

Abstract
As the resident macrophages of the brain's innate immune system, microglial cells are key modulators in the neurodegenerative disease Alzheimer's disease (AD). In particular, the activation and accumulation of microglial cells around amyloid plaques is considered to result in chronic neuroinflammation. Although the pathologic mechanism remains to be fully elucidated, inflammation has been shown to be critical in the pathogenesis of AD. The Gengnianchun (GNC) formula has long been used to treat perimenopausal syndrome clinically, and is particularly effective in improving learning ability and memory. Our previous study demonstrated that GNC formula had an anti-inflammatory effect and offered neuroprotection in animal experiments. In the present study, the anti-inflammatory properties of GNC and its underlying mechanism of action were examined in BV-2 microglial cells. Amyloid-β peptide (Aβ)-stimulated microglial cells were examined for the production of proinflammatory cytokines and the underlying signaling pathways. Compared with the normal control group, the protein expression levels of IL-1β and TNF-α were significantly increased following treatment with Aβ (P<0.01), but medicated rat serum containing GNC formula (MRS) could significantly attenuated the Aβ-induced secretion of these pro-inflammatory cytokines. It was identified by CCK-8 assay that the viability of the BV-2 cells was not reduced following treatment with various concentrations of MRS. The phosphorylation of factor-κB (NF-κB) and c-Jun N-terminal kinase (JNK) was markedly increased following treatment with Aβ, compared with the normal control group (P<0.01). However, treatment with MRS resulted in a significant reduction in the phosphorylation of NF-κB (P<0.05). These results suggested that MRS suppressed the Aβ-induced inflammatory response of microglial cells by inhibiting the NF-κB and JNK signaling pathways. These novel findings provide insights into the development of GNC formula as a therapeutic agent for the treatment of neurodegenerative disorders.
Keywords: Alzheimer's disease, Gengnianchun formula, amyloid β, microglial cells
Introduction
Alzheimer's disease (AD) is a chronic, progressive and irreversible neurodegenerative disease with clinical features of memory loss, dementia and cognitive impairment. It is the most common cause of dementia among the elderly, accounting for 50–70% of all cases worldwide (1–3). AD is a multifactorial disorder, involving several pathological mechanisms. It is histopathologically characterized by the presence of senile plaques, neurofibrillary tangles and neuron loss in the brain. In particular, the senile plaques are extracellular deposits of amyloid-β peptide (Aβ), which is cleaved from the amyloid precursor protein. However, the exact mechanism triggering neurodegeneration in AD remains to be fully elucidated, and there is no effective treatment to prevent or reverse AD at present (4,5).
The inflammatory response is a beneficial host immune response to tissue injury or infection. However, it may lead to potentially damaging consequences and result in several inflammatory diseases and types of cancer (6). In AD, the activation and accumulation of microglial cells around amyloid plaques has long been described and is considered to result in chronic neuroinflammation, which leads to the production of proinflammatory cytokines, including interleukin (IL)-1β and tumor necrosis factor (TNF)-α, followed by neurodegeneration (7,8). The different functions of microglial cells on the progression of AD have been considered to be a double-edged sword; as Aβ is able to activate microglia and initiate an inflammatory response, microglia may be important in the clearance of Aβ peptides in the early stages (9,10). However, the over stimulation of microglia cells can lead to the excessive secretion of pro-inflammation cytokines, thus forming a chronic neuro-inflammatory reaction in AD. Therefore, it is critical to maintain the beneficial functions of microglial cells and avoid neurotoxin production, which may offer useful therapeutic strategies for AD.
Nuclear factor (NF)-κB is a transcription factor, which is critical in regulating cellular proliferation, inflammatory responses and cell adhesion (11). Inactive NF-κB complexes are sequestered in the cytoplasm via binding to the inhibitory protein (IκB). Once activated, IκB proteins are phosphorylated rapidly, and the phosphorylated IκB proteins are targeted for ubiquitination and degradation. The free NF-κB then translocates into the nucleus where it regulates the production of various pro-inflammatory mediators (12,13). It has been shown that microglia express activated NF-κB in AD (14,15). Mitogen-activated protein kinase (MAPK), namely, the extracellular signal-regulated protein kinase, the c-Jun N-terminal kinase (JNK), p38 MAPK and atypical MAPKs, is one of the signal transduction cascades involved in the regulation of inflammatory mediators (16). The activation of MAPK signaling pathways has been reported to be involved in the development of several human diseases, including AD and Parkinson's disease (17–19). Therefore, the NF-κB and MAPK signaling pathways serve as important molecular targets for the development of potential anti-inflammatory drugs.
