated with poorer clinicopathological outcomes and can be used to predict recurrence independently; therefore, the BRAF mutation may serve as a promising marker for risk assessment of PTC (5–7). Stephens et al (8) investigated the loss of heterozygosity for three single nucleotide polymorphisms (SNPs; G691S, S904S, and L769L) of ret proto-oncogene (RET) in thyroid tumor and normal tissues, and demonstrated that RET SNPs may function in sporadic PTC. Salvatore et al (9) demonstrated that both RET/PTC rearrangements and the BRAF mutation are markers of PTC and can be utilized for fine-needle aspiration in adjunction with traditional cytology. Vascular endothelial growth factor regulates cancer-associated neo-angiogenesis and progression, and its expression and polymorphisms may indicate the aggressiveness behavior of PTC (10). The A-kinase anchoring protein 9-BRAF gene fusion, which is induced by the BRAF rearrangement through paracentric inversion of chromosome 7q, serves a role in activating the MAPK pathway in thyroid cancer (11). However, the molecular mechanisms of PTC have not yet been fully investigated.
RNA-sequencing (RNA-seq), which is a useful tool for transcriptome analysis, can be applied to reveal genomic structural variations, gene fusion events, novel genes and transcripts (12,13). By RNA-seq, Costa et al (14) identified new missense mutations in Casitas B-cell lymphoma gene; NOTCH1; phosphoinositide-3-kinase regulatory subunit 4 and SW/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily a, member 4 genes; somatic mutations in dicer 1, ribonuclease type III; met proto-oncogene (MET); and von Hippel-Lindau genes; and a new chimeric transcript induced by the WNK lysine deficient protein kinase 1-β1, 4-N-acetylgalactosaminyltransferase-3 gene fusion in PTC patients. Smallridge et al (15) used RNA-seq to analyze differentially expressed genes (DEGs) between BRAF wild-type and BRAF V600E mutation PTCs, demonstrating that the BRAF V600E mutation inhibits expression of immune/inflammatory response genes but promotes expression of chemokine (C-X-C motif) ligand 14 and human leukocyte antigen-G. However, the above studies did not perform comprehensive bioinformatics analysis. In the present study, the transcriptional profiles of thyroid tumor tissues and adjacent normal tissues were obtained by RNA-seq. Thereafter, SNPs were identified and functionally annotated. In addition, the DEGs were screened and further investigated by enrichment analysis, protein domain analysis and protein-protein interaction (PPI) network analysis. Furthermore, the potential gene fusion events were separately predicted for the tumor tissues and normal tissues. The flow chart of the bioinformatics analysis is presented in Fig. 1.
Figure 1.

Flow chart of the bioinformatics analysis. DEGs, differentially expressed genes; SNP, single nucleotide polymorphism; PPI, protein-protein interaction.
Materials and methods
Sample source and RNA sample preparation
Thyroid tumor tissues and adjacent normal tissues were collected from 5 PTC patients [2 women and 3 men; mean age, 37.4 years; 3 patients in stage I (T1N1aM0) and 2 patients in stage III (T1N1aM0)] from The Affiliated Hospital of Qingdao University (Qingdao, China). All patients were treated by thyroidectomy and lymphadenectomy; ultrasonography confirmed that the patients had no obvious abnormality after surgery. Total RNA was isolated from the tumor and normal tissues, using a TRIzol total RNA extraction kit (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The concentration of RNA was tested with Qubit® RNA Assay kit in Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Inc.). Next, the RNA integrity was assessed by spectrophotometry using NanoDrop™ 2000 (Thermo Fisher Scientific, Inc., Wilmington, DE, USA) at 260 and 280 nm and 1% (w/v) agarose gel electrophoresis at 37°C for 15 min with 95°C preheat for 5 min. The results were stained with ethidium bromide (0.50 µg/ml; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), followed by visualization using the Agilent 2100 Bioanalyzer system (Agilent Technologies, Inc., Santa Clara, CA, USA). The following primers were used in this study: P5 (forward, AAT GAT ACG GCG ACC ACC GAG A and reverse, CAA GCA GAA GAC GGC ATA CGA G); P7 (forward, ATC TCG TAT GCC GTC TTC TGC TTG and reverse, CAA GCA GAA GAC GGC ATA CGA GAT). The Research Ethics Committee at The Affiliated Hospital of Qingdao University gave the approval for this study, and all participants wrote informed consent.
cDNA library construction and Illumina RNA-seq
Library preparation and RNA-seq were performed by Beijing BerryGenomics Co., Ltd. (Beijing, China). Briefly, oligo(dT) magnetic beads were used to enrich mRNAs from total RNA, and then fragmentation buffer was applied to fragment the enriched mRNAs. Subsequently, the first cDNA strand was amplified using the mRNA templates and random hexamer primers. Subsequently, the second strand of cDNA was synthesized by adding DNA polymerase I (New England BioLabs, Inc., Ipswich, MA, USA), dNTPs (New England BioLabs, Inc.), RNase H (New England BioLabs, Inc.) and buffer. The double-stranded cDNA fragments were purified using QIAquick PCR Purification kit (Qiagen GmbH, Hilden, Germany) and eluted with elution buffer, followed by end repair and the ligation of sequencing adaptors. After the fragments with suitable size were selected by agarose gel electrophoresis, polymerase chain reaction (PCR) amplification was performed. Finally, the cDNA libraries were constructed and sequenced on an Illumina HiSeq 2500 platform according the manufacturer's protocol.
Sequencing data processing
After removing low-quality reads (exceeding 50 bp and with >50% of bases with Q-value ≤3) and those reads with >3% (N bases) of adaptors or unknown nucleotides, the clean reads were obtained from the raw reads.
