

Abstract
Colorectal cancer (CRC) is a major cause of mortality and morbidity. Chronic inflammation is closely associated with the development, progression and prognosis of the majority of intestinal malignancies. In recent years, targeting the nuclear factor (NF)-κB signaling pathway for CRC therapy has become an attractive strategy. Riccardin D, a novel macrocyclicbis (bibenzyl) compound, was isolated from the Chinese liverwort plant. Previous studies have suggested that Riccardin D exerted chemo-preventative effects against the intestinal malignancy formation. In the present study, cell counting kit-8, Hochest 33258 staining, mitochondria membrane permeability assay, western blotting analysis, reverse transcription-polymerase chain reaction, luciferase reporter gene assay and molecular modeling analysis were performed to detect the effect and mechanisms of Riccardin D on human colon cancer cells. The results demonstrated that Riccardin D significantly inhibited the growth of HT-29 cells. In addition, the cDNA expression of cyclooxygenase-2, and the protein expression and activity of NF-κB and tumor necrosis factor-α were downregulated; however, the protein expression of cleaved caspase-3 and −9, and cleaved poly (adenosine diphosphate-ribose) polymerase, and the B-cell lymphoma (Bcl)-2: Bcl-2-associated X protein ratio were upregulated. Furthermore, Auto Dock analysis identified binding sites between Riccardin D and NF-κB. These results indicated that Riccardin D may inhibit cell proliferation and induce apoptosis in HT-29 cells, which may be associated with the blocking of the NF-κB signaling pathway. Thus, Riccardin D should be investigated as an NF-κB inhibitor in cancer therapy.
Keywords: colorectal cancer, Riccardin D, nuclear factor-κB signaling, autodock analysis
Introduction
Colorectal cancer (CRC) is one of the leading causes of cancer mortalities worldwide, and is the third most common cancer type and the fifth leading cause of mortality in China (1–3). The majority of CRCs develop sporadically (70–80%), and the somatic loss of tumor suppressor gene, adenomatous polyposis coli (APC), was revealed to be present in 85% of patients with CRC and familial adenomatous polyposis (4–6). It has previously been demonstrated to have significant pro-apoptosis and anti-proliferative effects in several different types of cancers in vivo and in vitro (1–3). Recently, Riccardin D, a novel macrocyclicbis (bibenzyl) compound isolated from the Chinese liverwort plant Dumortiera hirsuta, has been observed to prevent the growth of intestinal polyps in adenomatous polyposis coli (APC)Min/+ mice, an animal model of human familial adenomatous polyposis, which spontaneously develops numerous polyps in the intestinal tract due to a mutation in the APC gene (6). This indicates that Riccardin D may serve an important role in the inhibition of CRC.
APC serves a critical role in regulating gene transcription through the Wnt signaling pathway (7). The APC gene mutation is the most common cause of colon cancer associated with the Wnt/β-catenin signaling pathway, which serves a critical role in the development of colon cancer (6–8). By contrast, the nuclear factor (NF)-κB-cyclooxygenase (COX)-2 signaling pathway has also been shown to affect the APC gene mutation in the human intestine and colon cells (9,10). In the APCMin/+ mouse, a higher expression of inflammation-associated cytokines has been reported (11,12). These results have suggested that there may be a connection between inflammation and tumor development. Our previous report also demonstrated that Riccardin D downregulated inflammatory factors, particularly those associated with the NF-κB signaling pathway, in the mouse model (6). NF-κB is hypothesized to promote tumorigenesis via pro-inflammatory transcription factors, which can be activated by bacterial products, such as lipopolysaccharides (LPS), and inflammatory cytokines. The transcriptional targets of NF-κB include genes encoding COX-2, intercellular adhesion molecule-1, vascular endothelial growth factor, and the inflammatory cytokines, interleukin (IL)-1β and tumor necrosis factor α (TNFα) (13,14).
Previous reports have indicated that regular use of nonsteroidal anti-inflammatory drugs lowers the mortality associated with sporadic colon cancer and results in the regression of adenomas in patients with familial adenomatous polyposis, who have inherited a mutation in the APC gene (15,16). Pro-inflammatory cytokines, including TNFα, IL-6 and IL-1β, or transcription factors that are required by these cytokines for signaling, such as NF-κB and signal transducer and activator of transcription, have been identified as potential targets for anticancer therapy (17). In the present study, the mechanism underlying the anti-inflammation effects of Riccardin D in vitro were investigated. The results indicated that Riccardin D may exert its modulatory effects by blocking NF-κB activity in colon cancer cells. To the best of our knowledge, these results show, for the first time, the evaluation of macrocyclicbis (bibenzyls) against CRC associated with the inflammation pathway, suggesting its potential in the therapeutic intervention of intestinal cancers as a novel NF-κB inhibitor.
