

Abstract
Preeclampsia is a pregnancy-specific disorder, which is a leading cause of maternal and perinatal mortality and morbidity. A lower increase of estrogen, compared with the increase in progesterone, is associated with pathogenesis of the disease during pregnancy. G-protein-coupled receptor 30 (GPR30) mediates the action of estrogen, however remains to be investigated in preeclampsia. The levels of GPR30 were measured in placentae from uncomplicated pregnancies and pregnancies complicated by preeclampsia using immunohistochemistry and western blotting. GPR30 expression was additionally measured in placental HTR8/SVneo cells following 17β-estrogen (E2) treatment in normal or hypoxia-reoxygenation conditions by western blotting. In addition, the outgrowth of HTR8/SVneo cells following E2 treatment in hypoxia-reoxygenation conditions was measured. Levels of GPR30 were significantly reduced in placentae from women with preeclampsia as compared with uncomplicated pregnancies. Treatment with E2 significantly increased the expression of GPR30 in HTR8/SVneo cells, in normal and hypoxia-reoxygenation conditions. Furthermore, treatment with E2 increased the outgrowth of HTR8/SVneo cells in hypoxia-reoxygenation conditions. The present study demonstrated lowered placental expression of GPR30 in preeclampsia. Estrogen treatment increases GPR30 expression in extravillous trophoblast and GPR30 may be involved in extravillous trophoblast invasion.
Keywords: preeclampsia, GPR30, extravillous trophoblast, estrogen, placenta
Introduction
Preeclampsia is a pregnancy-specific disorder which is a leading cause of maternal and perinatal mortality and morbidity (1). It is characterised by systemic endothelial cell activation and exaggerated inflammation. Although the pathogenesis of preeclampsia remains unclear, it is well documented that deficient placentation is associated with this disease; this starts from 6–18 weeks of gestation. Disruption to extravillous cytotrophoblast invasion and spiral artery remodelling, subsequently induces placental dysfunction and placental oxidative stress, which in turn contribute to the pathogenesis of the disease (2–4).
During pregnancy, levels of estrogen and progesterone are increased. In animal models, inhibition of estrogen production results in pregnancy loss, suggesting that estrogen may have a role in the maintenance of a healthy pregnancy (5,6). 17β-estradiol (E2) is the common form of estrogen in women and the levels of E2 are increased during pregnancy due to placental production (7). There is evidence to suggest that reduced levels of estrogen and increased levels of progesterone are associated with the pathogenesis of preeclampsia (8–10). The level of E2 has been also suggested to be reduced in preeclampsia (11). In addition, a previous study suggested that estrogen deficiency affects endothelial cell function (12).
The biological effects of estrogen are commonly mediated by estrogen receptors α (ERα) and β (ERβ) (13,14). However, a previous study suggested that factors other than ERα and ERβ are involved in the functions of estrogen in pregnancy (15). The G-protein-coupled receptor 30 (GPR30) was reported as a novel estrogen receptor in 2005 and is able to mediate estrogen action (16,17). Unlike ERα and ERβ that function as estrogen-activated transcription factors in the nucleus and do not influence gene transcription (18), GPR30 is a transmembrane estrogen receptor (19). The most potent estrogen produced in the body is E2 (7) and GPR30 is a specific receptor for E2 (19). GPR30 can additionally promote the ability of estrogen to inhibit apoptosis induced by oxidative stress in keratinocytes (20) and GPR30 mediates the proliferative effects of estrogen in endometrial cancer (21).
GPR30 mRNA is expressed in the placenta (19), however, the role of GPR30 in pregnancy or in pregnancy complications such as preeclampsia and fetal growth restriction (FGR) remains to be investigated. It has been previously reported that hypoxia-reoxygenation is a potential inducer of placental oxidative stress and thus the development of preeclampsia (22). Therefore, the present study aimed to investigate whether the expression of GPR30 in the placenta is altered in preeclampsia and whether hypoxia-reoxygenation conditions affects the expression of GPR30 in vitro. In addition, the potential function of GPR30 in pregnancy was investigated in vitro.