SNP identification and DEG screening
The clean reads were mapped to the hg19 human reference genome by the Burrows-Wheeler Aligner (bio-bwa.sourceforge.net/bwa.shtml) software (16), and then SNPs were identified by the SAMtools software (samtools.sourceforge.net) (17). Using ANNOVAR software (http://www.openbioinformatics.org/annovar/) (18), functional annotation was performed for the identified SNPs to investigate their genomic locations and variation information.
The DEGs in the thyroid tumor tissues compared with the adjacent normal tissues were screened using edgR software (bioconductor.org/packages/2.4/bioc/html/edgeR.html) (19). Based on the negative binomial model (20), the gene significance levels between the two groups were calculated. Following this, the adjusted P-value (the false discovery rate, FDR) of each gene was calculated by the Benjamini-Hochberg method (21). FDR<0.05 and |log2 fold-change (FC)|≥1 were considered thresholds.
Gene fusion analysis and enrichment analysis
The potential gene fusion events were predicted using FusionMap software (http://www.omicsoft.com/fusionmap/) (22), and then visualized using Circos software (http://www.circos.ca/) (23). The Gene Ontology (GO; www.geneontology.org/) database describes cellular component (CC), molecular function (MF), and biological process (BP) of gene products (24). The Kyoto Encyclopedia of Genes and Genomes (KEGG; www.genome.ad.jp/kegg/) database, which consists of known genes and their functions, can be used for pathway mapping (25). Using the Database for Annotation, Visualization and Integrated Discovery tool (DAVID; version 6.8; david.abcc.ncifcrf.gov/) software (26), GO functional and KEGG pathway enrichment analyses were conducted for the identified DEGs. P<0.05 was considered as the cut-off criterion.
PPI network analysis and protein domain analysis
The interactions among the proteins encoded by the DEGs were predicted using the Human Protein Reference Database (http://www.hprd.org) (27), and then PPI network was visualized using Cytoscape software (version 3.2.0; www.cytoscape.org/) (28). Using pfam_scan tool (http://www.ebi.ac.uk/Tools/pfa/pfamscan/) (29), the domains in the proteins containing risk SNP loci were predicted, and the domains in the proteins containing non-synonymous risk SNP loci were selected. In addition, the non-synonymous risk SNP loci in the interaction domains involved in the PPI pairs were identified using the Database of Protein Domain Interactions (domine.utdallas.edu/cgi-bin/Domine) (30).
Results
SNP analysis
Upon mapping the clean reads to human reference genome hg19, 70,172 and 83,869 SNPs separately were identified in all of the thyroid tumor tissues and all of the adjacent normal tissues. A Venn diagram demonstrated that 18,795 SNPs were specific in the thyroid tumor tissues and were considered risk SNPs for PTC (Fig. 2). Subsequently, functional annotation was performed for these risk SNPs. Among the 18,795 SNPs, 739 risk SNPs were located in the exon regions (Fig. 3A). The variation information of the risk SNPs located in the exon regions is presented in Fig. 3B.
Figure 2.
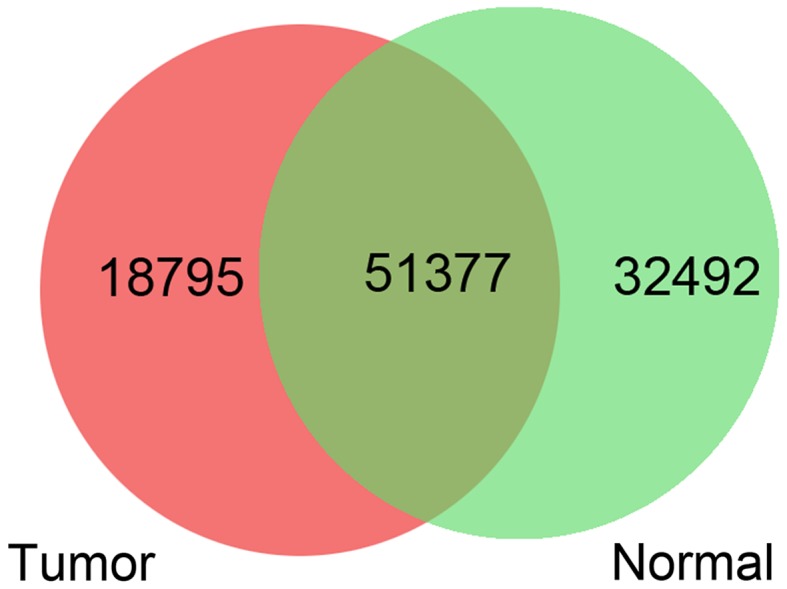
Venn diagram of the single nucleotide polymorphisms in the thyroid tumor tissues and adjacent normal tissues.
Figure 3.

Location statistics of the risk SNPs. (A) Genomic locations of all the risk SNPs. (B) Variation information of the risk SNPs located in the exon regions. UTR, untranslated region; SNV, single nucleotide variant; SNP, single nucleotide polymorphism.
Integrated analysis of SNPs and DEGs
With the thresholds of FDR<0.05 and|log2 FC|≥1, a total of 2,686 DEGs were screened in the thyroid tumor tissues compared with adjacent normal tissues, including 1,361 upregulated genes and 1,325 downregulated genes. The integrated analysis of SNPs and DEGs demonstrated that 12,528 risk SNPs were located in 4,317 genes (157 upregulated genes and 519 downregulated genes).
Using the pfam_scan tool, the domains in the proteins containing risk SNP loci or non-synonymous risk SNP loci were predicted. The results demonstrated that the risk SNPs had no significant effects on protein domains, whereas the non-synonymous risk SNPs may affect protein domains. The non-synonymous risk SNPs in the protein domains are listed in Table I.
Table I.