The Gengnianchun (GNC) formula has long been used in China to treat perimenopausal syndrome (PMS) clinically, and is particularly effective in improving learning ability and memory (20,21). Previous studies have shown the anti-inflammatory and neuroprotective effects of GNC (22,23). In the present study, the anti-inflammatory properties of GNC and its underlying molecular mechanism of action were investigated in BV-2 microglial cells.
Materials and methods
Materials and reagents
Synthetic Aβ (1–42) was purchased from AnaSpec, Inc. (San Jose, CA, USA). Minimum essential medium (MEM), fetal bovine serum (FBS) and phosphate-buffered saline (PBS) were purchased from Gibco; Thermo Fisher Scientific, Inc. (Waltham, MA, USA). Cytokine (IL-1β and TNF-α) enzyme-linked immunosorbent assay (ELISA) kits were purchased from R&D Systems, Inc. (Minneapolis, MN, USA). A Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Molecular Technologies, Inc. (Kumamoto, Japan). Antibodies against phosphorylated JNK (p-JNK) and p-p65 were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). All other chemicals were purchased from common commercial suppliers.
Preparation of medicated rat serum
A single unit of GNC formula was 138 g total weight, composed of 12 crude herbs: 15 g Radix Rehmanniae (Shengdihuang), 12 g of Herba Epimedium brevicornum (Yinyanghuo), Radix Paeoniae Alba (Baishao), Semen Cuscuta (Tusizi), Wolfberry fruit (Gouqizi), Radix Morindae officinalis (Bajitian) and Herba Cistanche (Roucongrong), 15 g of Tortoise shell (Guijia) and Rhizoma anemarrhenae (Zhimu), 9 g of Poria (Fuling) and Cortex Phellodendri (Huangbai), and 3 g of Rhizoma Coptidis (Huanglian). A total of 138 g of herb mixture was reduced to 25 g of concentrated powder by Jiangyin Tianjiang Pharmaceutical Co., Ltd. (Shanghai, China). A total of 40 female SD rats (3 months old and weighing 250±20 g) were obtained from the Experimental Animal Center of the Chinese Academy of Sciences [Shanghai, China, license no. SCXK (Hu) 2012–0002]. The rats were housed at 23–25°C with 55% humidity, a 12-h light/dark cycle and free access to food and tap water. All the rats were bilaterally ovariectomized following anaesthetization with an intraperitoneal injection with 4% chloral hydrate. After 7 days, the rats were randomly divided into two groups. One group was intragastrically administered with GNC (25 g powder dissolved in 100 ml normal saline) at a dose of 5 g/kg. The rats in the other group were administered with normal saline at the same volume for 5 days. At 1 h following the final administration, serum was obtained by centrifugation (3,000 × g for 10 min at 4°C). The serum was sterilized by vacuum filtration and stored at −20°C.
Cell culture and treatment
The murine microglia BV-2 cell line was purchased from the China Center for Type Culture Collection (Wuhan, China) and cultured in MEM supplemented with FBS (10%), 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C in a humidified cell incubator in a 95/5% (v/v) mixture of air and CO2. The BV-2 microglial cells, seeded in a 96-well plate at a density of 1×104/well, were pretreated with different concentrations of MRS (2.5, 5, 10 and 20%) for 2 h, and were then incubated with 5 µM Aβ (1–42) for another 12 h at 37°C. In the preliminary experiments to determine the effect of different concentrations of Aβ (1–42) on the secretion of IL-1β and TNF-α, the microglial cells were seeded at a density of 1×104/well in a 96-well plate and treated with Aβ (1–42) at different concentrations (2.5, 5, 10 and 20 µM). Subsequently, microglial cells seeded on a 96-well plate at a density of 1×104/well were treated with 5 µM Aβ (1–42) for the indicated periods of time. Following treatment, the supernatant was collected and stored at −80°C for measuring the protein levels of IL-1β and TNF-α using ELISA.
Cell viability
The cell viability was detected using a CCK-8. Briefly, 100 µl of BV2 cell suspension (1×104 cells/well) was seeded into 96-well plates overnight, following which the supernatant was removed and the cells were incubated in FBS-free medium for 12 h. MRS was then added at the various concentrations for 24 h. Following treatment, the cell viability was assessed using the CCK-8. The optical density was measured at 450 nm.
ELISA
The protein levels of IL-1β and TNF-α in the supernatant of the cultured BV2 microglial cells were measured using an ELISA kit according to the manufacturer's protocol.