Materials and methods
Drugs
Riccardin D, a novel macrocyclicbis (bibenzyl) compound, was extracted from the Chinese liverwort plant Dumortiera hirsuta (previously collected from the Guizhou region, China), and its structure was identified as reported previously (18). The purity of Riccardin D, as measured by high performance liquid chromatography (18), was 98.6%. The compound was dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at 20 mM as stock solution for the in vitro study (18).
Cell lines and cell culture
The human colon cancer cell line HT-29 with a mutant APC gene, expressed as two C-terminal-truncated APC proteins of 100 and 200 kDa, was purchased from American Type Cell Culture Collection (Manassas, VA, USA) (12). The HCT-8 cell line expressing normal APC proteins was also obtained from American Type Cell Culture Collection. Cancer cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), penicillin (100 IU/ml), streptomycin (100 µg/ml) and 10 mM HEPES buffer at 37°C in a humid atmosphere (5% CO2, 95% air).
Cell Counting kit (CCK)-8 assay
HCT-8 and HT-29 cells were seeded in 96-well plates (5×103 cells/well) and incubated with increasing concentrations (2.5, 5, 10, 20, 40 and 60 µM) of Riccardin D for 24, 48 and 72 h at 37°C, respectively. The control cells were treated with an equal volume of the drug's vehicle DMSO. The cell viability was then detected using a CCK-8 kit (Dojindo Molecular Technologies, Inc., Kumamoto, Japan).
Hoechst 33258 staining
HT-29 cells were seeded in 6-well plates (3×105 cells/well) and treated with 0, 5, 10 and 20 µM of Riccardin D for 24 h at 37°C; whereas control cells were treated with DMSO only. Cells were then fixed with 4% formaldehyde in phosphate-buffered saline (PBS) for 10 min, stained with Hoechst 33258 (10 mg/l) for 1 h at 37°C, and then subjected to fluorescence microscopy (Nikon TE2000; Nikon Corporation, Tokyo, Japan). These data were obtained by eye via counting the number of apoptotic cells in five different fields of view for each group.
Mitochondrial membrane permeability assay
The mitochondria membrane potential (MMP) was investigated using JC-1 dye (Beyotime Institute of Biotechnology, Shanghai, China) according to the manufacturer's protocol. The ratio of green to red fluorescence provides an estimate of the changes in MMP. Briefly, HT-29 cells seeded in 6-well plates (3×105 cells/well) were exposed to 0, 5, 10 and 20 µM Riccardin D for 24 h at 37°C; whereas control cells were treated with DMSO only. Cells were then incubated with an equal volume of JC-1 staining solution (5 µg/ml) at 37°C for 20 min and rinsed twice with PBS. MMPs were monitored by determining the relative quantity of dual emissions from mitochondrial JC-1 monomers or aggregates using an Olympus fluorescent microscope under Argon-ion 488 nm laser excitation. Mitochondrial depolarization is indicated by an increase in the green/red fluorescence intensity ratio. Experiments were performed at least three times.
Western blotting analysis
Western blotting analysis was employed to evaluate the protein expression associated with tumor growth in human colon cancer cells. HT-29 cells (3×105 cells/well) cultured in 6-well plates were incubated with 20 µM of Riccardin D for 48 h at 37°C. The cells were collected and washed thrice using PBS (Beyotime Institute of Biotechnology) to produce lysates using radioimmunoprecipitation assay lysis buffer (Beyotime Institute of Biotechnology), and concentrations of proteins were determined using a bicinchoninic acid assay kit purchased from Beyotime Institute of Biotechnology. Protein samples (30 µg/lane) were separated by 10% SDS-PAGE and then transferred to polyvinylidene fluoride (PVDF) membranes. Non-specific binding was blocked via incubation with 5% non-fat milk for 2 h at room temperature, and membranes were then incubated for 1 h at room temperature using the following primary antibodies: Anti-NF-κB (1:800; cat. no. sc-8008; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-phosho (p)-NF-κB Ser536 (1:800; cat. no. sc-33020; Santa Cruz Biotechnology, Inc.), anti-caspase-3 (1:1,000; cat. no. 9662; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-caspase-9 (1:1,000; cat. no. 9502; Cell Signaling Technology, Inc.), anti-cleaved poly(adenosine diphosphate-ribose) polymerase (PARP; 1:1,000; cat. no. 9541; Cell Signaling Technology, Inc.), anti-B-cell lymphoma (Bcl)-2 (1:1,000; cat. no. 2872; Cell Signaling Technology, Inc.), anti-Bcl-2-associated X protein (1:1,000; Bax; cat. no. 2772; Cell Signaling Technology, Inc.), TNFα (1:1,000; cat. no. 6945; Cell Signaling Technology, Inc.) and anti-β-actin (1:5,000; cat. no. ab6276; Abcam, Cambridge, MA, USA). The PVDF membranes were then washed with TBS containing 0.05% Tween-20 prior to incubation with horseradish peroxidase-conjugated secondary antibodies (1:1,000; cat. nos. ZDR-5306 and ZDR-5307; OriGene Technologies, Inc., Beijing, China) at room temperature for 1 h. The bound antibodies were detected using an enhanced chemiluminescence reagent (EMD Millipore, Billerica, MA, USA). Densitometry analysis was performed using an electrophoresis image analysis system (cat. no. FR980; Shanghai FuriScience & Technology Co., Ltd., Shanghai, China). Experiments were performed at least three times.