Non-synonymous risk SNPs in the protein domains.
| Gene | AA Change | SNP id | Domain start | Domain end | Hmm acc | Hmm name |
|---|---|---|---|---|---|---|
| CSGALNACT1 | S193N | rs7017776 | 168 | 507 | PF05679.13 | CHGN |
| IYD | C265R | rs612421 | 98 | 267 | PF00881.21 | Nitroreductase |
| ITGA7 | R655H | rs1800974 | 515 | 1,006 | PF08441.9 | Integrin_alpha2 |
| TMEM171 | R86G | rs637450 | 2 | 319 | PF15471.3 | TMEM171 |
| TMEM171 | N139K | rs636926 | 2 | 319 | PF15471.3 | TMEM171 |
| AGT | M268T | rs699 | 114 | 481 | PF00079.17 | Serpin |
| ATP11A | M317V | rs368865 | 100 | 377 | PF00122.17 | E1-E2_ATPase |
| CDT1 | C234R | rs507329 | 186 | 350 | PF08839.8 | CDT1 |
| CDT1 | T262A | rs480727 | 186 | 350 | PF08839.8 | CDT1 |
| CENPF | N3106K | rs7289 | 3,061 | 3,109 | PF10490.6 | CENP-F_C_Rb_bdg |
| COL1A1 | T1075A | rs1800215 | 1,019 | 1,077 | PF01391.15 | Collagen |
| EBI3 | V201I | rs4740 | 130 | 209 | PF00041.18 | fn3 |
| FN1 | T817P | rs2577301 | 812 | 889 | PF00041.18 | fn3 |
| HES4 | R44S | rs2298214 | 35 | 91 | PF00010.23 | HLH |
| LCP1 | K533E | rs4941543 | 520 | 621 | PF00307.28 | CH |
| LOXL2 | M570L | rs1063582 | 548 | 719 | PF01186.14 | Lysyl_oxidase |
| MCM4 | L650M | rs762679 | 447 | 769 | PF00493.20 | MCM |
| PLXND1 | H894R | rs2625962 | 891 | 976 | PF01833.21 | TIG |
| SLC17A9 | N228S | rs2427463 | 30 | 386 | PF07690.13 | MFS_1 |
| SPHK1 | A34T | rs346803 | 16 | 137 | PF00781.21 | DAGK_cat |
| SPTBN2 | S825G | rs4930388 | 749 | 849 | PF00435.18 | Spectrin |
| SYNC | R274Q | rs360042 | 187 | 456 | PF00038.18 | Filament |
| TNC | Q539R | rs1757095 | 532 | 558 | PF07974.10 | EGF_2 |
SNP, single nucleotide polymorphism; Hmm acc, accession number of hmmer.
Gene fusion analysis
The potential gene fusion events were predicted and are presented in Fig. 4. A total of 11 and 4 gene fusion events were identified in all of the thyroid tumor tissues and all of the normal tissues, respectively. A total of three gene fusion events were predicted in both thyroid tumor tissues and normal tissues, including RNA binding motif protein (RBM)14-RBM4, NK2 homeobox 1-surfactant associated 3 (NKX2-1-SFTA3), and chromosome 1 open reading frame 86 (C1orf86)-LOC100128003 gene fusions. The sodium channel, non-voltage gated 1 α-tumor necrosis factor receptor superfamily 1A gene fusion was predicted not in thyroid tumor tissues but in normal tissues. Furthermore, no gene fusion event was predicted in thyroid tumor tissues but not in normal tissues.
Figure 4.

Potential gene fusion events in the 5 matched samples. Red represents gene fusion events predicted only in thyroid tumor tissues. Grey indicates gene fusion events predicted only in adjacent normal tissues. Green represents gene fusion events predicted in both thyroid tumor tissues and adjacent normal tissues.
Enrichment analysis of the DEGs containing risk SNPs
The DEGs containing risk SNPs were investigated by functional and pathway enrichment analyses. The upregulated genes containing risk SNPs were significantly enriched in urogenital system development (GO_BP, P=1.38×103), extracellular region part (GO_CC, P=5.34×103), hormone binding (GO_MF, P=7.23×103), and pathways in cancer (pathway, P=1.28×102) (Table IIA). Meanwhile, the significant terms enriched for the downregulated genes containing risk SNPs included regulation of cell morphogenesis (GO_BP, P=4.41×109), plasma membrane part (GO_CC, P-value = 2.22×1011), calcium ion binding (GO_MF, P=6.18×107) and cell adhesion molecules (pathway, P=1.16×106; Table IIB).
Table II.
Significant GO and KEGG terms separately enriched for the upregulated and downregulated genes containing risk SNPs.
