

Abstract
As a novel reactive oxygen species (ROS) scavenger, deuterohemin His peptide-6 (DhHP-6) has been demonstrated to prolong the lifespan of Caenorhabditis elegans and has also exhibited protective effects in myocardial ischemia-reperfusion injury. Whether similar effects occur during cerebral ischemia-reperfusion (CIR) injury remains to be elucidated. The present study evaluated the function of DhHP-6 and its underlying mechanisms in a middle cerebral artery occlusion (MCAO) model in rats. The focal transient MCAO model was implemented using the Longa method of ischemia for 2 h followed by reperfusion for 22 h in male Wistar rats. DhHP-6 was administered at the onset of reperfusion via intraperitoneal injection. The infarct volume, brain edema, brain apoptosis and neurological function were evaluated 24 h following stroke. To further determine the role of DhHP-6 in CIR injury, the levels of ROS and malondialdehyde (MDA), the activities of superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GSH-Px), and the protein expression levels of B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax), cleaved caspase-3, cytochrome c, Bcl-2 and phosphorylated-Akt/Akt were measured in ischemic cortex tissues. The results demonstrated that DhHP-6 significantly improved infarct volume, brain edema and neurological deficits, and reduced the percentage of TUNEL-positive cells. The levels of ROS and MDA were decreased, whereas no significant changes in the activities of SOD, CAT and GSH-Px were observed. The levels of Bax, cleaved caspase-3, and cytochrome c were downregulated, whereas the levels of Bcl-2 and p-Akt/Akt were upregulated. The results of the present study indicated that DhHP-6 may offer therapeutic potential for cerebral ischemia. The neuroprotective effects of DhHP-6 maybe mediated by its anti-oxidative properties, anti-apoptotic activities, or activation of the phosphoinositide 3-kinase/Akt survival pathway.
Keywords: deuterohemin His peptide-6, cerebral ischemia, oxidative stress, neuroprotection, apoptosis
Introduction
Cerebrovascular disorders are the third leading cause of mortality worldwide, and the majority of affected patients exhibit cerebral ischemia (1). The recovery of blood flow to injured tissue has been demonstrated to be the most effective therapeutic strategy to relieve the clinical symptoms of cerebral ischemia, however, cerebral ischemia-reperfusion (CIR) injury can occur, which is a more serious clinical outcome. CIR injury presents a major medical challenge and requires extensive investigation, as there are currently few neuroprotective treatments (2,3).
Increasing evidence shows that oxidative stress is crucial in facilitating neuronal death during CIR. During CIR injury, abnormal mitochondrial activity produces high concentrations of reactive oxygen species (ROS), and leads to cell damage and eventual apoptosis or necrosis (4). Therefore, scavenging ROS may improve the outcomes of CIR injury in humans. Although several agents, including certain peptides and proteins, have shown promising in vitro activities, disadvantages, including hydrophobicity, antigenicity and large molecular size, impede the delivery of these substances to brain tissues and thereby limit their therapeutic benefits (5,6).
Deuterohemin His peptide-6 (DhHP-6; Dh-β-AHTVEK-NH2), is a novel microperoxidase mimetic with a molecular weight of 1,230 Da. It has been demonstrated that DhHP-6 can enter cells and can exhibit high enzyme-activity to scavenge free radicals effectively (7,8). Furthermore, DhHP-6 has been observed to increase survival rates in Caenorhabditis elegans by promoting the elimination of oxidative stress (9,10). The protective effects of DhHP-6 as a scavenger of ROS have also been observed in myocardial ischemia-reperfusion injury. The authors have observed that DhHP-6 also protects PC12 cells against H2O2-induced oxidative stress injury (Yang et al, unpublished data). However, whether DhHP-6 exhibits protective effects on CIR injury in vivo remains to be elucidated. The aim of the present study was to investigate the function of DhHP-6 and its underlying mechanisms using a CIR rat model.
Materials and methods
Materials
DhHP-6 was provided by Dr Li Wei of the Life Science College at Jilin University (Changchun, China) and had a purity of >99%. Primary polyclonal antibodies against B-cell lymphoma-2 (Bcl-2; cat. no. ab59348), cleaved caspase-3 (cat. no. ab2302), cytochrome c (cat. no. ab90529), Bcl-2-associated X protein (Bax; cat. no. ab77566) and p-Akt (cat. no. ab8932) were purchased from Abcam (Cambridge, UK). The biotinylated secondary antibodies (cat. no. sc2357) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The catalase (CAT; cat. no. A007-1-1), glutathione peroxidase (GSH-Px; cat. no. A005), superoxide dismutase (SOD; cat. no. A001-3) and malondialdehyde (MDA; cat. no. A003-1) assay kits were purchased from Jianchen Bioengineering Institute (Nanjing, China). The TUNEL kit was acquired from Maixin Bioengineering Institute (Fuzhou, China).