Materials and methods
Ethical approval and consent
The present study was approved by the Ethics Committee of Human Experimentation of the First Affiliated Hospital of Chongqing Medical University (Chongqing, China). For placentae collection, all patients gave written informed consent.
Study population for placenta collection
Placentae from women with preeclampsia (n=21, three with early onset <34 weeks and 18 with late on-set >34 weeks gestation) and from women whose pregnancies were uncomplicated (n=21) were collected after delivery between January 2013 and May 2014 at the First Affiliated Hospital of Chongqing Medical University (Chongqing, China). All women underwent caesarean section. The clinical details of the 42 women for placentae collection are summarised in Table I.
Table I.
Summary of clinical characteristics of patients with preeclampsia and normotensive controls.
| Preeclampsia (n=21) | Normotensive control (n=21) | P-value | |
|---|---|---|---|
| Maternal age [median age in years (range)] | 28 (23–33) | 28 (17–33) | P>0.05 |
| BMI [median BMI in kg/m2 (range)] | 23.9 (17–27.7) | 24 (18.4–28.1) | P>0.05 |
| Gestation weeks (+ days) at sampling [median (range)] | 35+3 (31–38+6) | 35+3 (33–37+6) | P>0.05 |
| Systolic blood pressure [median value in mmHg (range)] | 167 (149–178) | 121 (101–137) | P=0.0001 |
| Diastolic blood pressure [median value in mmHg (range)] | 97 (88–115) | 77 (66–89) | P=0.0001 |
| Birth weight [median value in g (range)] | 2,398 (1,985–2,958) | 3,097 (2,534–3,684) | P=0.0001 |
Preeclampsia was defined as a maternal systolic blood pressure ≥140 mmHg and/or diastolic blood pressure ≥90 mmHg measured on two occasions separated by at least 6 h following 20 weeks of gestation in accordance with the guidelines of the American College of Obstetricians and Gynaecologists (23). Three of the preeclampsia pregnancies were also complicated by FGR.
Reagents
E2, a general agonist for estrogen receptors, was purchased from Abcam (ab120657; Cambridge, UK). The selective GPR30 agonist G1 and the selective GPR30 antagonist G15 were purchased from Sigma-Aldrich; Merck Millipore (G6798 and G6748; Darmstadt, Germany).
Tissue preparation
Placentae were collected within 10 min of delivery and placental explants (collected from the maternal side of the placenta) and decidua were then harvested. All the tissues were snap frozen and stored at −80°C for western blotting or fixed in 4% formaldehyde (Shanghai Aladdin Bio-Chem Technology Co., Ltd., Shanghai, China) prior to embedding in paraffin for immunohistochemical analysis (IHC).
Cell culture and treatment
The human-transformed primary extravillous trophoblast cell line, HTR8/SVneo cell line was donated by Dr C.H. Graham (Queen's University, Kingston, ON, Canada). HTR8/SVneo cells were cultured were cultured at 20% O2 in RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10% fetal bovine serum (FBS; 10099; Gibco; Invitrogen). HTR-8/SVneo cells were growth in 12 well plates until confluent. Cells were then pretreated with E2 (100 nM) or G1 (1 µM) or G15 (2 µM) for 1 h under normal conditions. Pretreated cells were randomly divided into 2 groups: Cells in one group were cultured under normal conditions, whereas cells in the other were treated in hypoxia (1% O2) for 4 h and followed by 18 h of reoxygenation (20% O2). Cells were then harvested for further studies.
Placental explants culture and treatment
Placental explants were cultured on matrigel (10%) in 48-well plates. Explants were pre-treated with E2 (100 nM) or G1 (1 µM) or G15 (2 µM) for 1 h in normoxic conditions (20% O2). Explants were subsequently maintained in normoxic conditions for 24 h or subjected to hypoxia (1% O2) for 4 h followed by 18 h of reoxygenation (20% O2). Extravillous trophoblast sprouting and migration from the distal end of the villous tips was recorded. The extent of migration (i.e., the distance from the cell column base to the tip of the outgrowth) was measured at defined positions with Adobe Fireworks software version CS5 (Adobe Systems, Inc., San Jose, CA, USA).