| A, Significant GO and KEGG terms enriched for the upregulated genes containing risk SNPs | ||||
|---|---|---|---|---|
| Category | Term | Gene number | Gene symbol | P-value |
| GO_BP | GO:0001655~urogenital system development | 6 | ACE, AR, BMP2, PGF, WFS1, ZBTB16 | 1.38E-03 |
| GO_BP | GO:0055114~oxidation reduction | 13 | ALDH6A1, SORD, FOXRED2, OGDHL, AASS, CYP20A1, SOD3, IYD, ALDH1A1, ERO1LB, DIO2, BCO2, WWOX | 2.83E-03 |
| GO_BP | GO:0031667~response to nutrient levels | 7 | BMP2, GATM, MAP1LC3A, PPARG, LIPG, ADIPOR2, GHR | 3.56E-03 |
| GO_BP | GO:0001822~kidney development | 5 | ACE, BMP2, PGF, WFS1, ZBTB16 | 5.75E-03 |
| GO_BP | GO:0043434~response to peptide hormone stimulus | 6 | AR, EGR2, GATM, PPARG, FOXO1, GHR | 5.91E-03 |
| GO_CC | GO:0044421~extracellular region part | 18 | TG, MATN2, BMP2, PODN, SORD, PGF, MMP15, CCL28, SOD3, TNFSF10, ACE, FBLN2, EMID1, NUCB2, LIPG, ANGPTL1, MFAP4, GHR | 5.34E-03 |
| GO_CC | GO:0005794~Golgi apparatus | 16 | TG, SLC39A14, RAP1GAP, NCS1, ACACB, MAN1A1, SOD3, CSGALNACT1, AP1S3, SYNE1, EMID1, NUCB2, NDRG2, GRB14, WWOX, SMPD3 | 1.14E-02 |
| GO_CC | GO:0005925~focal adhesion | 5 | TNS3, PGM5, PRUNE, LAYN, LPP | 1.35E-02 |
| GO_CC | GO:0005924~cell-substrate adherens junction | 5 | TNS3, PGM5, PRUNE, LAYN, LPP | 1.53E-02 |
| GO_CC | GO:0031985~Golgi cisterna | 3 | CSGALNACT1, NUCB2, SMPD3 | 1.65E-02 |
| GO_MF | GO:0042562~hormone binding | 4 | ALDH1A1, AR, ADIPOR2, GHR | 7.23E-03 |
| GO_MF | GO:0005096~GTPase activator activity | 7 | ALDH1A1, AGAP11, RAP1GAP, TBC1D4, OPHN1, RGS16, STARD13 | 8.86E-03 |
| GO_MF | GO:0008289~lipid binding | 10 | ALDH1A1, ALDH6A1, AR, MAP1LC3A, SDPR, OSBPL1A, FAAH, PPARG, CYTH3, PLEKHA2 | 1.06E-02 |
| GO_MF | GO:0005543~phospholipid binding | 6 | MAP1LC3A, SDPR, OSBPL1A, FAAH, CYTH3, PLEKHA2 | 1.44E-02 |
| GO_MF | GO:0050662~coenzyme binding | 6 | ALDH6A1, ERO1LB, SORD, OGDHL, FOXRED2, WWOX | 1.57E-02 |
| Pathway | hsa05200: Pathways in cancer | 8 | AR, BMP2, PGF, PPARG, FOXO1, ZBTB16, TCF7L1, DAPK1 | 1.28E-02 |
| B, Significant GO and KEGG terms enriched for the downregulated genes containing risk SNPs | ||||
| Category | Term | Gene number | Gene symbol | P-value |
| GO_BP | GO:0022604~regulation of cell morphogenesis | 20 | PALM, LST1, LZTS1, PTPRF, LIMK1, PLXNB2, CDH2, CDH4, TTL, TGFβ 2, NRCAM, FYN, TIAM1,MYH14, CDC42EP3, ARAP1, RASA1, MYH10, CDC42EP5, FN1 | 4.41E-09 |
| GO_BP | GO:0007155~cell adhesion | 48 | MPZL3, NRP2, CLDN4, POSTN, CD151, CDSN, CTNNB1, NRCAM, PCDH1, WISP1, CD44 | 2.56E-08 |
| GO_BP | GO:0022610~biological adhesion | 48 | MPZL3, NRP2, CLDN4, POSTN, CD151, CDSN, CTNNB1, NRCAM, PCDH1, WISP1, CD44 | 2.63E-08 |
| GO_BP | GO:0043062~extracellular structure organization | 20 | MPZL3, MYO6, ADAMTS14, LGALS3, SPOCK2, TNC, TGFβ R1, POSTN, CDH2, COL5A2, CTNNB1, ANXA2, TGFβ2, DVL1, NRCAM, AGT, COL1A2, AGRN, COL1A1, ADAMTS2 | 1.63E-07 |
| GO_BP | GO:0009611~response to wounding | 36 | DCBLD2, TNC, CLU, TLR2, CDH3, TNFRSF4, TGFβ2, CCL22, HRH1, NOD1, CD44, MAP3K1, SERPINE1, ITGB6, CFI, BLNK, RAB27A, FN1, NOX4, CIITA, NFKBIZ, LYN, OLR1, CFB, IL1RN, ANXA1, CHST2, NFAM1, CD55, BAX, VCAN, CTSB, PARP4, PROS1, ACVR1, MYH10 | 2.51E-06 |
| GO_MF | GO:0005509~calcium ion binding | 55 | PXDN, LTBP1, S100A5, LDLR, ARSJ, EFHD2, PCDH1, GALNTL6, PADI2, ACTN1, NPTXR, CAPN12 | 6.18E-07 |
| GO_MF | GO:0030695~GTPase regulator activity | 31 | TBC1D9, RASGEF1A, IQGAP1, PLEKHG2, RINL, TIAM1, RAPGEF5, RAP1GAP2, DOCK10, RASA1, RHOBTB3, NGEF, ARHGEF2, ABR, TNIK, PSD3, DOCK9, SIPA1L2, ARHGAP23, DOCK3, ANXA2, MAP4K4, CDC42BPG, RASGRF1, RGS4, RIN1, SYTL2, SH3BP1, LRRK2, SYTL1, ARAP1 | 2.83E-06 |