Western blot analysis
To confirm the effects of MRS on the Aβ (1–42)-induced activation of NF-κB and JNK in the BV2 microglia cells, protein extracts from the cell cultures were obtained using a commercial extract kit (Active Motif, Carlsbad, CA, USA) and analyzed using western blot analysis according to standard protocols. The cells were lysed in lysis buffer (Biyuntian Biotech Co., Ltd., Shanghai, China) following the manufacturer's protocols. A bicinchoninic acid assay (Biyuntian Biotech Co., Ltd., Shanghai, China) was used to determine concentration of the protein lysate. The extracts, containing 30 µg proteins, were loaded on a 10% SDS-PAGE gel. Following electrophoresis, the proteins were transferred onto a polyvinylidene fluoride (PVDF) membrane (Invitrogen; Thermo Fisher Scientific, Inc.). Following blocking with 5% BSA for 1 h at room temperature, the PVDF membrane was incubated with the p-p65 antibody (3033; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA A), p-JNK antibody (4668; 1:1,000; Cell Signaling Technology, Inc.), and β-actin antibody (ab8229; 1:1,000; Abcam, Cambridge, UK) at 4°C overnight. Subsequently, the membrane was incubated with a horseradish peroxidase-conjugated secondary antibody (SA00001-15; 1:5,000; Proteintech Group, Inc., Chicago, IL, USA) for 1 h at room temperature. Following three washes, the immune complexes were detected using an electrochemiluminescence system. Images were captured using the ImageQuant LAS 4000 mini analyzer (General Electric Company, Fairfield, CT, USA).
Statistical analysis
Each experiment was repeated at least three times. The data are expressed as the mean ± standard deviation. The statistical significance was analyzed by one-way analysis of variance followed by Fisher's LSD-t post hoc test. Statistical analysis was performed using SPSS software (version 16.0; SPSS, Inc., Chicago, IL, USA) to determine significant differences. P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of Aβ on the secretion of pro-inflammatory cytokines in BV-2 microglial cells
In order to assess the pro-inflammatory properties of Aβ in BV-2 microglial cells, the present study measured the effects of Aβ on the production of pro-inflammatory cytokines. As shown in Fig. 1A and B, treatment with 2.5–20 µM Aβ dose-dependently induced the release of IL-1β and TNF-α.
Figure 1.
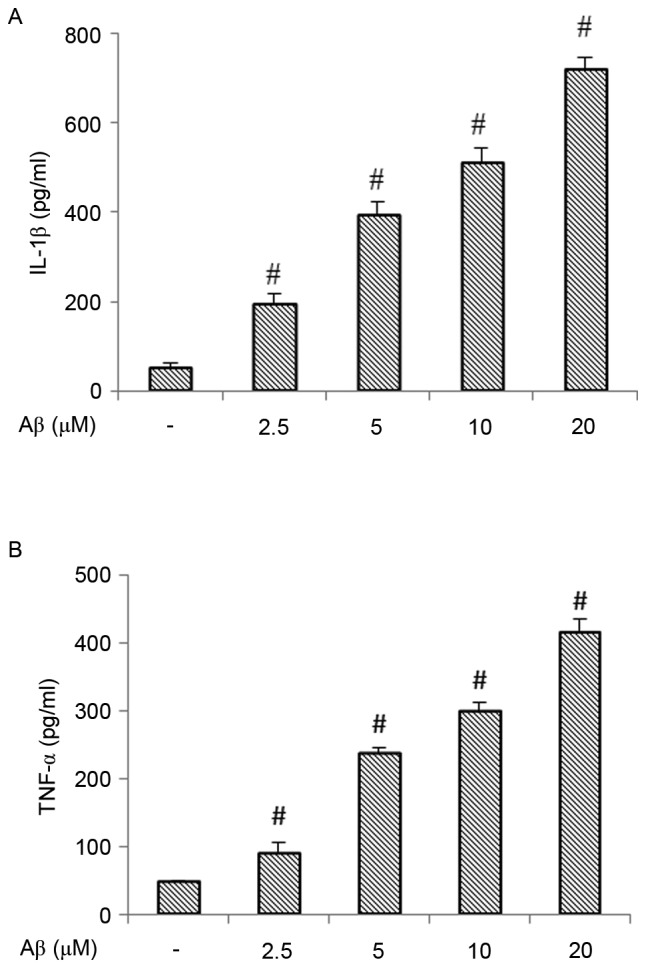
Effect of Aβ (1–42) on the production of IL-1β and TNF-α. (A) Production of IL-1β in BV-2 cells treated with different concentrations of Aβ (1–42) for 12 h. (B) Production of TNF-α in BV-2 cells treated with different concentrations of Aβ (1–42) for 12 h. Each value indicates the mean ± standard deviation from three independent experiments. #P<0.01, vs. untreated control cells. Aβ, amyloid β; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor-α.