Animals
Male Wistar rats (6–8 weeks of age; 250–280 g) were obtained from the Center of Laboratory Animal Science of Jilin University. The rats were maintained in an animal house at a temperature of 22±2°C in a 12-h light/dark cycle. The animals were provided with food and water ad libitum. The protocol used in the present study strictly adhered to the rules of the Jilin University Animal Care and Use Committee for all procedures and followed the guidelines outlined in the Principles of Laboratory Nursing of Animals (11).
Experimental groups and drug administration
A total of 80 male rats were used in the present study. The rats were divided equally into two groups. One group was used to measure neurological deficits and infarct area, and the other group was used to evaluate ROS levels, MDA content, and activities of CAT, SOD and GSH-Px. This group was also used for performing TUNEL staining and western blot analysis. In each group, 40 rats were randomly separated into four subgroups (n=10): Sham-operated group (Sham), ischemia-reperfusion group (I/R), I/R+DhHP-6 (1 mg/kg) group (I/R+DH) and I/R+DhHP-6 (0.1 mg/kg) group (I/R+DL). DhHP-6 was administered intraperitoneally at the onset of reperfusion and it was only given once. The animals in the sham and I/R groups were treated with saline in parallel.
Establishment of focal cerebral ischemia-reperfusion
The occlusion and reperfusion model of the middle cerebral artery (MCA) in rats was performed using the Longa method, which has been described previously (12). In brief, 20% urethane (5 ml/kg) was used to anesthetize the animals. The left common carotid artery, the internal carotid artery (ICA) and the external carotid artery were carefully exposed. A 3-0 monofilament nylon suture was inserted into the left ICA, which was inserted ~18.0 mm from the carotid artery bifurcation to obstruct the MCA. At 2 h post-MCAO, reperfusion was performed by withdrawal of the nylon suture. During surgery, the rectal temperature of the animals was maintained at 37.0±0.5°C. The animals in the sham group underwent the same surgical procedure as those in the other animal groups, but did not receive the nylon suture.
Neurological deficit assessment
Behavioral tests were performed 24 h following MCAO by two investigators who were blinded to the experimental groups. The scoring system was as follows: 0, no manifestation of neurological dysfunction; 1, failure to fully stretch the forepaw contralateral to the injured side, and failure to stretch and turn the trunk towards the injured side when held by the tail; 2, spinning motion toward the uninjured side; 3, failure to bear weight on the injured side; and 4, inability to walk normally. When the neurological deficit scores were higher, the disorders in exercise behavior were more severe (13,14).
Evaluation of infarct area
The brains of the rats anesthetized with 20% urethane (5 ml/kg i.p.) were removed and immediately placed on a frozen surface for 5 min. Each brain was then cut into five 2-mm-thick coronal slices and stained at 37°C for 30 min in 2% triphenyltetrazolium chloride (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Infarct volume was detected using image analysis software (Image Pro plus 6.0 system; Media Cybernetics, Inc., Rockville, MD, USA). To avoid data errors caused by brain edema, the following formula was used to evaluate infarct area: Infarct volume (%) = (contralateral volume - ipsilateral non-infarct volume)/contralateral volume.
Assessment of brain edema
The wet-dry method was used to assess brain edema. Briefly, fresh tissue was weighed immediately following isolation (termed wet weight). The tissues were then dried at 100°C in the oven for 24 h, followed by weighing again (termed dry weight). The percentage of brain water content was evaluated using the following formula: Brain water content (%) = (wet weight - dry weight)/wet weight × 100% (1).
Evaluation of ROS levels in the brain
The tissue samples from the ischemic cerebral cortex were homogenized in cold isolation buffer containing 0.25 M sucrose, 10 mM Tris-HCl, 0.5 M EDTA and 250 µg/ml BSA (pH 7.1). The cell fragments and nuclei were centrifuged at 4,000 × g for 3 min at 4°C and then at 1,000 × g for 5 min at 4°C. The supernatants were then centrifuged again for 15 min at 10,000 × g at 4°C. Mitochondria were suspended in the isolation buffer. Mitochondria isolated from different groups (0.5 mg protein, measured with biocinchoninic acid protein assay kit) were incubated with 10 µM DCFH-DA (Beyotime Institute of Biotechnology, Shanghai, China) at 37°C for 15 min in the dark. The levels of intracellular ROS were evaluated using flow cytometry (BD Biosciences, Franklin Lakes, NJ, USA) at an emission wavelength of 525 nm and an excitation wavelength of 488 nm (15,16).
Measurement of MDA content and activities of SOD, CAT and GSH-Px
Tissue samples of the ischemic cortex were homogenized in ice-cold saline and then centrifuged at 2,000 × g for 15 min at 4°C to obtain 10% homogenates. The concentration of MDA was determined using an assay kit (Jiancheng Biotechnology Institute, Nanjing, China) according to the manufacturer's protocol. The activities of CAT, SOD and GSH-Px were also determined using kits (Jiancheng Biotechnology Institute). The protein concentrations were evaluated using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific, Inc., Walham, MA, USA).