Western blotting
HTR-8/SVneo cells that had been treated were lysed with RIPA buffer (P0013; Beyotime Institute of Biotechnology, Haimen, China). Protein concentration was determined by the BCA Protein Assay kit (P0010; Beyotime Institute of Biotechnology) according to the manufacturer's instructions. Protein samples (30 µg) were loaded in 10% SDS-polyacrylamide gels, resolved by electrophoresis and transferred to polyvinylidene difluoride membranes (PVDF; 0.22 mm; EMD Millipore, Billerica, MA, USA). Immunoblotting was performed using primary antibodies against GPR30 (1:1,000, ab154069; Abcam) and β-actin (1:1,000, TA-09; OriGene Technologies, Inc., Rockville, MD, USA). Following incubation with the goat anti-rabbit (1:1,000, A0208; Beyotime Institute of Biotechnology) or goat anti-mouse (1:1,000, A0192; Beyotime Institute of Biotechnology) horseradish peroxidase-conjugated secondary antibodies, the bands of specific proteins on the membranes were developed with BeyoECL Plus (P0018; Beyotime Institute of Biotechnology). The levels of proteins were quantified by a ChemiDoc image analyzer (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The expression of β-actin was used as a loading control.
IHC
IHC was performed as previously described (15). Briefly, 5 µm paraffin-embedded tissue sections were deparaffinized in xylene, then rehydrated in a serial gradient of ethanol and washed in PBS. Tissue sections were further quenched sequentially by the use of 3% hydrogen peroxide for 15 min and incubated in 10% normal goat serum (Sigma-Aldrich; Merck Millipore) for 45 min at room temperature. The slides were then incubated at 4°C overnight with polyclonal rabbit anti-GPR30 antibody (1:150, ab39742; Abcam) or polyclonal rabbit anti-CD31 antibody (1:100, cat. no. GTX110602; GeneTex, Inc., Irvine, CA, USA). The slides were rinsed with PBS, and then incubated with a horseradish peroxidase-conjugated goat anti-rabbit IgG for 30 min at 37°C. 3,3′-Diaminobenzidine (chromogenic reagent; OriGene Technologies, Inc.) was used as the chromogen, and hematoxylin (Sigma-Aldrich; Merck Millipore) was used for nuclear counterstaining. For the negative controls, the primary antibodies were omitted. Experiments were repeated at least three times.
Statistical analysis
Data are presented as the mean ± standard deviations. The statistical significance of the results was assessed by a Mann-Whitney U test or a Kruskal-Wallis test using Graphpad Prism software (version 5.0; GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
GPR30 expression was significantly lower in placenta and decidua from preeclampsia pregnancies
IHC analysis demonstrated that GPR30 was expressed in vascular endothelial cells and syncytiotrophoblasts in placentae from uncomplicated pregnancies (Fig. 1). GPR30 was also expressed in vascular endothelial cells and in the stroma in decidua from uncomplicated pregnancies (Fig. 1). However, GPR30 expression was reduced in the placenta and decidua from pregnancies complicated by preeclampsia (Fig. 1). To quantify the IHC results, semi-quantitative analysis of the western blots was performed. The protein levels of GPR30 relative to β-actin were significantly lower in preeclampsia placentas (Fig. 2A; P=0.0078) and decidua (Fig. 2B; P=0.0163), as compared with tissues from uncomplicated pregnancies.
Figure 1.

Immunohistochemistry staining in placenta/villous (A-D) and decidua (E-H). Reduced expression of GPR30 was observed in (B) preeclampsia placentae and (F) decidua compared with (A) placentae and (E) decidua from uncomplicated pregnancies. (C and G) Indicated CD31 staining for endothelial cells, whiles (D and H) were the NC. Scale bar, 200 µm. GPR30, G-protein coupled receptor 30; PE, preeclampsia placentae; NC, negative control; SC, syncytiotrophoblast; EC, vascular endothelial cell.
Figure 2.