MRS inhibits the release of pro-inflammatory cytokines in β-stimulated BV-2 microglial cells
Stimulation of the BV-2 microglial cells with Aβ increased the expression of pro-inflammatory cytokines. As shown in Fig. 2, the protein expression levels of IL-1β and TNF-α were significantly increased following treatment with Aβ, compared with the levels in the normal control group (P<0.01). However, treatment with MRS significantly attenuated the Aβ-induced secretion of these pro-inflammatory cytokines. At concentrations of 10–20%, MRS significantly attenuated the Aβ-induced protein expression of IL-1β (Fig. 2A). The increase in the protein expression of TNF-α was also reduced significantly following treatment with MRS at concentrations of 5–20% (Fig. 2B).
Figure 2.
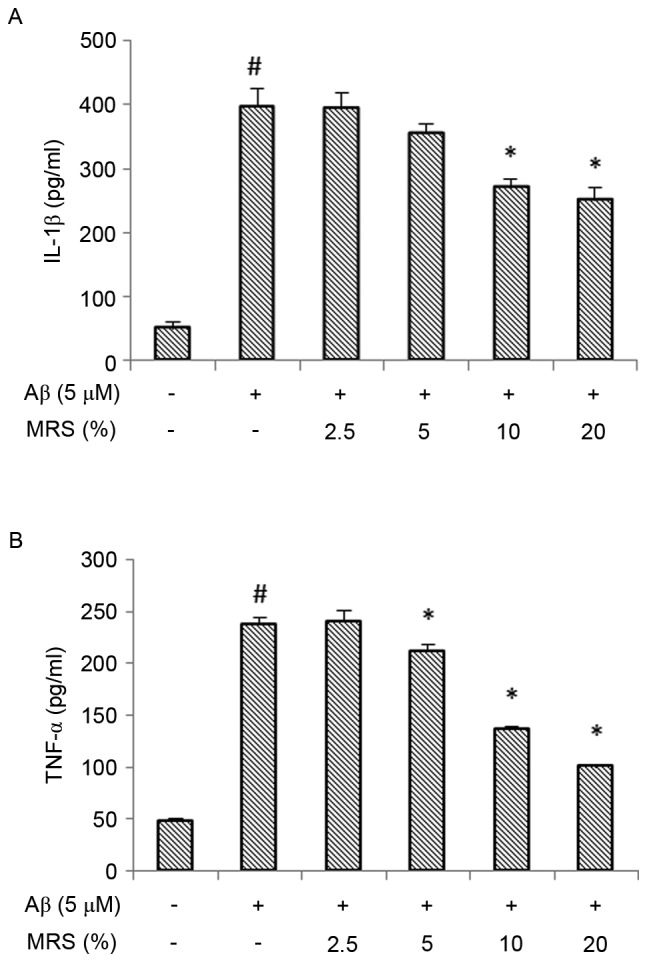
Effect of MRS on the Aβ (1–42)-induced production of IL-1β and TNF-α. The BV-2 cells were pretreated with different concentrations of MRS for 2 h, followed by co-treatment with 5 µM Aβ (1–42) for another 12 h. The culture supernatants were collected and assayed for (A) IL-1β and (B) TNF-α. Each value indicates the mean ± standard deviation from three independent experiments. #P<0.01, vs. untreated control cells. *P<0.05, vs. cells treated with Aβ (1–42) alone. MRS, medicated rat serum containing Gengnianchun formula; Aβ, amyloid β; IL-1β, interleukin-1β; TNF-α, tumor necrosis factor-α.
Cell viability
To confirm that the anti-inflammatory property of MRS was not due to cytotoxic effects on the BV-2 microglial cells, a CCK-8 assay was performed. As shown in Fig. 3, the viability of the BV-2 cells was not reduced following treatment with various concentrations of MRS, suggesting that the inhibitory effects of the Aβ-induced inflammatory response did not result from its cytotoxic action.
Figure 3.