| GO_MF | GO:0060589~nucleoside-triphosphatase regulator activity | 31 | TBC1D9, RASGEF1A, IQGAP1, PLEKHG2, RINL, TIAM1, RAPGEF5, RAP1GAP2, DOCK10, RASA1, RHOBTB3, NGEF, ARHGEF2, ABR, TNIK, PSD3, DOCK9, SIPA1L2, ARHGAP23, DOCK3, ANXA2, MAP4K4, CDC42BPG, RASGRF1, RGS4, RIN1, SYTL2, SH3BP1, LRRK2, SYTL1, ARAP1 | 4.39E-06 |
| GO_MF | GO:0019899~enzyme binding | 34 | CDH2, IQGAP1, CTNNB1, MAP3K1, SERPINE1, CDK5RAP2, FAS, DOCK10, INSR, RASA1, CDC42EP5, RHOBTB3, FMNL2, ARHGEF2, LYN, TGFβR1, DOCK9, DOCK3, FLNA, DVL1, ANXA2, NRIP1, RFC5, MICALCL, CCND1, MAST2, CCND3, CCND2, IGF2R, SYTL2, PARP4, CACNA1C, SYTL1, ADAMTS4 | 2.76E-05 |
| GO_MF | GO:0008092~cytoskeletal protein binding | 32 | SHROOM4, FXYD5, SDC4, KLHL2, NRCAM, FRMD3, CORO2A, FRMD5, MACF1, CDK5RAP2, MSN, CDC42EP3, RAB27A, FMNL2, ARHGEF2, MYO6, HTT, MYO1E, MYO1D, MYO1G, ACTN1, FLNA, ANXA2, FYN, ARPC5L, SPTBN2, FLII, MYH14, DST, FHOD1, LCP1, MYH10 | 7.90E-05 |
| GO_CC | GO:0044459~plasma membrane part | 119 | MICB, SRCIN1, PTGS2, ATP1B3, GABRB2, TLR2, CTNNB1, NRCAM, ST3GAL5, CD44, KCNK5 | 2.22E-11 |
| GO_CC | GO:0005886~plasma membrane | 171 | MICB, ATP1B3, PTGS2, SRCIN1, GABRB2, TLR2, IQGAP1, CTNNB1, NRCAM, MCOLN3 | 2.42E-10 |
| GO_CC | GO:0031012~extracellular matrix | 34 | ADAMTS17, LTBP1, ADAMTS14, SPOCK2, TNC, ADAMTSL5, POSTN, TGFβ2, TIMP1, LAMB3, CD44, FBN3, AGRN, FBN2, FGF1, LOXL1, FN1, LGALS3, COL13A1, PAPLN, SPARC, EMILIN2, COL5A2, ANXA2, ADAMTS9, BGN, NAV2, COL1A2, LAMC2, VCAN, COL1A1, DST, ADAMTS2, ADAMTS4 | 3.48E-09 |
| GO_CC | GO:0005578~proteinaceous extracellular matrix | 32 | ADAMTS17, LTBP1, ADAMTS14, SPOCK2, TNC, ADAMTSL5, POSTN, TIMP1, LAMB3, FBN3, AGRN, FBN2, FGF1, LOXL1, FN1, LGALS3, COL13A1, PAPLN, SPARC, EMILIN2, COL5A2, ANXA2, ADAMTS9, BGN, NAV2, COL1A2, LAMC2, VCAN, COL1A1, DST, ADAMTS2, ADAMTS4 | 7.79E-09 |
| GO_CC | GO:0005887~integral to plasma membrane | 71 | MICB, LDLR, CLDN4, SLC20A1, ATP1B3, CORIN, GABRB2, TLR2, GABBR2, CD151, SDC4 | 2.00E-08 |
| Pathway | hsa04514: Cell adhesion molecules (CAMs) | 19 | CLDN16, ICAM1, CLDN4, PTPRF, CDH2, HLA-B, CDH3, SDC4, CDH4, NRCAM, ITGA9, ITGB8, CD58, CLDN1, HLA-DPA1, VCAN, CNTNAP1, HLA-DOA, HLA-DRA | 1.16E-06 |
| Pathway | hsa04512: ECM-receptor interaction | 15 | TNC, ITGA11, ITGA3, SDC4, COL5A2, ITGA9, LAMB3, CD44, ITGB8, ITGB6, COL1A2, LAMC2, AGRN, COL1A1, FN1 | 1.68E-06 |
| Pathway | hsa04510: Focal adhesion | 22 | TNC, MET, ITGA11, ACTN1, ITGA3, COL5A2, FLNA, CTNNB1, ITGA9, LAMB3, CCND1, CCND3, CCND2, ITGB8, RASGRF1, FYN, ITGB6, COL1A2, LAMC2, COL1A1, SHC3, FN1 | 1.17E-05 |
| Pathway | hsa05412: Arrhythmogenic right ventricular cardiomyopathy (ARVC) | 12 | ITGA9, DSG2, PKP2, ITGB8, ITGB6, ITGA11, CACNB1, ACTN1, ITGA3, CDH2, CACNA1C, CTNNB1 | 8.78E-05 |
| Pathway | hsa04115: p53 signaling pathway | 11 | CCNE2, CCND1, TNFRSF10B, CCND3, CCND2, ZMAT3, BAX, SERPINE1, DDB2, FAS, PERP | |
GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; SNP, single nucleotides polymorphism; BP, biological process; CC, cellular component; MF, molecular function.
PPI network analysis
The PPI network constructed for the DEGs consisted of 716 nodes (273 upregulated genes and 443 downregulated genes) and 1,011 interactions (Fig. 5). Importantly, fibronectin 1 (FN1, degree=31) and transforming growth factor β (TGFβ) receptor 1 (TGFβR1, degree=22) had higher degrees in the PPI network. Thereafter, the non-synonymous risk SNP loci in the interaction domains involved in the PPI pairs were identified. A total of 7 PPI pairs containing the non-synonymous risk SNP loci in the interaction domains were identified (Table III). Particularly, the interaction domains involved in the interactions of FN1 and 5 other proteins (such as tenascin C, TNC) contained non-synonymous risk SNP loci.
Figure 5.

Protein-protein interaction network constructed for the differentially expressed genes.
Table III.