Western blotting indicated that the levels of GPR30 were lower in PE compared with (A) placentae and (B) decidua from uncomplicated pregnancies. Semi-quantitative analysis of western blotting confirmed the significantly reduced levels of GPR30 in (A) PE and (B) decidua (*P<0.05 vs. control). GPR30, G-protein coupled receptor 30; PE, preeclampsia placentae.
Expression of GPR 30 in HTR-8/SVneo was increased by E2
Whether E2 could enhance the in vitro expression of GPR30 was investigated. GPR30 expression was significantly increased by treatment with E2 in HTR-8/SVneo cells (Fig. 3; P=0.03). The expression of GPR30 in HTR8/SVneo cells was additionally significantly increased by pre-treatment with the GPR30 agonist G1 (Fig. 3; P=0.03). The increased expression of GPR30 induced by E2 was inhibited by pre-treatment with G15, a GPR30 selective antagonist (Fig. 3; P=0.04). Treatment of HTR-8/SVneo cells with G15 did not change GPR30 expression (Fig. 3; P=0.87).
Figure 3.

(A) Western blotting and (B) semi-quantitative analysis indicating that levels of GPR30 were significantly increased in HTR8/SVneo cells that had been treated with E2 or G1 in normal conditions. The increased levels of GPR30 induced by E2 were inhibited by G15. (C) Western blotting and (D) semi-quantitative analysis indicated that levels of GPR30 were significantly increased in HTR8/SVneo cells that had been treated with E2 or G1 under conditions of H/R. The increased levels of GPR30 induced by E2 were inhibited by G15. GPR30, G-protein coupled receptor 30; E2, 17β-estrogen; H/R, hypoxia/reoxygenation; DMSO, dimethyl sulfoxide.
There were no significant differences in GPR30 expression under hypoxia/reoxygenation conditions compared with normoxic conditions (Fig. 3; P=0.57). However, GPR30 expression was significantly increased by E2 treatment under hypoxia/reoxygenation conditions (Fig. 3; P=0.01) and this increase was inhibited by pre-treatment with G15 (Fig. 3; P=0.025). When HTR-8/SVneo cells were pre-treated with E2 or G1, the expression of GPR30 was significantly reduced in hypoxia/reoxygenation conditions compared with normoxic conditions (Fig. 4; P=0.03 or P=0.02). There was no significant difference in GPR30 expression in normoxic compared with hypoxia/reoxygenation conditions in HTR-8/SVneo cells that were treated with G15 (P=0.18).
Figure 4.
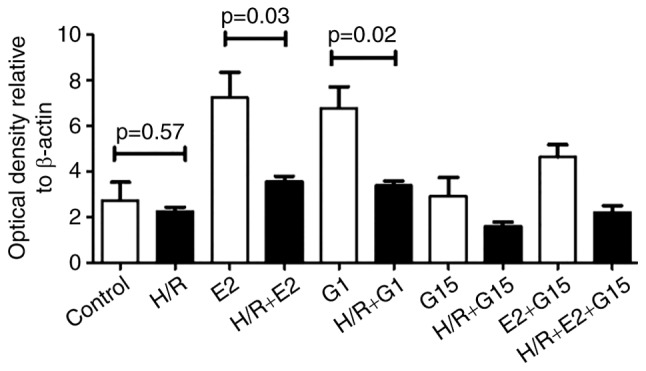
Semi-quantitative analysis of western blotting showing that levels of GPR30 were significantly lower in HTR8/SVneo cells that had been treated with E2 or G1 in conditions of H/R (black bar) as compared with normoxic conditions (white bar). GPR30, G-protein coupled receptor 30; E2, 17β-estrogen; H/R, hypoxia/reoxygenation.
Treatment with E2 or G1 enhances placental explants outgrowth
In order to investigate whether GPR30 is involved in extravillous trophoblast invasion, the outgrowth of placental explants in hypoxia/reoxygenation conditions. Notably, it was identified that the outgrowth of placental explants was significantly reduced in hypoxia/reoxygenation compared with normoxic conditions (Fig. 5). A trend towards an E2-induced increase in the reduced outgrowth did not reach statistical significance (P=0.08); in contrast, the reduced outgrowth was significantly increased by treatment with G1 (Fig. 5; P=0.038).