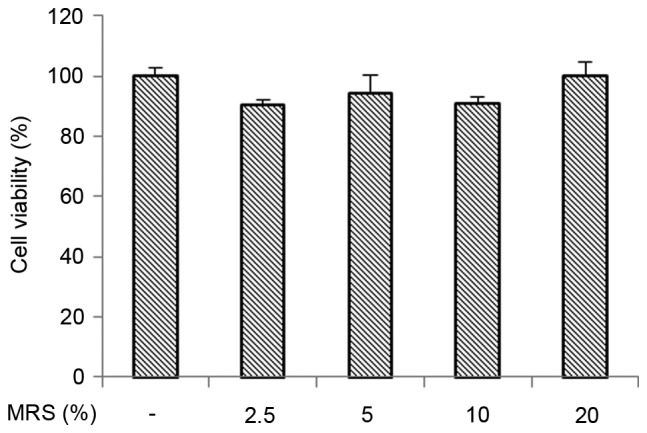
Effect of MRS on the viability of BV-2 cells. Each value indicates the mean ± standard deviation from three independent experiments. MRS, medicated rat serum containing Gengnianchun formula.
MRS inhibits the Aβ-induced activation of NF-κB and JNK in BV-2 microglial cells
As NF-κB is known to be key in the inflammatory response, the present study investigated whether NF-κB is involved in suppression of the inflammatory response induced by MRS. The effect of MRS on Aβ-induced NF-κB activation was examined using western blot analysis. It is known that the phosphorylation of NF-κB is considered a marker of NF-κB activation. As shown in Fig. 4, the phosphorylation of NF-κB was markedly increased following treatment with Aβ, compared with that in the normal control group (P<0.01). No marked reduction in the Aβ-induced phosphorylation of NF-κB was observed in the non-medicated rat serum (NRS) group, however, treatment with MRS resulted in a significant reduction in the phosphorylation of NF-κB (P<0.05). As there is a crosstalk between NF-κB and MAPK, and this crosstalk is important for maximal activation of genes associated with inflammation, the present study examined the effect of MRS on MAPK activity, which regulates the function of NF-κB. As shown in Fig. 5, the phosphorylation level of JNK was markedly increased following treatment with Aβ, compared with that in the normal control group (P<0.01), whereas treatment with MRS significantly inhibited Aβ-induced JNK activation (P<0.05). Again, the NRS group had no effect on reducing the Aβ-induced phosphorylation of JNK. These data suggested that the inhibitory effect of MRS on the Aβ-induced inflammatory response occurred through suppressing the activation of NF-κB and JNK in the BV-2 microglial cells.
Figure 4.
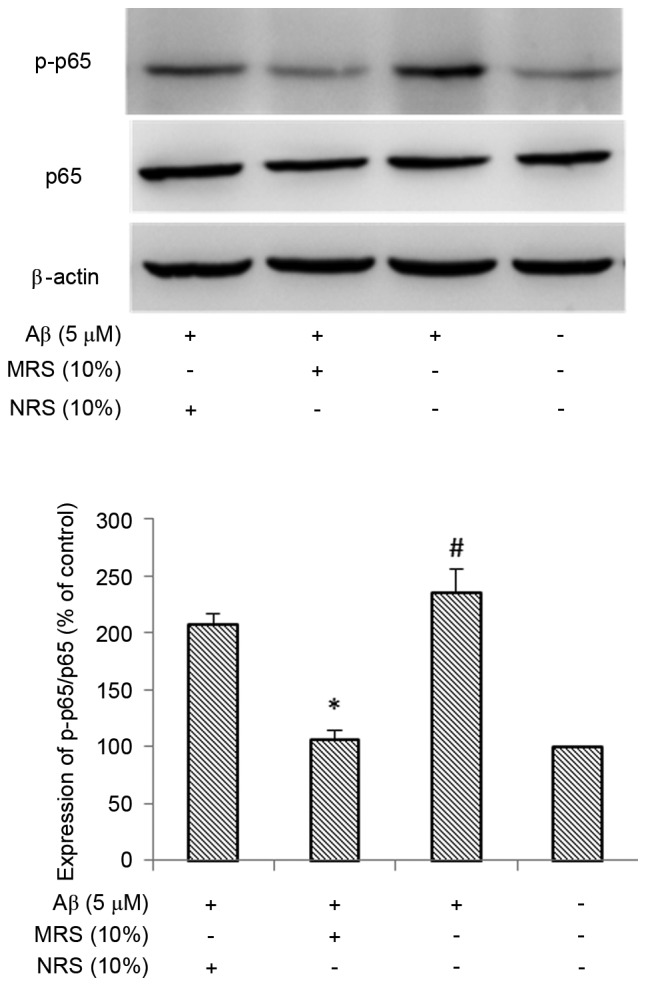
Effects of MRS and NRS on Aβ (1–42)-induced nuclear factor-κB p65 activity. The BV-2 cells were pretreated with 10% MRS or NRS for 1 h, followed by co-treatment with 5 µM Aβ (1–42) for another 4 h. The extracts from cell cultures were prepared and analyzed. Each value indicates the mean ± standard deviation from three independent experiments. #P<0.01, vs. untreated control cells. *P<0.05, vs. cells treated with Aβ (1–42) alone. MRS, medicated rat serum containing Gengnianchun formula; NRS, non-medicated rat serum; Aβ, amyloid β; p-, phosphorylated.