Protein-protein interaction pairs containing the non-synonymous risk single nucleotide polymorphism loci in the interaction domains.
| Protein-1 | Protein-2 | Domain-1 | Domain-2 |
|---|---|---|---|
| TNC | FN1 | PF00041 | PF00041 |
| FN1 | COL1A1 | PF00041 | PF01391 |
| FN1 | COL1A2 | PF00041 | PF01391 |
| COL1A1 | COL7A1 | PF01391 | PF01391 |
| FN1 | COL7A1 | PF00041 | PF01391 |
| FN1 | COL13A1 | PF00041 | PF01391 |
| MCM4 | MCM2 | PF00493 | PF00493 |
TNC, tenascin C; FN1, fibronectin 1; MCM, minichromosome maintenance; COL, collagen type.
Discussion
There were some limitations in the present study (low case number, no own in vivo or in vitro experiments), thus the findings in five PTC cases had the characteristics of an advanced case report. In this study, a total of 70,172 and 83,869 SNPs were identified in all of the thyroid tumor tissues and all of the normal tissues, respectively. A Venn diagram demonstrated that 18,795 risk SNPs were specific in the thyroid tumor tissues. There were more SNPs in the normal tissues than in the thyroid tumor tissues; however, only the 18,795 SNPs specific in the thyroid tumor tissues were potential risk SNPs for PTC. Total 2,686 DEGs were screened in the thyroid tumor tissues, including 1,361 upregulated genes and 1,325 downregulated genes. The integrated analysis of SNPs and DEGs demonstrated that 12,528 risk SNPs were located in 4,317 genes (157 upregulated and 519 downregulated genes).
A previous study demonstrated that the activity of the TGFβ/mothers against decapentaplegic (Smad)-dependent signaling pathway is associated with nodal metastasis, local invasion and BRAF-mutated PTCs (31). TGFβ1 has been identified as a key factor in PTC cells that affects the activation of stromal fibroblasts in a paracrine manner. Furthermore, the activation of the TGFβ/Smad3 and Notch signaling pathways can impact tumor growth (32). Choe et al (33) investigated the association between SNPs (−1444C/G, Asn389Asn, −834 G/A) of TGFβR2 and PTC development, and the SNPs and clinicopathological characteristics of PTC (including lymph node metastasis, location, size, number and extrathyroidal invasion), and demonstrated that TGFβR2 may serve a role in PTC progression in the Korean population. These studies suggested that TGFβR had a correlation with the pathogenesis of PTC. In the present study, FN1 (degree=31) and TGFβR1 (degree=22) had higher degrees in the PPI network. Thus, TGFβR1 may serve an important role in the progression of PTC.
A total of 7 PPI pairs containing the non-synonymous risk SNP loci in the interaction domains were identified. Particularly, the interaction domains involved in the interactions of FN1 and 5 other proteins (such as TNC) contained non-synonymous risk SNP loci. Prasad et al (34) hypothesized that an immunohistochemical panel containing FN1, Hector Battifora mesothelial cell 1 and galectin-3 may contribute to the diagnosis of thyroid tumors derived from follicular cells. Using reverse transcription-quantitative polymerase chain reaction, da Silveira Mitteldorf et al (35) demonstrated that FN1, MET, glutaminyl-peptide cyclotransferase, and UDP-galactose-4-epimerase were significantly upregulated in patients with PTC. TNC re-expression can be detected by immunohistochemistry in papillary and medullary thyroid carcinomas supplemented by the analysis of two TNC mRNA splice variants; thus TNC may be synthesized by tumor cells (36). The above studies indicated that FN1 is involved in PTC, and TNC is also associated with tumor. Therefore, FN1 and TNC containing the non-synonymous risk SNP loci might serve a role in PTC by interacting with each other.
Furthermore, 11 and 4 gene fusion events were identified in all of the thyroid tumor tissues and all of the normal tissues, respectively. A total of three gene fusion events were predicted in both normal tissues and thyroid tumor tissues, including RBM14-RBM4, NKX2-1-SFTA3 and C1orf86-LOC100128003 gene fusions. Through NKX2-1 and forkhead box E1 (FOXE1) genotyping, Matsuse et al (37) revealed that both NKX2-1 and FOXE1 can increase the risk of Japanese sporadic PTC. The missense mutation (1016C>T) in homeobox transcription factor TTF1 (TITF-1)/NKX2-1 is the first germline mutation detected in multinodular goiter (MNG)/PTC patients, which increases the susceptibility for PTC and/or MNG and contributes to the development of PTC (38,39). Impaired paired box 8 and TITF-1/NKX2-action may be correlated with the expression changes of type 1 and type 2 iodothyronine 5′ deiodinases in PTC (40). NKX2-1 is important for thyroid organogenesis and controls thyroid functions, and its inactivation is implicated in epigenetics in thyroid carcinomas; thus, TTF-1 may serve as a therapeutic target via epigenetic modification (41). These previous studies demonstrated that NKX2-1 also acted in the mechanisms of PTC. Therefore, the NKX2-1-SFTA3 gene fusion might be involved in PTC. However, small sample size, as well as the lack of experimental validation and the basic molecular analysis of RAS and BRAF status were the limitations of the present study. Furthermore, the definitive pathophysiological role and underlying mechanism of the TGFβR1/NKX2-1-SFTA3 gene fusion as well as non-synonymous risk SNP loci of FN1 and TNC in PTC remain unclear. Thus, further confirmation of these results is required.
In conclusion, the current study compared the transcriptional profiles of thyroid tumor tissues and normal tissues. A total of 18795 risk SNPs and 2686 DEGs were identified in the thyroid tumor tissues. Therefore, TGFβR1 and the NKX2-1-SFTA3 gene fusion might be implicated in PTC. Additionally, FN1 and TNC containing the non-synonymous risk SNP loci might serve a role in PTC by interacting with each other.
Acknowledgements
The present study was supported by the Qingdao Municipal Science and Technology Bureau (grant no. 13-1-4-166-jch).