TUNEL staining
TUNEL-staining was performed using an in situ apoptosis detection kit (Maixin Biotechnology, Fuzhou, China) according to the manufacturer's protocol. The tissue sections were incubated with proteinase K (20 µg/ml) for 15 min and 0.3% H2O2 for 30 min at 37°C. The sections were then treated with terminal deoxyribonucleotidy ltransferase enzyme at 37°C for 1 h and peroxidase-conjugated antibody for at 37°C for 30 min. To terminate the reaction, the sections were incubated in a reaction termination buffer for 5 min at room temperature. The sections were visualized following staining with diaminobenzidinete trahydrochloride. Apoptotic cells exhibited brown particles in the nuclei. A total of five visual fields in the cerebral cortex were analyzed from each section. TUNEL-positive cells were counted using an optical microscope (Nikon Ti; Nikon Corporation, Tokyo, Japan). The total number of cells and the number of TUNEL-positive cells were assessed in each field. The percentage of TUNEL-positive cells was calculated as follows: TUNEL-positive cells (%) = number of positive cells/number of total cells × 100% (17).
Western blot analysis
Protein samples were prepared from the ischemic cortex by homogenization in ice-cold lysate buffer (Beyotime Institute of Biotechnology). The mitochondrial and cytosolic proteins were prepared as previously described (18). To detect the expression of Bax, cytochrome c and Bcl-2, the mitochondrial and cytosolic protein extracts were evaluated independently. The expression of cleaved caspase-3 was detected using the total protein lysates. The total protein samples (60 µg) were separated on 12% SDS polyacrylamide gels and were transferred onto polyvinylidenedifluoride membranes. The membranes were blocked with 5% BSA for 2 h at room temperature, and then were incubated overnight at 4°C with rabbit polyclonal antibodies Bcl-2 (1:1,000), Bax (1:1,000), cytochrome c (1:1,000), cleaved caspase-3 (1:500) and p-Akt (1:500) antibodies, (all from Santa Cruz Biotechnology, Inc.). The membranes were then incubated with secondary antibody (goat anti-rabbit; 1:1,000; Santa Cruz Biotechnology, Inc.) for 2 h at room temperature. The immunoreactive bands were visualized using enhanced chemiluminescence. Images of the protein bands were captured using an imaging densitometer and were quantified using image analysis software (Quantity One; version 4.62; Bio-Rad Laboratories, Inc., Hercules, CA, USA). The protein values were normalized to those of β-actin in the same lane.
Statistical analysis
All values are presented as the mean ± standard deviation. Significant differences between groups were performed using one-way analysis of variance followed by the Tukey's post hoc test. GraphPad Prism statistical software (version 5.0; GraphPad Software, Inc., La Jolla, CA, USA) was used for statistical analysis. P<0.05 was considered to indicate a statistically significant difference.
Results
DhHP-6 reduces infarct volume
MCAO produced a well-defined infarct, which included the cortex and striatum (Fig. 1A and B). In the I/R group, 2 h of cerebral ischemia and 22 h of reperfusion caused infarction, which involved 35.28±4.13% of the ipsilateral cerebral hemisphere. The infarct volume in the I/R+DH group was significantly reduced to 23.56±3.47% (P<0.05). The I/R+DL group also exhibited a reduced infarct volume, however, this was not a significant difference (P>0.05).
Figure 1.

Effect of DhHP-6 on cerebral ischemia induced by 2 h of middle cerebral artery occlusion and 22 h of reperfusion. (A) Triphenyltetrazolium chloride staining of coronal brain sections 22 h following reperfusion. Infarct brain tissues appeared unstained. Tissues in the I/R+DH group, but not the I/R+DL group, demonstrated significant reductions in (B) infarct volume, (C) neurological deficits and (D) brain edema, in a dose-dependent manner. Data are shown as the mean ± standard deviation. One-way analysis of variance and Tukey's post hoc test were performed. ###P<0.001 and ##P<0.01, vs. Sham group; *P<0.05, vs. I/R group (n=10). I/R, ischemia/reperfusion; DhHP-6, deuterohemin His peptide-6; DH, 1 mg/kg/day DhHP-6; DL, 0.1 mg/kg/day DhHP-6.
DhHP-6 decreases neurological dysfunction
Neurological symptoms were evaluated 22 h following reperfusion (Fig. 1C). Normal reflexes were present in all animals in the sham group (score 0), where as animals in the I/R group presented with severe neurological deficits (2.34±0.15; P<0.001). Compared with the I/R group, the neurological deficit scores were markedly reduced to 1.57±0.28 in the I/R+DH group (P<0.05), but did not differ significantly from those in the I/R+DL group (P>0.05).
DhHP-6 decreases brain edema
As shown in Fig. 1D, MCAO led to an increase in brain water content in the right hemisphere of the I/R group, however, the level of brain water content was significantly reduced in the I/R+DH group (P<0.05) in comparison.