Figure 5.

Placental explants were cultured on Matrigel under various culture conditions. (A) The growth of placental explants was captured under a light microscope (a-1, b-1, c-1, d-1). Scale bar, 100 µm. GPR30 expression was detected using immunohistochemical staining in placental explants (a-2, b-2, c-2, d-2). Scale bar, 100 µm. (a-1 and a-2) Control; (b-1 and b-2) H/R; (c-1 and c-2) H/R+E2; (d-1 and d-2) H/R+G1. (B) When compared with normoxic conditions (control), the outgrowth of placental explants was significantly reduced in H/R conditions. Treatment with G1 significantly increased the outgrowth of placental explants in H/R conditions. H/R, hypoxia/reoxygenation; GPR30, G-protein coupled receptor 30; E2, 17β-estrogen.
Discussion
The present study was, to the best of our knowledge, the first to report that the expression of GPR30 is reduced in preeclamptic placentae. The in vitro study further demonstrated that supplementation of E2 enhanced the GPR30 expression in trophoblasts in normal and hypoxia/reperfusion conditions, in addition to enhancing trophoblast outgrowth under conditions of hypoxia/reperfusion.
Although the pathogenesis of preeclampsia is unclear, placental hypoxia/reperfusion injury may be involved in the development of preeclampsia. During pregnancy, both maternal estrogen and progesterone is rapidly increased in order to maintain a successful pregnancy. Estrogen is a critical hormone during pregnancy, exerting its effect both at the transcriptional level and the level of intracellular signaling through secondary messengers (24). The effect of estrogens is mediated by the classical receptors, ERα and ERβ (13,14). However, in 2005, studies identified that estrogen also binds to GPR30 (16,17).
GPR30 is widely expressed in a number of tissues including the placenta and ovary (25,26), and promotes estrogen-mediated inhibition of oxidative stress induced apoptosis (20), which is one of the mechanisms implicated in the development of preeclampsia. However the expression of GPR30 in preeclampsia has not previously been investigated. The present study indicated, to the best of our knowledge for the first time, that expression of GPR30 was significantly reduced in trophoblasts and vascular endothelial cells from placentae of preeclampsia as compared with uncomplicated pregnancies using IHC and western blotting. Consistent with earlier reports (15), it was observed that GPR30 was expressed in decidual stroma and vascular endothelial cells. GPR30 expression in decidual stroma and vascular endothelial cells was also significantly reduced in preeclampsia.
Higher levels of estrogen are suggested to exert anti-inflammatory effects and the reduction of estrogen has been reported in women with preeclampsia (11). Estrogen such as E2, the most common form of estrogen, exerts its action either by binding to ERα/ERβ receptors of estrogen or by activating GPR30 (27,28). Therefore, it was investigated whether exogenous estrogen could alter the expression of GPR30 in trophoblasts. In the present study it was identified that treatment with E2 significantly increased the expression of GPR30 in trophoblasts in vitro. The increased expression of GPR30 in trophoblasts induced by E2 was blocked by the specific antagonist of GPR30, G15. The in vitro study demonstrated that E2 supplementation enhanced trophoblast GPR30 expression in normoxic and hypoxia/reperfusion conditions, in addition to trophoblast outgrowth in hypoxia/reperfusion conditions.