References
- 1.Carling T, Udelsman R. Thyroid cancer. Annu Rev Med. 2014;65:125–137. doi: 10.1146/annurev-med-061512-105739. [DOI] [PubMed] [Google Scholar]
- 2.Vecchia CL, Malvezzi M, Bosetti C, Garavello W, Bertuccio P, Levi F, Negri E. Thyroid cancer mortality and incidence: A global overview. Int J Cancer. 2015;136:2187–2195. doi: 10.1002/ijc.29251. [DOI] [PubMed] [Google Scholar]
- 3.Dinets A, Hulchiy M, Sofiadis A, Ghaderi M, Höög A, Larsson C, Zedenius J. Clinical, genetic, and immunohistochemical characterization of 70 Ukrainian adult cases with post-Chornobyl papillary thyroid carcinoma. Eur J Endocrinol. 2012;166:1049–1060. doi: 10.1530/EJE-12-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mitchell RS, Kumar V, Abbas AK, Fausto N. Robbins Basic Pathology. 8 edition. Vol. 8. Philadelphia: Saunders; 2007. pp. 345–355. [Google Scholar]
- 5.Xing M, Alzahrani AS, Carson KA, Shong YK, Kim TY, Viola D, Elisei R, Bendlová B, Yip L, Mian C, et al. Association between BRAF V600E mutation and recurrence of papillary thyroid cancer. J Clin Oncol. 2015;33:42–50. doi: 10.1200/JCO.2015.61.4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, Pai S, Bishop J. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J Clin Oncol. 2014;32:2718–2726. doi: 10.1200/JCO.2014.55.5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lesueur F, Corbex M, McKay JD, Lima J, Soares P, Griseri P, Burgess J, Ceccherini I, Landolfi S, Papotti M, et al. Specific haplotypes of the RET proto-oncogene are over-represented in patients with sporadic papillary thyroid carcinoma. J Med Genet. 2002;39:260–265. doi: 10.1136/jmg.39.4.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stephens LA, Powell NG, Grubb J, Jeremiah SJ, Bethel JA, Demidchik EP, Bogdanova TI, Tronko MD, Thomas GA. Investigation of loss of heterozygosity and SNP frequencies in the RET gene in papillary thyroid carcinoma. Thyroid. 2005;15:100–104. doi: 10.1089/thy.2005.15.100. [DOI] [PubMed] [Google Scholar]
- 9.Salvatore G, Giannini R, Faviana P, Caleo A, Migliaccio I, Fagin JA, Nikiforov YE, Troncone G, Palombini L, Basolo F, Santoro M. Analysis of BRAF point mutation and RET/PTC rearrangement refines the fine-needle aspiration diagnosis of papillary thyroid carcinoma. J Clin Endocrinol Metab. 2004;89:5175–5180. doi: 10.1210/jc.2003-032221. [DOI] [PubMed] [Google Scholar]
- 10.Salajegheh A, Smith RA, Kasem K, Gopalan V, Nassiri MR, William R, Lam AK. Single nucleotide polymorphisms and mRNA expression of VEGF-A in papillary thyroid carcinoma: Potential markers for aggressive phenotypes. Eur J Surg Oncol. 2011;37:93–99. doi: 10.1016/j.ejso.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Ciampi R, Knauf JA, Kerler R, Gandhi M, Zhu Z, Nikiforova MN, Rabes HM, Fagin JA, Nikiforov YE. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J Clin Invest. 2005;115:94–101. doi: 10.1172/JCI23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Clark AG. Using next-generation RNA sequencing to identify imprinted genes. Heredity (Edinb) 2014;113:156–166. doi: 10.1038/hdy.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Klerk E, den Dunnen JT, 't Hoen PA. RNA sequencing: From tag-based profiling to resolving complete transcript structure. Cell Mol Life Sci. 2014;71:3537–3551. doi: 10.1007/s00018-014-1637-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Costa V, Esposito R, Ziviello C, Sepe R, Bim LV, Cacciola NA, Decaussin-Petrucci M, Pallante P, Fusco A, Ciccodicola A. New somatic mutations and WNK1-B4GALNT3 gene fusion in papillary thyroid carcinoma. Oncotarget. 2015;6:11242–11251. doi: 10.18632/oncotarget.3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smallridge RC, Chindris AM, Asmann YW, Casler JD, Serie DJ, Reddi HV, Cradic KW, Rivera M, Grebe SK, Necela BM, et al. RNA sequencing identifies multiple fusion transcripts, differentially expressed genes, and reduced expression of immune function genes in BRAF (V600E) mutant vs BRAF wild-type papillary thyroid carcinoma. J Clin Endocrinol Metab. 2014;99:E338–E347. doi: 10.1210/jc.2013-2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abuín JM, Pichel JC, Pena TF, Amigo J. BigBWA: Approaching the Burrows-Wheeler aligner to Big Data technologies. Bioinformatics. 2015;31:4003–4005. doi: 10.1093/bioinformatics/btv506. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. 1000 Genome Project Data Processing Subgroup: The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang H, Wang K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc. 2015;10:1556–1566. doi: 10.1038/nprot.2015.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robinson MD, McCarthy DJ, Smyth GK. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park BJ, Lord D, Hart J. Bias properties of Bayesian statistics in finite mixture of negative binomial regression models in crash data analysis. Accid Anal Prev. 2010;42:741–749. doi: 10.1016/j.aap.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 21.Haynes W. Encyclopedia of Systems Biology. Springer; 2013. Benjamini-Hochberg Method; pp. 78–78. [DOI] [Google Scholar]