DhHP-6 reduces the oxidative stress caused by MCAO
DhHP-6 acts as a free radical scavenger (9). Therefore, the present study aimed to determine whether the levels of free radicals and oxidative stress were reduced by DhHP-6 treatment following I/R. To evaluate this, the levels of ROS and MDA, which is a lipid peroxidation product caused by oxidative stress, were examined. The activities of antioxidant enzymes, including CAT, SOD and GSH-Px, were also measured.
The ROS level was determined as the percentage of fluorescence intensity, compared with that in the sham group (Fig. 2A). The mitochondrial DCF fluorescence of the I/R group exceeded that of the shamgroup (P<0.01). The levels of ROS in the mitochondria were decrease by 21.34% in the I/R+DH group. Compared with the I/R group, the I/R+DH group showed a significant decrease in mitochondrial ROS (P<0.05).
Figure 2.

Effect of DhHP-6 on oxidative stress induced by 2 h of middle cerebral artery occlusion and 22 h of reperfusion. (A) ROS levels in brain mitochondria are expressed as the percentage of fluorescence intensity relative to that in the Sham group. DhHP-6 attenuated ROS formation in a dose-dependent manner. Compared with the I/R group, the ROS level was significantly reduced in the I/R+DH group. (B) MDA content was significantly reduced in the I/R+DH group, compared with that in the I/R group. Data are shown as the mean ± standard deviation. One-way analysis of variance and Tukey's post hoc test were performed. ##P<0.01, vs. Sham group; *P<0.05, vs. I/R group (n=10). I/R, ischemia/reperfusion; DhHP-6, deuterohemin His peptide-6; DH, 1 mg/kg/day DhHP-6; DL, 0.1 mg/kg/day DhHP-6, ROS, reactive oxygen species; MDA, malondialdehyde.
As exhibited in Fig. 2B, I/R increased the formation of MDA. Compared with the level in the I/R group, MDA was significantly reduced to 5.40±0.96 nmol/mg (P<0.05) in the I/R+DH group.
As demonstrated in Table I, CIR injury resulted in a significant decrease in the activities of CAT, GSH-Px and SOD. Treatment with DhHP-6 appeared to elevate the activities of these antioxidant enzymes; however, no significant differences were observed between the I/R group and the DhHP-6 treatment group (P>0.05).
Table I.
Effect of DhHP-6 on antioxidant enzyme activities following 22 h of 12 reperfusion.
| Group | Dose (mg/kg) | CAT (U/mg protein) | SOD (U/mg protein) | GSH-Px (U/mg protein) |
|---|---|---|---|---|
| Sham | – | 65.46±9.25 | 11.58±2.75 | 46.32±5.45 |
| I/R | – | 28.38±7.62a | 5.90±2.19b | 27.04±4.24b |
| I/R+DH | 1 | 35.69±6.81c | 8.58±2.23c | 35.96±5.08c |
| I/R+DL | 0.1 | 30.10±5.36 | 6.03±1.91 | 26.76±4.15 |
All values are presented as the mean ± standard deviation (n=10 rats per group).
P<0.001
P<0.01, vs. Sham group
P<0.05, vs. I/R group. DhHP-6, 15 deuterohemin His peptide-6; CAT, catalase; SOD, superoxide dismutase; GSH-Px, 16 glutathione peroxidase; I/R, ischemia/reperfusion; DH, 1 mg/kg/day DhHP-6; DL, 0.1 mg/kg/day DhHP-6.
Together, these data indicated that DhHP-6 reduced oxidative stress, however, the effect was not mediated by enhancing the activities of endogenous CAT, GSH-Px or SOD antioxidant enzymes.
To examine the effects of DhHP-6 on apoptosis, apoptotic cells were identified using TUNEL staining in the parietal cortex 24 h post-MCAO. There were few TUNEL-positive nuclei in the brain sections from animals in the sham group; however, the number of TUNEL-positive cells was significantly increased in the I/R group. DhHP-6 prevented the increase in TUNEL-positive cells. As shown in Fig. 3A and B, the proportion of TUNEL-positive cells was significantly reduced from 73.5±6.09% in the I/R group to 52.83±5.89% in the I/R+DH group (P<0.05).
Figure 3.

Effect of DhHP-6 on neuronal apoptosis of the ischemic cortex in rats 22 h post-reperfusion. (A) TUNEL staining was used to identify apoptotic cells in the parietal cortex. Arrows indicate TUNEL-positive cells (magnification, ×200). (B) As demonstrated in the bar graphs, the apoptotic index indicates the percentage of TUNEL-positive cells in the ischemic cortex. The percentage of TUNEL-positive cells in the I/R+DH group was significantly lower, compared with that in the I/R group. Data are demonstrated as the mean ± standard deviation. One-way analysis of variance and Tukey's post hoc test were performed. ###P<0.001, vs. Sham group; *P<0.05, vs. I/R group (n=5). DhHP-6, deuterohemin His peptide-6; DH, 1 mg/kg/day DhHP-6; DL, 0.1 mg/kg/day DhHP-6.