There is much evidence for increased placental oxidative stress in preeclampsia and for hypoxia-reoxygenation as a possible mechanism for inducing the oxidative stress (22). Hypoxia-reoxygenation induces apoptotic changes in syncytiotrophoblasts (22). Increasing evidence suggests that hypoxia-reoxygenation within the placenta leads to production of reactive oxidative species (ROS) (29,30) and G-protein coupled pathways are involved in ROS production (31). Increased levels of ROS may result in reduced GPR expression. The in vitro study using 1% O2 as a hypoxic condition and 20% O2 as a normoxic condition, the data demonstrated that treatment with E2 increased trophoblast GPR30 expression in hypoxia-reoxygenation (Fig. 3), although there was no statistical difference between trophoblasts that were cultured in normal conditions and in hypoxia-reoxygenation conditions. Notably, it was identified that compared with trophoblasts that were treated with E2 under normoxic conditions, trophoblast GPR30 expression following E2 treatment was significantly reduced under hypoxia-reoxygenation conditions (Fig. 4). This suggests that hypoxia-reoxygenation may alter the positive regulation of GPR30 expression by E2. It is well documented that reduced trophoblast invasion is associated with the pathogenesis of preeclampsia and that hypoxia-reoxygenation is involved in the inhibition of trophoblast invasion (29), however the potential mechanism is unclear. In the present study using 1% O2 as a hypoxic condition and 20% O2 as a normoxic condition for placental explants culture, it was confirmed that extravillous trophoblast invasion was significantly reduced in hypoxia-reoxygenation condition. However, E2 treatment did not significantly increase trophoblast invasion in the hypoxia-reoxygenation conditions. This may be due to the fact that E2 treatment in hypoxia-reoxygenation conditions was unable to increase GPR30 expression to the levels that result from E2 treatment in normoxic conditions (Fig. 4). Therefore, the data suggest that the action of GPR30 in trophoblasts may not promote estrogen-mediated inhibition of apoptosis induced by oxidative stress alone (20), however may additionally be involved in trophoblast invasion during placentation.
Previous studies indicate that there are distinct vascular adaptation profiles in early onset and late onset preeclampsia, and that the placental pathology in early onset preeclampsia is significantly different from that in late onset preeclampsia (32,33). Due to the sample size in the present study (n=21), it was not possible to investigate GPR30 expression differences in severe and mild preeclampsia or early onset and late onset preeclampsia. Future studies to investigate whether the expression of GPR30 is associated with the severity or time of onset in preeclampsia is warranted. In addition, numerous women with preeclampsia additionally present with growth restricted fetuses. There were three cases of FGR in the present study, and it was not possible to investigate associations between GPR30 and FGR. However, preeclampsia and FGR share common mechanisms (deficient placentation) thus there may be also an association between GPR30 and FGR.
It remains unclear whether the low levels of estrogen cause the reduction of GPR30 expression in preeclampsia or whether the lower expression of GPR30 affect the reduction of estrogen action in preeclampsia. Further study is required.
In conclusion, it was demonstrated that placental GPR30 levels were significantly reduced in preeclampsia. Treatment with one of the common forms of estrogen, E2, increased GPR30 in conditions of normoxia and hypoxia/reoxygenation. However, treatment with E2 in hypoxia-reoxygenation conditions did not increase GPR30 levels to those in normoxic conditions. It was further identified that GPR30 may also be involved in extravillous trophoblast invasion. The present study may suggest that the promotion of placental development through increased levels of estrogen in normal pregnancy is at least partially mediated by GPR30, and that aberrant levels of E2 in preeclampsia contribute to the pathogenesis of the disease through this mechanism.
Acknowledgements
The authors would like to thank Dr. Joanna Stanley, from The University of Auckland (Auckland, New Zealand) for editing this manuscript. The present study was supported by National Natural Science Foundation of China (grant nos. 81370732 and 81571453).