Reverse transcription-semi-quantitative polymerase chain reaction (RT-sqPCR)
A RT-sqPCR assay was performed to analyze the expression of COX-2 in colon cancer cells. HT-29 cells (3×105 cells/well) cultured in 6-well plates were administrated 0, 10 and 20 µM Riccardin D for 24 h at 37°C. Total RNA was extracted using an RNAeasy kit (Sangon Biotech Co., Ltd., Shanghai, China) according to the manufacturer's instructions. cDNA was synthesized from RNA using the First Strand cDNA Synthesis kit (Toyobo Life Science, Osaka, Japan). The following primers (Gene Core Biotech Co., Ltd., Shanghai, China) were used for amplification: COX-2 forward, 5′-TTCAAATGAGATTGTGGGAAAATTGCT-3′ and reverse, 5′-AGATCATCTCTGCCTGAGTATCTT-3′; and β-actin forward, 5′-GGGTCAGAAGGATTCCTATG-3′ and reverse, 5′-GGTCTCAAACATGATCTGGG-3′. The reverse transcription reaction was performed at 37°C for 15 min and 98°C for 5 min; and PCR was performed for 40 cycles at 92°C for 30 sec, 56°C for 30 sec and 68°C for 1 min. PCR products were run on 2% agarose gels containing 0.51 g/ml of ethidium bromide and then photographed using an ultraviolet transilluminator (cat. no. FR980; Shanghai FuriScience & Technology Co., Ltd.). β-actin was used as the internal control and experiments were performed at least three times. Alpha Ease FC software was used to analyze relative light intensities (version 4.0.0.34; Protein Simple, San Jose, CA, USA).
Luciferase reporter gene assay
The effect of Riccardin D on NF-κB-dependent reporter gene transcription induced by LPS was analyzed by NF-κB-luciferase assay using a Firefly Luciferase Reporter Gene Assay kit (Beyotime Institute of Biotechnology). Briefly, HT-29 cells (3×105 cells/well) were plated in 6-well plates and transiently transfected using Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.) method with the pNF-κB-luc plasmid reporter gene (1 µg; Beyotime Institute of Biotechnology) containing four NF-κB binding motifs (5′-GGGAATTTCC-3′) and β-galactosidase (90 ng; BioVector NTCC Inc., Beijing, China). Following 24 h post-transfection, cells were treated with Riccardin D and 10 µg/ml LPS for a further 20 h. Cells were then harvested in order to measure β-galactosidase and luciferase activity. Relative luciferase activity was normalized to the value of β-galactosidase to correct the transfection efficacy (β-galactosidase Assay kit; Beyotime Institute of Biotechnology). Triplicate experiments with triplicate samples were performed.
Molecular modeling analysis
Auto Dock Vina (version 4.0; Molecular Graphics Laboratory, The Scripps Research Institute, La Jolla, CA, USA) has previously been demonstrated to locate docking modes that are consistent with X-ray crystal structures (19). Auto Dock simulates interactions between substrates or drug candidates as ligands, and their macromolecular receptors with known three dimensional structures, allowing ligand flexibility described to a full extent. In order to investigate the binding mode of Riccardin D, a molecular model of Riccardin D docked into NF-κB was produced using SYBYL-X Suite by Certara USA, Inc. (Princeton, NJ, USA). The X-ray crystal structure of NF-κB was taken from the Protein Data Bank (1K3Z) and was used for docking studies. Computer software was used in the present study to mimic the structure of Riccardin D and NF-κB, in order to identify the main binding sites between Riccardin D and NF-κB through docking simulation. The default parameters as described in the Sybyl manual were used.