DhHP-6 increases the Bcl-2/Bax ratio
Bcl-2 is a key mitochondrial protein, which contributes to cell survival. In the present study, the levels of Bcl-2 were reduced in the I/R group, compared with those in the sham group 24 h post-MCAO (Fig. 4). However, the important apoptotic protein, Bax, was increased significantly in the I/R group. The ratio of Bcl-2/Bax was decreased significantly following I/R (P<0.001), however, there was an increase in the Bcl-2/Bax ratio when DhHP-6 was administered at a dose of 1 mg/kg (P<0.05). These data indicated that maintaining the balance between Bcl-2 and Bax was dependent on DhHP-6.
Figure 4.
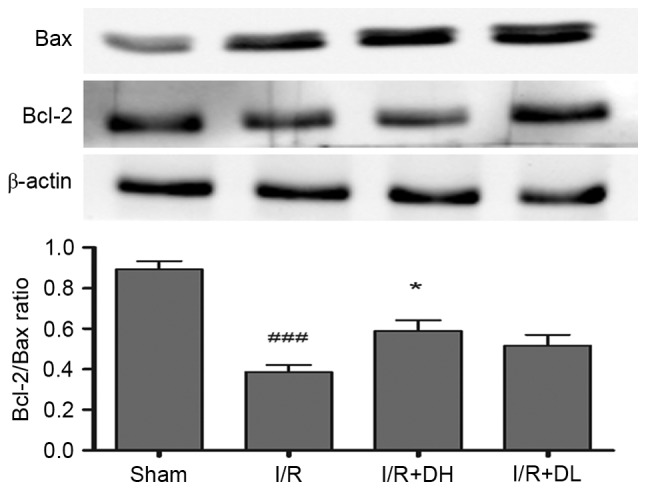
Effects of DhHP-6 on expression levels of Bcl-2 and Bax in the rat brain 22 h post-reperfusion. The protein expression levels of Bcl-2 and Bax in subcellular fractions were examined using western blot analysis. Scanning and quantification of the intensity of protein bands were performed using image analysis software. Representative bands from the Sham, I/R, and DhHP-6 (I/R+DH and I/R+DL)-treated groups, and the corresponding β-actin bands (loading control) are demonstrated. Middle cerebral artery occlusion mediated a decrease in Bcl-2, and an increase in Bax led to a significantly decreased Bcl-2/Bax ratio in mitochondria, compared with the ratio in the Sham group. Compared with the I/R group, the ratio of Bcl-2/Bax in mitochondria was significantly higher in the I/R+DH group. Data are shown as the mean ± standard deviation. One-way analysis of variance and Tukey's post hoc test were used. ###P<0.001, vs. Sham group; *P<0.05, vs. I/R group (n=3). DhHP-6, deuterohemin His peptide-6; DH, 1 mg/kg/day DhHP-6; DL, 0.1 mg/kg/day DhHP-6; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X protein.
DhHP-6 inhibits the activation of caspase-3 and release of cytochrome c
As demonstrated in Fig. 5A, in the I/R group, an increased level of cytochrome c was released into the cytoplasm from the mitochondria, and this effect was significantly reduced by treatment with DhHP-6 (P<0.05). The levels of cleaved caspase-3 were also investigated. DhHP-6 reduced the expression of cleaved caspase-3. These effects were significant in the I/R+DH group (P<0.05; Fig. 5B).
Figure 5.

Effects of DhHP-6 on the rate of cytochrome c release and the activation of caspase-3 22 h post-reperfusion. The contents of cytochrome c in the subcellular fractions were examined using western blot analysis. (A) The ratio of cytochrome c content in the cytosolic and mitochondrial fractions was increased significantly in the I/R group, but the ratio was decreased markedly in the I/R+DH group. (B) Additionally, the expression level of cleaved caspase-3 was increased in the I/R group at 22 h post-reperfusion, but significantly decreased in the I/R+DH group, compared with the I/R group. Data are shown as the mean ± standard deviation. One-way analysis of variance and Tukey's post hoc test were performed. ###P<0.001 and ##P<0.01, vs. Sham group; *P<0.05, vs. I/R group (n=3). DhHP-6, deuterohemin His peptide-6; DH, 1 mg/kg/day DhHP-6; DL, 0.1 mg/kg/day DhHP-6; mito, mitochondria; cyto, cytosol; I/R, ischemia/reperfusion.
DhHP-6 increases the expression of p-Akt/Akt
As demonstrated in Fig. 6, compared with the I/R group, treatment with DhHP-6 (1 mg/kg) led to a significant increase in the expression of p-Akt/Akt (P<0.05).
Figure 6.