Figure 5.
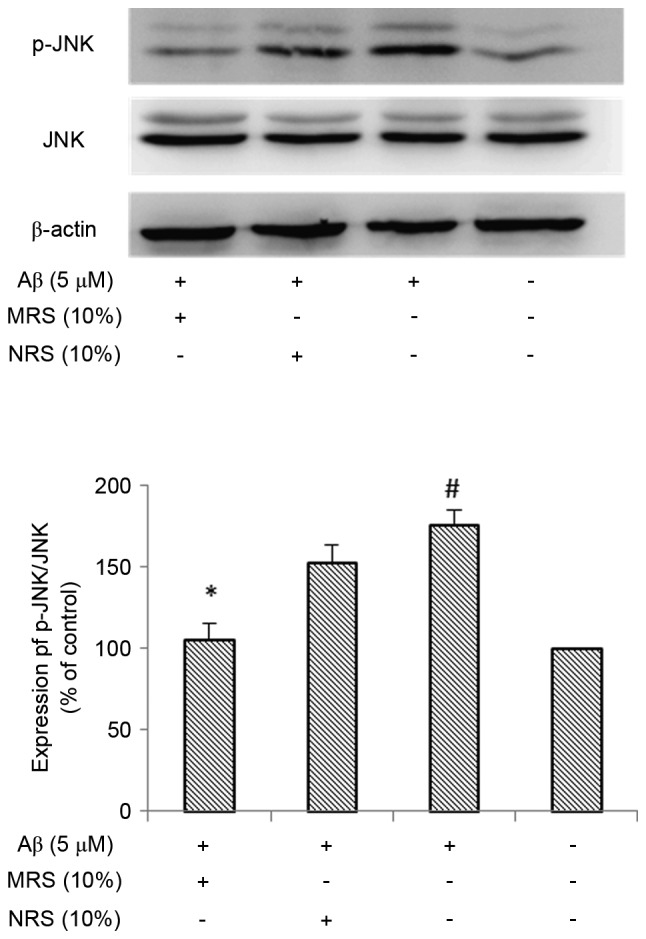
Effects of MRS and NRS on Aβ (1–42)-induced JNK activity. The BV-2 cells were pretreated with 10% MRS or NRS for 1 h, followed by co-treatment with 5 µM Aβ (1–42) for another 4 h. The extracts from cell cultures were prepared and analyzed. Each value indicates the mean ± standard deviation from three independent experiments. #P<0.01, vs. untreated control cells. *P<0.05, vs. cells treated with Aβ (1–42) alone. MRS, medicated rat serum containing Gengnianchun formula; NRS, non-medicated rat serum; Aβ, amyloid β; JNK, c-Jun N-terminal kinase; p-JNK, phosphorylated JNK.
Discussion
AD is a chronic, progressive and irreversible neurodegenerative disease, with characteristics of memory loss and cognitive impairment. Although the exact pathophysiologic mechanism of AD remains to be elucidated, there is accumulating evidence that neuroinflammation is critical in the pathogenesis of AD (5). AD is characterized by senile plaques, neurofibrillary tangles, and loss of neurons and synapses. Aβ peptide accumulation is a major pathological hallmark of AD. In the AD brain, microglial cells are found in close association with Aβ deposits. It is widely accepted that inflammation in the central nervous system is characterized by the activation of microglial cells, and the production of pro-inflammatory cytokines and chemokines. Furthermore, the release of neuroinflammatory cytokines is harmful to neighboring healthy cells. Over previous years, the process of inflammation has increased in interest in AD, not only for its potential role in neurodegenerative disease but as a potential therapeutic approach. Therefore, regulating microglia-mediated neuroinflammation and neurotoxicity is considered to be an important therapeutic target in the treatment of neurodegenerative disorders (24). However, the exact mechanism underlying the release of these pro-inflammatory cytokines in activated microglial cells remains to be elucidated.