Statistical analysis
Statistical analysis was performed using PASW Statistics Windows 18 download (version 15.0; SPSS, Inc., Chicago, IL, USA). Data were presented as the mean ± standard deviation and were analyzed by Student's t-test or one-way analysis of variance followed by a Student-Newman-Keuls post hoc test for multiple comparisons. P<0.05 was considered to indicate a statistically significant difference. Triplicate experiments were performed using triplicate samples.
Results
Inhibition of colon cancer cell growth by Riccardin D
Human colon cancer HT-29 and HCT-8 cells were exposed to Riccardin D for 24, 48 and 72 h, and were then subjected to a proliferation assay. The inhibition rate (%) was calculated as (OD450 value of control group-OD450 value of drug treated group)/OD450 value of control group ×100. As shown in Fig. 1, Riccardin D, in a concentration range of 2.5–40 µM, inhibited HT-29 and HCT-8 cell growth in a dose-dependent manner. Notably, the anti-proliferation effects of Riccardin D on human colon cancer cells was more significant in HT-29 cells with the APC mutation (HT-29 vs. HCT-8: at 2.5 µM for 24 h, and at 60 µM for 48 and 72 h; P>0.05). These experiments also indirectly revealed the chemopreventative effects of Riccardin D on intestinal adenoma formation in APCMin/+ mice; thus, the subsequent experiments were carried out using HT-29 cells only.
Figure 1.

Inhibition of the human colon cancer cell lines HT-29 and HCT-8 induced by treatment with Riccardin D. HT-29 and HCT-8 cells were exposed to increasing concentrations of Riccardin D or an equal volume of the drug's vehicle (DMSO) for 24, 48 and 72 h. Viable cells were evaluated by a Cell Counting kit-8 assay and were recorded as a percentage of the untreated controls at the corresponding time point. The results are presented as the mean ± standard deviation of 3 repeated experiments. #P<0.05 HT-29 vs. HCT-8 cells.
Riccardin D induces HT-29 cell apoptosis
The induction of apoptosis was detected to evaluate the inhibitory effect of Riccardin D via a number of different assays. As indicated by Hochest 33258 analysis in Fig. 2A, the number of apoptotic cells increased following 24 h incubation with 20 µM of Riccardin D. Treatment with Riccardin D, also revealed morphological changes using Hochest 33258 staining, including decreased cell size and nuclear chromatin condensation.
Figure 2.
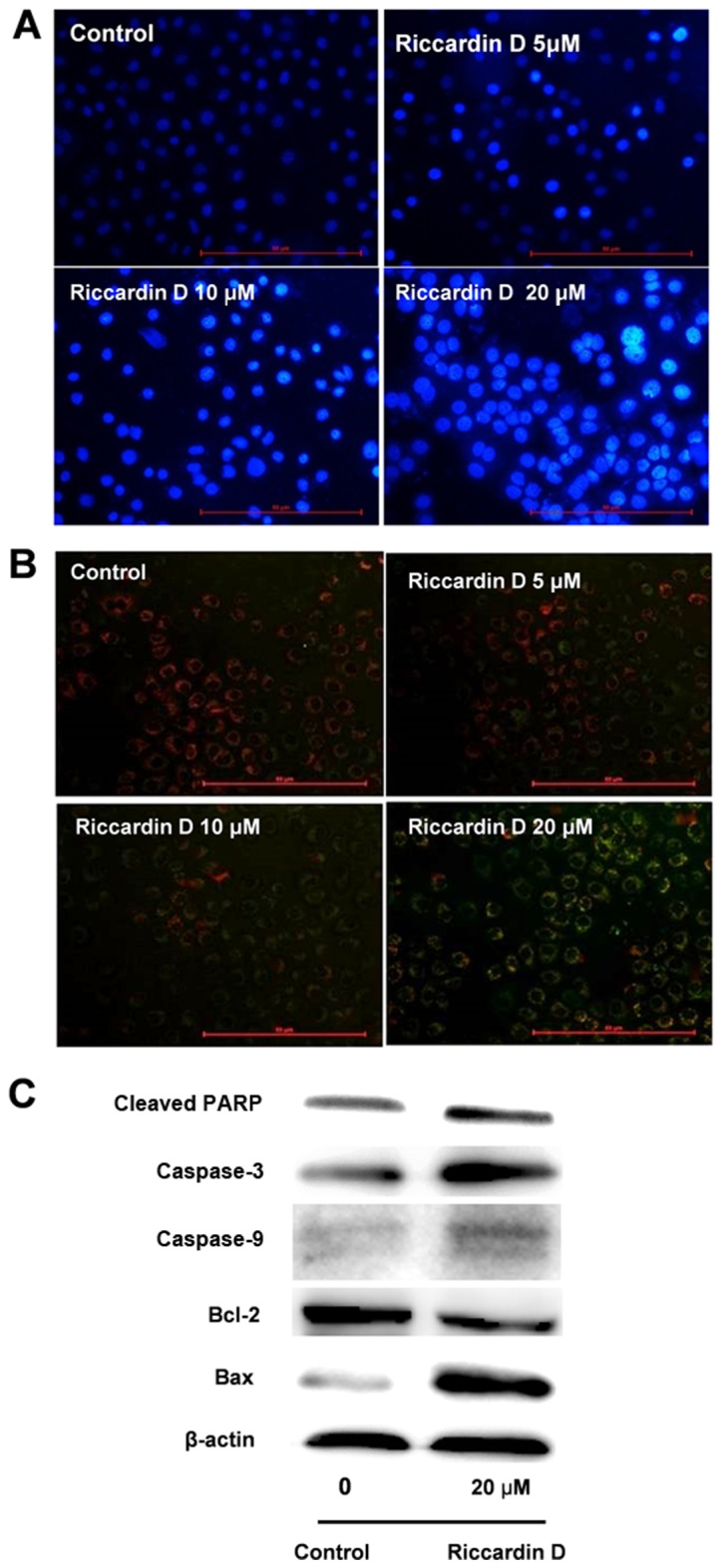