References
- 1.Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. 2005;365:785–799. doi: 10.1016/S0140-6736(05)71003-5. [DOI] [PubMed] [Google Scholar]
- 2.Hubel CA. Oxidative stress in the pathogenesis of preeclampsia; Proc Soc Exp Biol Med; 1999; pp. 222–235. [DOI] [PubMed] [Google Scholar]
- 3.Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308:1592–1594. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 4.Roberts JM, Hubel CA. Is oxidative stress the link in the two-stage model of pre-eclampsia? Lancet. 1999;354:788–789. doi: 10.1016/S0140-6736(99)80002-6. [DOI] [PubMed] [Google Scholar]
- 5.Bonney EA. Demystifying animal models of adverse pregnancy outcomes: Touching bench and bedside. Am J Reprod Immunol. 2013;69:567–584. doi: 10.1111/aji.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jukic AM, Weinberg CR, Wilcox AJ, Baird DD. Effects of early pregnancy loss on hormone levels in the subsequent menstrual cycle. Gynecol Endocrinol. 2010;26:897–901. doi: 10.3109/09513590.2010.487601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heldring N, Pike A, Andersson S, Matthews J, Cheng G, Hartman J, Tujague M, Ström A, Treuter E, Warner M, Gustafsson JA. Estrogen receptors: How do they signal and what are their targets. Physiol Rev. 2007;87:905–931. doi: 10.1152/physrev.00026.2006. [DOI] [PubMed] [Google Scholar]
- 8.Jobe SO, Tyler CT, Magness RR. Aberrant synthesis, metabolism, and plasma accumulation of circulating estrogens and estrogen metabolites in preeclampsia implications for vascular dysfunction. Hypertension. 2013;61:480–487. doi: 10.1161/HYPERTENSIONAHA.111.201624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamimi R, Lagiou P, Vatten LJ, Mucci L, Trichopoulos D, Hellerstein S, Ekbom A, Adami HO, Hsieh CC. Pregnancy hormones, pre-eclampsia, and implications for breast cancer risk in the offspring. Cancer Epidemiol Biomarkers Prev. 2003;12:647–650. [PubMed] [Google Scholar]
- 10.Zeisler H, Jirecek S, Hohlagschwandtner M, Knöfler M, Tempfer C, Livingston JC. Concentrations of estrogens in patients with preeclampsia. Wien Klin Wochenschr. 2002;114:458–461. [PubMed] [Google Scholar]
- 11.Hertig A, Liere P, Chabbert-Buffet N, Fort J, Pianos A, Eychenne B, Cambourg A, Schumacher M, Berkane N, Lefevre G, et al. Steroid profiling in preeclamptic women: Evidence for aromatase deficiency. Am J Obstet Gynecol. 2010;203:477.e1–e9. doi: 10.1016/j.ajog.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 12.Lee SJ, Lee DW, Kim KS, Lee IK. Effect of estrogen on endothelial dysfunction in postmenopausal women with diabetes. Diabetes Res Clin Pract. 2001;54(Suppl 2):S81–S92. doi: 10.1016/S0168-8227(01)00339-4. [DOI] [PubMed] [Google Scholar]
- 13.Maruyama A, Nakayama T, Sato N, Mizutani Y, Furuya K, Yamamoto T. Association study using single nucleotide polymorphisms in the estrogen receptor beta (ESR2) gene for preeclampsia. Hypertens Res. 2004;27:903–909. doi: 10.1291/hypres.27.903. [DOI] [PubMed] [Google Scholar]
- 14.Molvarec A, Vér A, Fekete A, Rosta K, Derzbach L, Derzsy Z, Karádi I, Rigó J., Jr Association between estrogen receptor alpha (ESR1) gene polymorphisms and severe preeclampsia. Hypertens Res. 2007;30:205–211. doi: 10.1291/hypres.30.205. [DOI] [PubMed] [Google Scholar]
- 15.Kolkova Z, Noskova V, Ehinger A, Hansson S, Casslén B. G protein-coupled estrogen receptor 1 (GPER, GPR 30) in normal human endometrium and early pregnancy decidua. Mol Hum Reprod. 2010;16:743–751. doi: 10.1093/molehr/gaq043. [DOI] [PubMed] [Google Scholar]
- 16.Revankar CM, Cimino DF, Sklar LA, Arterburn JB, Prossnitz ER. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science. 2005;307:1625–1630. doi: 10.1126/science.1106943. [DOI] [PubMed] [Google Scholar]