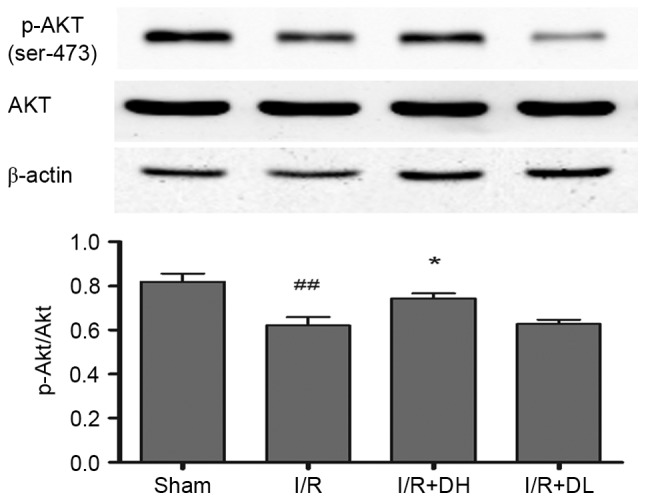
Effect of DhHP-6 on p-Akt/Akt 22 h post-reperfusion. To determine whether the activation of Akt contributes to DhHP-6-dependent protection against apoptosis induced by I/R injury, levels of p-Akt and Akt were examined using western blot analysis. The histograms demonstrate the level of p-Akt relative to that of Akt. Compared with the I/R group, the p-Akt/Akt ratio was significantly increased in the I/R+DH group. Data are shown as the mean ± standard deviation. One-way analysis of variance and Tukey's post hoc test were performed. ##P<0.01, vs. Sham group; *P<0.05, vs. I/R group (n=3). DhHP-6, deuterohemin His peptide-6; DH, 1 mg/kg/day DhHP-6; DL, 0.1 mg/kg/day DhHP-6; p-, phosphorylated; I/R, ischemia/reperfusion; Akt, AKT serine/threonine kinase.
Discussion
In the present study, the function of DhHP-6 in focal CIR injury was investigated in a rat model. The administration of DhHP-6 at 1 mg/kg was sufficient to reduce neurological deficits, brain edema and infarct volumes; however, a dose of 0.1 mg/kg did not exert protective effects. These data indicated that the beneficial effects of DhHP-6 occur in a dose-dependent manner and also suggested that the effective dose may be between 1 and 0.1 mg/kg. These results indicate a potential novel application for DhHP-6 as a potent neuroprotectant in cerebral ischemic disorder (19,20).
During I/R, ROS generation is enhanced and antioxidant defenses in brain tissues are weakened; therefore, an imbalance between oxidants and antioxidants occurs. The brain isvulnerable to oxidative stress as it contains high levels of unsaturated fatty acids, which can be oxidized leading to lipid peroxidation (21,22). ROS are produced primarily by mitochondria and are generated in I/R injury. DhHP-6 is a novel free radical scavenger and is synthesized as a microperoxidase mimetic. It was previously demonstrated that DhHP-6 produces antioxidant effects in Caenorhabditis elegans (9). Another study showed that DhHP-6 treatment caused complete reduction in reactive oxygen (10). In the present study, the levels of ROS in mitochondria were markedly increased following I/R injury, and this increase was significantly attenuated by DhHP-6 treatment. It was shown that oxidative injury to cell membrane lipids was caused by free radicals, which can produce MDA. The level of MDA in the I/R group was 2.18-fold higher, compared with that of the sham group and was decreased significantly in the DhHP-6 group (Table I). The human body has different mechanisms to decrease the impact of oxidative injury. The primary defenses against oxidative injury are antioxidant enzymes, including GSH-Px, SOD and CAT. However, the antioxidant effects of DhHP-6 in the present study were not mediated through increasing the activities of endogenous GSH-Px, SOD or CAT enzymes (Table I) (22,23).
The uncontrolled accumulation of ROS can disturb mitochondrial function and lead to cell apoptosis. Mitochondria are important in caspase-dependent and caspase-independent apoptotic processes. Mitochondria can induce the opening of the mitochondrial permeability transition pore and thereby lead to the release of cytochrome c. The Bcl-2 family of proteins, which are expressed on outer mitochondrial membranes, are also critical in mitochondria-mediated apoptosis (24). In the present study, Bax was identified in cytosolic brain tissue extracts. When Bax is activated, it translocates to mitochondria, where it then stimulates mitochondria to release cytochrome c. Bcl-2 can inhibit the activation of Bax (25,26), however, the present study showed that significantly higher levels of Bax were expressed in the mitochondria of rat brains subjected to MCAO; this effect was attenuated by treatment with DhHP-6. Therefore, DhHP-6 inhibited the release of cytochrome c andinhibited apoptosis by ameliorating the CIR-induced degradation of the Bcl-2/Bax ratio, and by retaining the normal balance between Bcl-2 and Bax. In the present study, it was also observed that TUNEL-positive cells increased in the cortex 10-fold following 22 h of brain reperfusion. This increase was significantly reduced in the I/R+DH group. Therefore, these results indicated that DhHP-6 inhibited the mitochondria-initiated apoptotic pathway and thereby improved neuronal survival (27,28).