Riccardin D induces HT-29 cell apoptosis via a caspase-dependent pathway. (A) Morphology was observed in HT-29 cells with Riccardin D treatment induced apoptosis. Cells were treated with 0 (control), 5, 10 and 20 µM Riccardin D treated. Arrows indicate apoptotic characteristics in treated cells (scale bar: 50 µm). (B) Riccardin D induced a reduction in the mitochondrial membrane potential of HT-29 cells. A decrease in the red fluorescence ratio indicated a shift associated with a reduction in mitochondrial depolarization following treatment with 5–20 µM Riccardin D (scale bar: 50 µm). (C) Riccardin D triggers HT-29 cell apoptosis via the caspase-dependent pathway. The protein expression levels of cleaved caspase-3, caspase-9 and PARP in HT-29 cells were increased by Riccardin D treatment. In addition, Bcl-2 protein expression was decreased and Bax expression was increased in cancer cells treated with 20 µM Riccardin D (n=3 repeated experiments). PARP, poly (adenosine diphosphate-ribose) polymerase; Bcl-2, B-cell lymphoma 2; BAX, Bcl-2-associated X protein.
The JC-1 fluorescence probe demonstrated that the MMP in HT-29 cells was markedly reduced following treatment with Riccardin D. As shown in Fig. 2B, the red fluorescence of JC-1 gradually decreased and the green fluorescence was correspondingly increased following Riccardin D administration. In the range of 5–20 µM, the ratio of green to red fluorescence increased in a dose-dependent manner. The results revealed that treatment with Riccardin D in HT-29 cells reduced the MMP.
Further studies of the apoptotic proteins in HT-29 cells were also undertaken. Riccardin D treatment activated the caspase cascade pathway, in agreement with in vivo data from our previous research in the APCMin/+ mouse (6). As shown in Fig. 2C, the levels of cleaved caspase-3, caspase-9 and PARP were markedly increased in Riccardin D treated cells. Riccardin D also increased the Bax:Bcl-2 ratio, as the protein level of Bax was markedly increased, which was accompanied by a notable decrease in Bcl-2 in HT-29 cells treated with 20 µM Riccardin D.
Suppression of inflammation via a decrease in COX-2 and NF-κB expression
The results revealed that Riccardin D decreased the protein expression of NF-κB. Fig. 3A indicated that the level of total NF-κB p65 protein in HT-29 cells was markedly decreased by 53.4% (P<0.01) when compared with control. Further analysis of p-NF-κB Ser536 demonstrated that the active form of NF-κB, was markedly decreased by 47.2% (P<0.01; Fig. 3A) compared with the control.
Figure 3.