- 22.Ge H, Liu K, Juan T, Fang F, Newman M, Hoeck W. FusionMap: Detecting fusion genes from next-generation sequencing data at base-pair resolution. Bioinformatics. 2011;27:1922–1928. doi: 10.1093/bioinformatics/btr310. [DOI] [PubMed] [Google Scholar]
- 23.An J, Lai J, Sajjanhar A, Batra J, Wang C, Nelson CC. J-Circos: An interactive Circos plotter. Bioinformatics. 2015;31:1463–1465. doi: 10.1093/bioinformatics/btu842. [DOI] [PubMed] [Google Scholar]
- 24.Tweedie S, Ashburner M, Falls K, Leyland P, McQuilton P, Marygold S, Millburn G, Osumi-Sutherland D, Schroeder A, Seal R, et al. FlyBase: Enhancing Drosophila gene ontology annotations. Nucleic Acids Res. 2009;37(Database issue):D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kotera M, Moriya Y, Tokimatsu T, Kanehisa M, Goto S. KEGG and GenomeNet, New Developments, Metagenomic Analysis. In: Nelson KE, editor. Encyclopedia of Metagenomics. Springer; New York, NY: 2015. pp. 329–339. [Google Scholar]
- 26.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- 27.Goel R, Harsha HC, Pandey A, Prasad TSK. Human Protein Reference Database and Human Proteinpedia as resources for phosphoproteome analysis. Mol Biosyst. 2012;8:453–463. doi: 10.1039/C1MB05340J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohl M, Wiese S, Warscheid B. Cytoscape: Software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- 29.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yellaboina S, Tasneem A, Zaykin DV, Raghavachari B, Jothi R. DOMINE: A comprehensive collection of known and predicted domain-domain interactions. Nucleic Acids Res. 2011;39(Database issue):D730–D735. doi: 10.1093/nar/gkq1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma S, Wang Q, Ma X, Wu L, Guo F, Ji H, Liu F, Zhao Y, Qin G. FoxP3 in papillary thyroid carcinoma induces NIS repression through activation of the TGF-β1/Smad signaling pathway. Tumor Biol. 2016;120:34–35. doi: 10.1007/s13277-015-3848-6. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J, Wang Y, Li D, Jing S. Notch and TGF-β/Smad3 pathways are involved in the interaction between cancer cells and cancer-associated fibroblasts in papillary thyroid carcinoma. Tumor Biol. 2014;35:379–385. doi: 10.1007/s13277-013-1053-z. [DOI] [PubMed] [Google Scholar]
- 33.Choe BK, Kim SK, Park HJ, Park HK, Kwon KH, Lim SH, Yim SV. Polymorphisms of TGFBR2 contribute to the progression of papillary thyroid carcinoma. Mol Cellular Toxicol. 2012;8:1–8. doi: 10.1007/s13273-012-0001-0. [DOI] [Google Scholar]
- 34.Prasad ML, Pellegata NS, Huang Y, Nagaraja HN, de la Chapelle A, Kloos RT. Galectin-3, fibronectin-1, CITED-1, HBME1 and cytokeratin-19 immunohistochemistry is useful for the differential diagnosis of thyroid tumors. Mod Pathol. 2005;18:48–57. doi: 10.1038/modpathol.3800235. [DOI] [PubMed] [Google Scholar]
- 35.da Silveira Mitteldorf CA, de Sousa-Canavez JM, Leite KR, Massumoto C, Camara-Lopes LH. FN1, GALE, MET and QPCT overexpression in papillary thyroid carcinoma: Molecular analysis using frozen tissue and routine fine-needle aspiration biopsy samples. Diagn Cytopathol. 2011;39:556–561. doi: 10.1002/dc.21423. [DOI] [PubMed] [Google Scholar]
- 36.Tseleni-Balafouta S, Gakiopoulou H, Fanourakis G, Voutsinas G, Balafoutas D, Patsouris E. Tenascin-C protein expression and mRNA splice variants in thyroid carcinoma. Exp Mol Pathol. 2006;80:177–182. doi: 10.1016/j.yexmp.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 37.Matsuse M, Takahashi M, Mitsutake N, Nishihara E, Hirokawa M, Kawaguchi T, Rogounovitch T, Saenko V, Bychkov A, Suzuki K, et al. The FOXE1 and NKX2-1 loci are associated with susceptibility to papillary thyroid carcinoma in the Japanese population. J Med Genet. 2011;48:645–648. doi: 10.1136/jmedgenet-2011-100063. [DOI] [PubMed] [Google Scholar]
- 38.Ngan ES, Lang BH, Liu T, Shum CK, So MT, Lau DK, Leon TY, Cherny SS, Tsai SY, Lo CY, et al. A germline mutation (A339V) in thyroid transcription factor-1 (TITF-1/NKX2. 1) in patients with multinodular goiter and papillary thyroid carcinoma. J Natl Cancer Inst. 2009;101:162–175. doi: 10.1093/jnci/djn471. [DOI] [PubMed] [Google Scholar]
- 39.Gilbert-Sirieix M, Makoukji J, Kimura S, Talbot M, Caillou B, Massaad C, Massaad-Massade L. Wnt/β-catenin signaling pathway is a direct enhancer of thyroid transcription factor-1 in human papillary thyroid carcinoma cells. PLoS One. 2011;6:e22280. doi: 10.1371/journal.pone.0022280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ambroziak M, Pachucki J, Stachlewska-Nasfeter E, Nauman J, Nauman A. Disturbed expression of type 1 and type 2 iodothyronine deiodinase as well as titf1/nkx2-1 and pax-8 transcription factor genes in papillary thyroid cancer. Thyroid. 2005;15:1137–1146. doi: 10.1089/thy.2005.15.1137. [DOI] [PubMed] [Google Scholar]
- 41.Kondo T, Nakazawa T, Ma D, Niu D, Mochizuki K, Kawasaki T, Nakamura N, Yamane T, Kobayashi M, Katoh R. Epigenetic silencing of TTF-1/NKX2-1 through DNA hypermethylation and histone H3 modulation in thyroid carcinomas. Lab Invest. 2009;89:791–799. doi: 10.1038/labinvest.2009.50. [DOI] [PubMed] [Google Scholar]