The NF-κB and MAPK signaling pathways are known to be crucial in inflammatory gene regulation and are considered to have an important function in the production of pro-inflammatory cytokines induced by Aβ. Regarding the NF-κB signaling pathway, various irritants, including Aβ, can activate it. A number of studies have reported that NF-κB is a major signaling molecule involved in inflammatory response (11,12). The MAPK signaling pathway has been shown to be involved in the Aβ-mediated production of several inflammatory genes, which are important for the activation of NF-κB. It is known that there is crosstalk between the NF-κB and MAPK signaling pathways, which is important for maximum activation of genes associated with inflammation. The present study provided definitive evidence that the NF-κB and JNK signaling pathways were activated following Aβ treatment in microglial cultures. It was found that Aβ treatment significantly increased the phosphorylation of NF-κB p65 and JNK, which are considered to be markers of NF-κB and JNK activation, upregulating the expression of pro-inflammatory cytokines, including IL-1β and TNF-α. Therefore, NF-κB and JNK may be considered as targets for the molecular therapy of AD.
The GNC formula, as a traditional Chinese decoction, has long been used in China to treat PMS clinically, and is particularly effective in improving learning ability and memory (20,21). Previous investigations have demonstrated the anti-inflammatory and neuroprotective effects of GNC (22,23). Similarly, the results of the present study showed that serum containing GNC formula significantly reduced the expression levels of IL-1β and TNF-α, accompanied by the inhibition of NF-κB and JNK activation in microglial cultures treated with Aβ. The present study also showed that the inhibitory effect of MRS on the expression levels of IL-1β and TNF-α was not due to its direct toxicity on microglial cells, as the different concentrations (2.5–20%) of GNC formula in the serum did not affect microglial cell viability.
Microglial cells are the primary immune effector cells of the brain, with an integral role in maintaining brain homeostasis and protecting the brain from infections. However, the persistent stimulation of microglial cells is considered to contribute to neurodegenerative disease through increasing the production of pro-inflammatory cytokines, including IL-1β and TNF-α. Several inflammatory mediators, including IL-1β and TNF-α, are considered to be critical in the inflammatory process of AD. There is an increase in the release of these pro-inflammatory cytokines from microglial cells following exposure to Aβ. Therefore, decreasing these pro-inflammatory cytokines offers an alternative therapy for AD. However, the inflammatory mediators, including pro-inflammatory cytokines, represent downstream molecules only, and the NF-κB and MAPK signaling pathways are the common pathways in the inflammatory cascade. Therefore, in order to obtain optimal therapeutic benefits, therapies interfering with key components of the inflammatory cascade, including NF-κB and JNK, may be more efficient. Therefore, GNC may be a potential drug in the treatment of AD as the therapeutic targets of GNC are critical and multiple.
In conclusion, the present study showed that GNC formula mediated its anti-inflammatory effects through decreasing pro-inflammatory cytokines and by suppressing the activation of NF-κB and JNK in Aβ-stimulated microglial cells. The treatment of AD remains a challenge, however, these findings suggested that GNC inhibits the release of neurotoxic products in the central nervous system. Therefore, GNC formula may be a useful therapeutic drug to prevent AD.
Acknowledgements
This study was supported by National Natural Sciences Foundation of China (grant no. 81273956) and Scientific Research Project of the Shanghai Committee of Science and Technology, China (grant no. 15401931800) awarded to Dr Wen-Jun Wang.