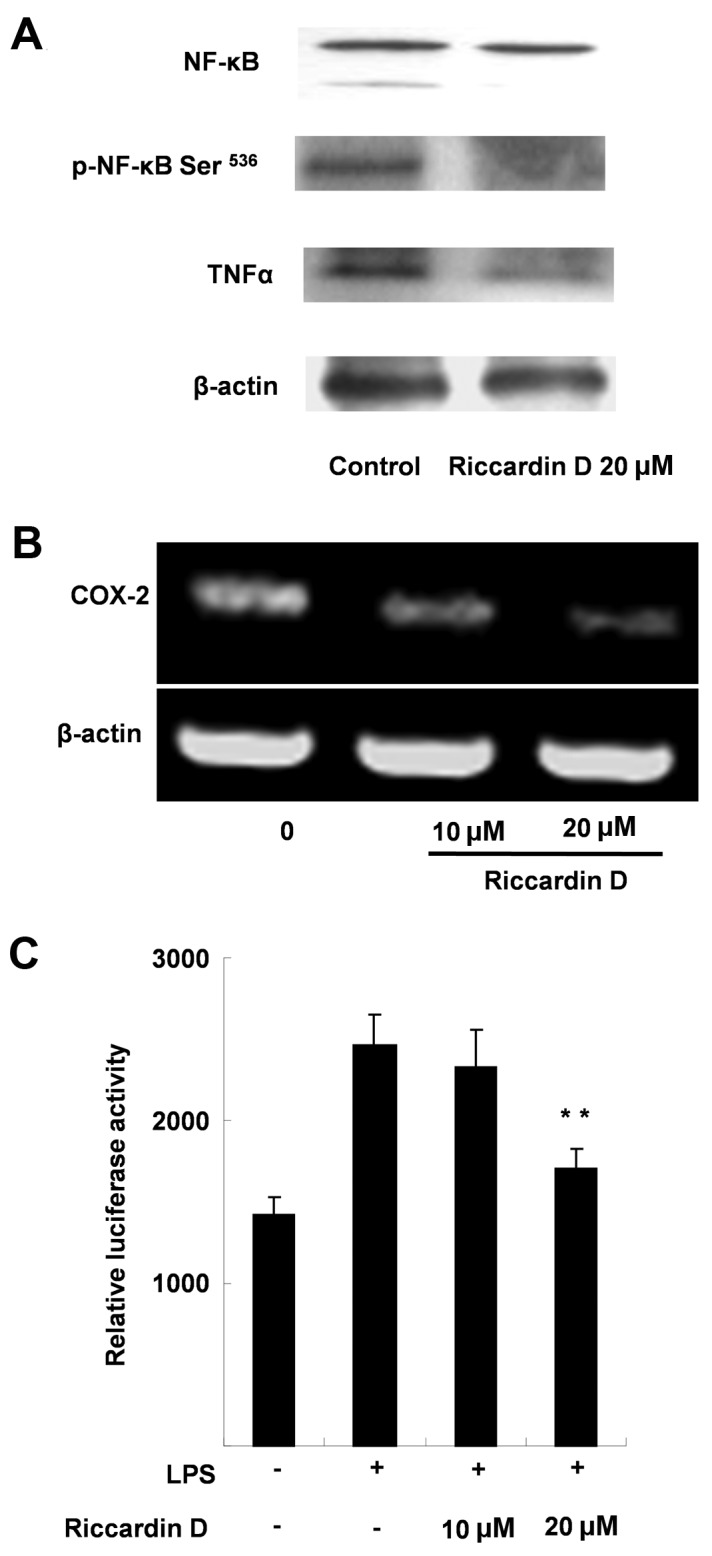
Riccardin D suppressed inflammation by decreasing COX-2 and NF-κB expression. (A) Western blotting demonstrated that Riccardin D treatment inhibited the protein expression of NF-κB and p-NF-κB Ser536 in HT-29 cells. (B) Reverse transcription-polymerase chain reaction revealed that COX-2 cDNA expression decreased in HT-29 cells treated with Riccardin D. (C) A luciferase reporter gene assay demonstrated that Riccardin D inhibits NF-κB transactivation activity induced by LPS. **P<0.01 vs. control (+ LPS with 0 µM Riccardin D). COX-2, cyclooxygenase-2; NF-κB, nuclear factor-κB; p-, phosphorylated; TNFα, tumor necrosis factor-α; LPS, lipopolysaccharide.
The inhibitory effect of Riccardin D on COX-2 cDNA expression was also shown in HT-29 cells by RT-PCR analysis. As shown in Fig. 3B, following 24 h exposure with 10 and 20 µM Riccardin D, the levels of COX-2 mRNA were decreased by 35.0 and 66.7%, respectively (Fig. 3B). These results suggested that Riccardin D treatment reduced COX-2 expression at the DNA level.