The activation of phosphoinositide 3-kinase (PI3K) and its downstream factor, Akt has been demonstrated to prevent apoptosis and promote cell survival. Increasing evidence has indicated that ROS-induced apoptosis correlates with the PI3K/Akt pathway in SH-SY5Y cells (29). In focal CIR injury models, p-Akt is usually dephosphorylated at Ser473. It has been demonstrated that, following activation, Akt can promote cell survival and subsequently inactivate apoptosis-inducing factors, including GSK3β (30). A previous study also demonstrated that GSK3β exerts its pro-apoptotic effects by regulating Bax localization to mitochondria (31,32). The results of the present study indicated thatDhHP-6 markedly enhanced the phosphorylation of Akt in rats following 2 h of MCAO and 22 h of reperfusion, compared with the results observed for the I/R group.
Taken together, the findings of the present study demonstrated that DhHP-6 reduced the levels of brain edema, infarct volumes and neurological deficits in CIR. The protective mechanisms of DhHP-6 in CIR injury included scavenging of radicals and inhibiting the mitochondrial apoptotic-signaling pathway, which may involve activation of the PI3K/Akt signaling pathway. Therefore, the beneficial effects of DhHP-6 on CIR investigated in the present study indicated that DhHP-6 may be a promising therapeutic candidate for protecting against cell death resulting from ischemic damage.
Acknowledgements
The authors would like to thank Dr Li Wei of the Life Science College at Jilin University for the provision of DhHP-6.
References
- 1.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marlangue C Charriaut, Remolleau S, Zouaoui D Aggoun, Ben-Ari Y. Apoptosis and programmed cell death: A role in cerebral ischemia. Biomed Pharmacother. 1998;52:264–269. doi: 10.1016/S0753-3322(98)80012-7. [DOI] [PubMed] [Google Scholar]
- 3.Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke. 2009;4:461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 5.Egleton RD, Davis TP. Bioavailability and transport of peptides and peptide drugs into the brain. Peptides. 1997;18:1431–1439. doi: 10.1016/S0196-9781(97)00242-8. [DOI] [PubMed] [Google Scholar]
- 6.Navaratna D, Guo S, Arai K, Lo EH. Mechanisms and targets for angiogenic therapy after stroke. Cell Adh Migr. 2009;3:216–223. doi: 10.4161/cam.3.2.8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong QG, Zhang Y, Wang MS, Feng J, Zhang HH, Wu YG, Gu TJ, Yu XH, Jiang CL, Chen Y, et al. Improvement of enzymatic stability and intestinal permeability of deuterohemin-peptide conjugates by specific multi-site N-methylation. Amino Acids. 2012;43:2431–2441. doi: 10.1007/s00726-012-1322-y. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Sun Y, Guo W, Fang C, Fawcett JP, Li W, Gao Y, Yang Y, Gu J. Determination of a deuterohemin-peptide conjugate in rat plasma by liquid chromatography-tandem mass spectrometry and application to a preclinical pharmacokinetic study. J Pharm Biomed Anal. 2014;98:401–406. doi: 10.1016/j.jpba.2014.06.026. [DOI] [PubMed] [Google Scholar]
- 9.Huang L, Li P, Wang G, Guan S, Sun X, Wang L. DhHP-6 extends lifespan of Caenorhabditis elegans by enhancing nuclear translocation and transcriptional activity of DAF-16. Free Radic Res. 2013;47:316–324. doi: 10.3109/10715762.2013.773588. [DOI] [PubMed] [Google Scholar]
- 10.Guan S, Li P, Luo J, Li Y, Huang L, Wang G, Zhu L, Fan H, Li W, Wang L. A deuterohemin peptide extends lifespan and increases stress resistance in Caenorhabditis elegans. Free Radic Res. 2010;44:813–820. doi: 10.3109/10715762.2010.485991. [DOI] [PubMed] [Google Scholar]
- 11.Xue H, Ji Y, Wei S, Yu Y, Yan X, Liu S, Zhang M, Yao F, Lan X, Chen L. HGSD attenuates neuronal apoptosis through enhancing neuronal autophagy in the brain of diabetic mice: The role of AMP-activated protein kinase. Life Sci. 2016;153:23–34. doi: 10.1016/j.lfs.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.STR.20.1.84. [DOI] [PubMed] [Google Scholar]
- 13.Yenari MA, Xu L, Tang XN, Qiao Y, Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: Improvement by minocycline in vivo and in vitro. Stroke. 2006;37:1087–1093. doi: 10.1161/01.STR.0000206281.77178.ac. [DOI] [PubMed] [Google Scholar]