Glossary
Abbreviations
- AD
Alzheimer's disease
- Aβ
amyloid-β peptide
- GNC
Gengnianchun formula
- MRS
medicated rat serum containing GNC
- NRS
non-medicated rat serum
- IL-1β
interleukin-1β
- TNF-α
tumor necrosis factor-α
- JNK
c-Jun N-terminal kinase
- NF-κB
nuclear factor-κB
References
- 1.Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 3.Wortmann M. Dementia: A global health priority-highlights from an ADI and World Health Organization report. Alzheimers Res Ther. 2012;4:40. doi: 10.1186/alzrt143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iqbal K, Grundke-Iqbal I. Alzheimer's disease, a multifactorial disorder seeking multitherapies. Alzheimers Dement. 2010;6:420–424. doi: 10.1016/j.jalz.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez-Vita J, Lawrence T. The resolution of inflammation and cancer. Cytokine Growth Factor Rev. 2010;21:61–65. doi: 10.1016/j.cytogfr.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 7.Liu L, Chan C. The role of inflammasome in Alzheimer's disease. Ageing Res Rev. 2014;15:6–15. doi: 10.1016/j.arr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mandrekar-Colucci S, Landreth GE. Microglia and inflammation in Alzheimer's disease. CNS Neurol Disord Drug Targets. 2010;9:156–167. doi: 10.2174/187152710791012071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solito E, Sastre M. Microglia function in Alzheimer's disease. Front Pharmacol. 2012;3:14. doi: 10.3389/fphar.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prokop S, Miller KR, Heppner FL. Microglia actions in Alzheimer's disease. Acta Neuropathol. 2013;126:461–477. doi: 10.1007/s00401-013-1182-x. [DOI] [PubMed] [Google Scholar]
- 11.Makarov SS. NF-kappaB as a therapeutic target in chronic inflammation: Recent advances. Mol Med Today. 2000;6:441–448. doi: 10.1016/S1357-4310(00)01814-1. [DOI] [PubMed] [Google Scholar]
- 12.Roshak AK, Callahan JF, Blake SM. Small-molecule inhibitors of NF-kappaB for the treatment of inflammatory joint disease. Curr Opin Pharmacol. 2002;2:316–321. doi: 10.1016/S1471-4892(02)00165-0. [DOI] [PubMed] [Google Scholar]
- 13.Mattson MP. NF-kappaB in the survival and plasticity of neurons. Neurochem Res. 2005;30:883–893. doi: 10.1007/s11064-005-6961-x. [DOI] [PubMed] [Google Scholar]
- 14.Boissiere F, Hunot S, Faucheux B, Duyckaerts C, Hauw JJ, Agid Y, Hirsch EC. Nuclear translocation of NF-kappaB in cholinergic neurons of patients with Alzheimer's disease. Neuroreport. 1997;8:2849–2852. doi: 10.1097/00001756-199709080-00009. [DOI] [PubMed] [Google Scholar]
- 15.Mattson MP, Camandola S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J Clin Invest. 2001;107:247–254. doi: 10.1172/JCI11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harper SJ, Wilkie N. MAPKs: New targets for neurodegeneration. Expert Opin Ther Targets. 2003;7:187–200. doi: 10.1517/14728222.7.2.187. [DOI] [PubMed] [Google Scholar]
- 17.Mehan S, Meena H, Sharma D, Sankhla R. JNK: A stress-activated protein kinase therapeutic strategies and involvement in Alzheimer's and various neurodegenerative abnormalities. J Mol Neurosci. 2011;43:376–390. doi: 10.1007/s12031-010-9454-6. [DOI] [PubMed] [Google Scholar]
- 18.Correa SA, Eales KL. The role of p38 MAPK and its substrates in neuronal plasticity and neurodegenerative disease. J Signal Transduct. 2012;2012:649079. doi: 10.1155/2012/649079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao H, Wang SL, Qian L, Jin JL, Li H, Xu Y, Zhu XL. Diammonium glycyrrhizinate attenuates Aβ(1–42)-induced neuroinflammation and regulates MAPK and NF-κB pathways in vitro and in vivo. CNS Neurosci Ther. 2013;19:117–124. doi: 10.1111/cns.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suyu J, Meijuan W. Clinical observation of ‘climacterium capsule’ in improving the memory of climacterium women. Shanghai Zhong Yi Yao Za Zhi. 2001;35:32–33. (In Chinese) [Google Scholar]
- 21.Zhao FG, Wang WJ, Zhou WJ. Effect of Gengnianchun recipe on learning memory function and hippocampal cholinergic system in ovariectmized rats. Zhongguo Zhong Xi Yi Jie He Za Zhi. 2008;28:234–237. (In Chinese) [PubMed] [Google Scholar]
- 22.Li J, Li B, Zhou WJ, Zhao FG, Li DJ, Wang WJ. Medicated rat serum containing Gengnianchun decoction reduces apoptosis of pheochromocytoma cells insulted by amyloid beta protein. Zhong Xi Yi Jie He Xue Bao. 2010;8:472–479. doi: 10.3736/jcim20100512. [DOI] [PubMed] [Google Scholar]
- 23.Jun L, Wenjun W, Dajin L, Wenjiang Z. Gengnianchun recipe inhibits apoptosis of pheochromocytoma cells from beta-amyloid 25–35 insult, better than monotherapies and their compounds. Neur Regen Res. 2011;6:2815–2821. [Google Scholar]
- 24.Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, Hong JT. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]