To further substantiate the finding that Riccardin D may target NF-κB, the present study also investigated the effects of Riccardin D on NF-κB-mediated transcriptional activity using HT-29 cells, wherein 20 µM of Riccardin D reduced luciferase activity by 45.2% (P<0.01) compared with control cells, which were treated with LPS and 0 µM Riccardin D (Fig. 3C). These results provided additional insight into the suppression of intestinal tumorigenesis by Riccardin D.
In addition, the theoretical binding mode of Riccardin D to NF-κB-p65 is presented in Fig. 4. Two binding sites were identified between Riccardin D and NF-κB-p65 protein.
Figure 4.
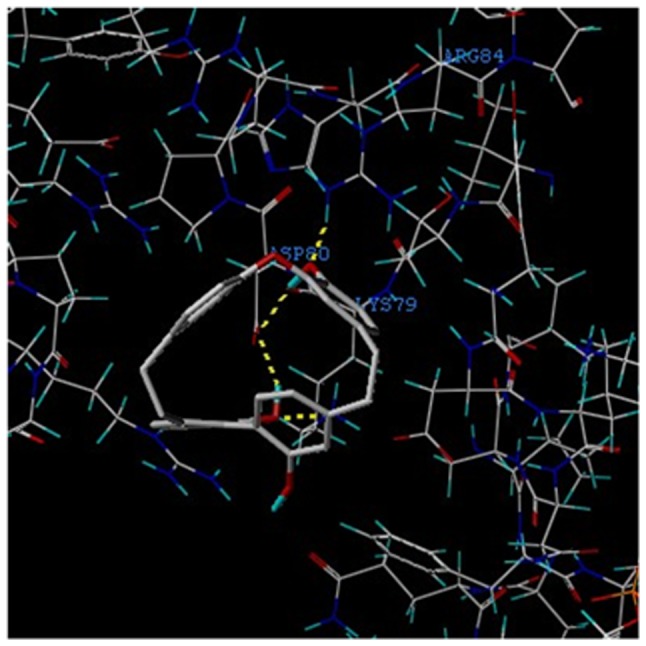
Theoretical binding mode of Riccardin D to the NF-κB-p65 protein. Superimposition of the native crystal structure of Riccardin D (grey) and the best docked conformation obtained by AutoDock Vina software (red). Representation of the 2 competitive binding sites, ASP-80 and LYS-79 (blue), between Riccardin D and NF-κB-p65 as obtained from AutoDock analysis. The dashed yellow lines represent the hydrogen bonds of the compound with residues of surrounding amino acids. NF-κB, nuclear factor-κB; p-, phosphorylated.
Discussion
Patients with chronic inflammatory bowel disease are more susceptible to developing CRC, and a number of studies have indicated that chronic inflammation may affect intestinal tumorigenesis (20–22). It is also believed that aberrant Wnt/β-catenin signaling following the loss of APC function may initiate colon adenoma formation (21,23). The APC gene mutation mouse model (APCMin/+) has been considered to be the standard experimental model for research into intestinal carcinogenesis as tumors grow spontaneously in the intestinal tract (4). Our previous study demonstrated that the administration of Riccardin D caused the inhibition of polyps in the intestine of APCMin/+ mice through its anti-proliferative, apoptotic and anti-inflammatory effects (6). Therefore, the present study investigated the anti-cancer effects of Riccardin D using human colon cancer cell lines in vitro and aimed to further understand the underlying mechanisms involved in the NF-κB signaling pathway in HT-29 cells with the APC mutation. The results revealed that Riccardin D markedly inhibited the proliferation of the human colon cancer cell line HT-29 with a mutant car boxy-truncated APC gene.
The NF-κB transcription factor pathway is a crucial regulator of a number of normal cellular functions. Under physiological conditions, NF-κB may coordinate the transcription of cytokines including COX-2 (24), cyclin D1 (25) and numerous other factors (26). The constitutive activation of NF-κB contributes to multiple cellular outcomes and pathophysiological conditions, including rheumatoid arthritis, asthma, inflammatory bowel disease, acquired immunodeficiency syndrome and cancer, through interactions with multiple pathways including the cell cycle, apoptosis and proliferation (27,28). Aberrant NF-κB signaling has been identified in a number of different types of cancer; NF-κB regulates the transcription of target genes that promote cell survival and proliferation, inhibit apoptosis, and mediate invasion and metastasis (29). Loss of APC function can lead to the activation of β-catenin signaling, which is the first step in the oncogenic pathway leading to CRC development (30). It has been reported that TNF receptor superfamily member 19 (TNFRSF19) is a β-catenin target gene and TNFRSF19 receptor molecule-associated activation of NF-κB signaling has demonstrated that β-catenin may regulate NF-κB activity via TNFRSF19; activation of NF-κB activity has also been observed in the APCMin+ mice model, which was inhibited by Riccardin D as shown in our previous study (4). NF-κB inhibition is also considered to be an important therapeutic target in CRC (14,31). Thus, it may be hypothesized that the inhibition of NF-κB by Riccardin D maybe a pivotal mechanism of its effects in chemotherapy for CRC with the APC mutation. The present study then detected the expression and activity of NF-κB, revealing that Riccardin D markedly reduced NF-κB protein expression in HT-29 cells; the active form of NF-κB, p-NF-κBSer536, was also markedly suppressed.