- 17.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146:624–632. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 18.Szego CM, Davis JS. Adenosine 3′,5′-monophosphate in rat uterus: Acute elevation by estrogen; Proc Natl Acad Sci USA; 1967; pp. 1711–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prossnitz ER, Arterburn JB, Sklar LA. GPR30: A G protein-coupled receptor for estrogen. Mol Cell Endocrinol 265–266. 2007:1–142. doi: 10.1016/j.mce.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kanda N, Watanabe S. 17beta-estradiol inhibits oxidative stress-induced apoptosis in keratinocytes by promoting Bcl-2 expression. J Invest Dermatol. 2003;121:1500–1509. doi: 10.1111/j.1523-1747.2003.12617.x. [DOI] [PubMed] [Google Scholar]
- 21.Vivacqua A, Bonofiglio D, Recchia AG, Musti AM, Picard D, Andò S, Maggiolini M. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17beta-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol Endocrinol. 2006;20:631–646. doi: 10.1210/me.2005-0280. [DOI] [PubMed] [Google Scholar]
- 22.Hung TH, Skepper JN, Charnock-Jones DS, Burton GJ. Hypoxia-reoxygenation: A potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ Res. 2002;90:1274–1281. doi: 10.1161/01.RES.0000024411.22110.AA. [DOI] [PubMed] [Google Scholar]
- 23.American College of Obstetricians and Gynecologists; Task Force on Hypertension in Pregnancy: Hypertension in pregnancy. report of the american college of obstetricians and gynecologists' task force on hypertension in pregnancy. Obstet Gynecol. 2013;122:1122–1131. doi: 10.1097/01.AOG.0000437382.03963.88. [DOI] [PubMed] [Google Scholar]
- 24.Prossnitz ER, Oprea TI, Sklar LA, Arterburn JB. The ins and outs of GPR30: A transmembrane estrogen receptor. J Steroid Biochem Mol Biol. 2008;109:350–353. doi: 10.1016/j.jsbmb.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takada Y, Kato C, Kondo S, Korenaga R, Ando J. Cloning of cDNAs encoding G protein-coupled receptor expressed in human endothelial cells exposed to fluid shear stress. Biochem Biophys Res Commun. 1997;240:737–741. doi: 10.1006/bbrc.1997.7734. [DOI] [PubMed] [Google Scholar]
- 26.Carmeci C, Thompson DA, Ring HZ, Francke U, Weigel RJ. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics. 1997;45:607–617. doi: 10.1006/geno.1997.4972. [DOI] [PubMed] [Google Scholar]
- 27.Mårtensson UE, Salehi SA, Windahl S, Gomez MF, Swärd K, Daszkiewicz-Nilsson J, Wendt A, Andersson N, Hellstrand P, Grände PO, et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology. 2009;150:687–698. doi: 10.1210/en.2008-0623. [DOI] [PubMed] [Google Scholar]
- 28.Kumar R, Balhuizen A, Amisten S, Lundquist I, Salehi A. Insulinotropic and antidiabetic effects of 17β-estradiol and the GPR30 agonist G-1 on human pancreatic islets. Endocrinology. 2011;152:2568–2579. doi: 10.1210/en.2010-1361. [DOI] [PubMed] [Google Scholar]
- 29.Hung TH, Burton GJ. Hypoxia and reoxygenation: A possible mechanism for placental oxidative stress in preeclampsia. Taiwan J Obstet Gynecol. 2006;45:189–200. doi: 10.1016/S1028-4559(09)60224-2. [DOI] [PubMed] [Google Scholar]
- 30.Hung TH, Skepper JN, Burton GJ. In vitro ischemia-reperfusion injury in term human placenta as a model for oxidative stress in pathological pregnancies. Am J Pathol. 2001;159:1031–1043. doi: 10.1016/S0002-9440(10)61778-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burton GJ, Hempstock J, Jauniaux E. Oxygen, early embryonic metabolism and free radical-mediated embryopathies. Reprod Biomed Online. 2003;6:84–96. doi: 10.1016/S1472-6483(10)62060-3. [DOI] [PubMed] [Google Scholar]
- 32.Nelson DB, Ziadie MS, McIntire DD, Rogers BB, Leveno KJ. Placental pathology suggesting that preeclampsia is more than one disease. Am J Obstet Gynecol. 2014;210:66.e1–e7. doi: 10.1016/j.ajog.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 33.Stergiotou I, Crispi F, Valenzuela-Alcaraz B, Bijnens B, Gratacos E. Patterns of maternal vascular remodeling and responsiveness in early-versus late-onset preeclampsia. Am J Obstet Gynecol. 2013;209:558.e1–558.e14. doi: 10.1016/j.ajog.2013.07.030. [DOI] [PubMed] [Google Scholar]