- 14.Wang N, Zhang Y, Wu L, Wang Y, Cao Y, He L, Li X, Zhao J. Puerarin protected the brain from cerebral ischemia injury via astrocyte apoptosis inhibition. Neuropharmacology. 2014;79:282–289. doi: 10.1016/j.neuropharm.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Yuan X, Liu T, Liu L, Hu Y, Wang Z, Zheng Q. Neuroprotective activity of lavender oil on transient focal cerebral ischemia in mice. Molecules. 2012;17:9803–9817. doi: 10.3390/molecules17089803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon SH, Hong SI, Kim JA, Jung YH, Kim SY, Kim HC, Lee SY, Jang CG. The neuroprotective effects of Lonicera japonica THUNB. against hydrogen peroxide-induced apoptosis via phosphorylation of MAPKs and PI3K/Akt in SH-SY5Y cells. Food Chem Toxicol. 2011;49:1011–1019. doi: 10.1016/j.fct.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 17.Yu SM, Kim SJ. Thymoquinone-induced reactive oxygen species causes apoptosis of chondrocytes via PI3K/Akt and p38kinase pathway. Exp Biol Med (Maywood) 2013;238:811–820. doi: 10.1177/1535370213492685. [DOI] [PubMed] [Google Scholar]
- 18.Kim DW, Jeong HJ, Kang HW, Shin MJ, Sohn EJ, Kim MJ, Ahn EH, An JJ, Jang SH, Yoo KY, et al. Transduced human PEP-1-catalase fusion protein attenuates ischemic neuronal damage. Free Radic Biol Med. 2009;47:941–952. doi: 10.1016/j.freeradbiomed.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 19.Pan R, Rong Z, She Y, Cao Y, Chang LW, Lee WH. Sodium pyruvate reduced hypoxic-ischemic injury to neonatal rat brain. Pediatr Res. 2012;72:479–489. doi: 10.1038/pr.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang F, Wang S, Signore AP, Chen J. Neuroprotective effects of leptin against ischemic injury induced by oxygen-glucose deprivation and transient cerebral ischemia. Stroke. 2007;38:2329–2336. doi: 10.1161/STROKEAHA.107.482786. [DOI] [PubMed] [Google Scholar]
- 21.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.STR.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 22.Keller JN, Kindy MS, Holtsberg FW, St Clair DK, Yen HC, Germeyer A, Steiner SM, Bruce-Keller AJ, Hutchins JB, Mattson MP. Mitochondrial manganese superoxide dismutase prevents neural apoptosis and reduces ischemic brain injury: Suppression of peroxynitrite production, lipid peroxidation, and mitochondrial dysfunction. J Neurosci. 1998;18:687–697. doi: 10.1523/JNEUROSCI.18-02-00687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM. Mechanisms of cell death inoxidative stress. Antioxid Redox Signal. 2007;9:49–89. doi: 10.1089/ars.2007.9.49. [DOI] [PubMed] [Google Scholar]
- 24.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J NeuroSci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 26.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 27.Xing Y, Zhang X, Zhao K, Cui L, Wang L, Dong L, Li Y, Liu Z, Wang C, Zhang X, et al. Beneficial effects of sulindac in focal cerebral ischemia: A positive role in Wnt/β-catenin pathway. Brain Res. 2012;1482:71–80. doi: 10.1016/j.brainres.2012.08.057. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell. 1998;94:739–750. doi: 10.1016/S0092-8674(00)81733-X. [DOI] [PubMed] [Google Scholar]
- 29.González-Sarrías A, Núñez-Sánchez MÁ, Tomás-Barberán FA, Espín JC. Neuroprotective effects of bioavailable polyphenol-derived metabolites against oxidative stress-induced cytotoxicity in human neuroblastoma SH-SY5Y Cells. J Agric Food Chem. 2017;65:752–758. doi: 10.1021/acs.jafc.6b04538. [DOI] [PubMed] [Google Scholar]
- 30.Li F, Omori N, Jin G, Wang SJ, Sato K, Nagano I, Shoji M, Abe K. Cooperative expression of survival p-ERK and p-Akt signals in rat brain neurons after transient MCAO. Brain Res. 2003;962:21–26. doi: 10.1016/S0006-8993(02)03774-5. [DOI] [PubMed] [Google Scholar]
- 31.Friguls B, Justicia C, Pallàs M, Planas AM. Focal cerebischemia causes two temporal waves of Akt activation. Neuroreport. 2001;12:3381–3384. doi: 10.1097/00001756-200110290-00046. [DOI] [PubMed] [Google Scholar]
- 32.Sun B, Feng M, Tian X, Lu X, Zhang Y, Ke X, Huang S, Cao J, Ding X. DL-3-n-Butylphthalide protects rat bone marrow stem cells against hydrogen peroxide-induced cell death through antioxidation and activation of PI3K-Akt pathway. Neurosci Lett. 2012;516:247–252. doi: 10.1016/j.neulet.2012.04.003. [DOI] [PubMed] [Google Scholar]