The NF-κB signaling pathway is a complex network that regulates cellular pathways involved in a myriad of physiological and pathological conditions. In addition, the accumulation of nuclear NF-κB increases the aberrant activation of the COX-2 gene (14). In the present study, Riccardin D inhibited the expression of NF-κB, COX-2 and TNFα, as well as the transcriptional activity of NF-κB induced by LPS. Riccardin D may also be directly associated with the transcriptional activity of NF-κB. One potential mode of this action is that Riccardin D may serve as a NF-κB activation suppressor, which is supported by Auto Dock molecular analysis; however, further confirmation via X-ray crystal structure analysis is required.
It is also believed that the NF-κB signaling pathway may be a critical regulator of apoptosis, as NF-κB can block apoptosis by regulating anti-apoptosis proteins, and apoptotic signals can be activated following NF-κB inhibition during the cancer treatment (32,33). Suppression of NF-κB is also thought to limit the proliferation of cancer cells (34). NF-κB regulates a number of anti-apoptotic genes, particularly the TNF receptor-associated factors (TRAFs), TRAF1 and TRAF2 (35,36). The present study revealed that Riccardin D induced HT-29 cell apoptosis as indicated by the reduction in MMP, the activation of cleaved caspase-3, caspase-9 and PARP, and the increase in the Bax/Bcl-2 ratio of Riccardin D-treated cells; this indicated that the intrinsic pathway may have been triggered by Riccardin D treatment. Therefore, these results suggested that Riccardin D, following NF-κB inhibition, may induce colon cell apoptosis by modulating the mitochondria-mediated intrinsic apoptosis signaling pathway.
The results of the present study demonstrated that Riccardin D inhibits cell proliferation and induces apoptosis in HT-29 cells, which may be associated with inhibiting the NF-κB signaling pathway. Previous studies have revealed that Riccardin D inhibits breast cancer growth through the suppression of telomerase (37). However, some telomeric proteins, such as Ras-related protein RAP1, have been shown to serve a role as a modulator of the NF-κB-mediated signaling pathway (38). Therefore, Riccardin D may have inhibitory effects on the NF-κB pathway through the suppression of telomerase. However, the underlying mechanisms require further investigation.
In conclusion, the results of the present study indicated that human colon cancer cell death induced by Riccardin D may be associated with the inhibition of NF-κB signaling and that Riccardin D may be a novel NF-κB inhibitor, inhibiting colon cancer cell proliferation and inducing cell apoptosis. The critical role of regulating the NF-κB signaling pathway in apoptosis, tumor promotion and tumor maintenance, suggests that Riccardin D maybe a promising candidate in cancer therapy. Further studies investigating the mechanism of Riccardin D and its therapeutic effects on clinical tumors are still required. However, based on these results, Riccardin D may be an effective NF-κB inhibitor in cancer therapy.
Acknowledgements
The authors are grateful to the staff at the Central Research Laboratory, the Second Hospital of Shandong University (Shandong, China) for their technical assistance and support.
Funding
The present study was supported by the Natural Science Foundation of China (grant no. 81402962).
Availability of data and materials
All of the materials used in the present study are commercially available and all data included in the present study were obtained by the co-authors.
Authors' contributions
HL, DS, BZ, FK and RW designed the study. HL, XX, JW and FC performed the experiments. HL, GL, XX, YL and WJ analyzed the data. HL, GL and XX wrote the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
References
- 1.Wu Y, Yang L, Zhao J, Li C, Nie J, Liu F, Zhuo C, Zheng Y, Li B, Wang Z, Xu Y. Nuclear-enriched abundant transcript 1 as a diagnostic and prognostic biomarker in colorectal cancer. Mol Cancer. 2015;14:191. doi: 10.1186/s12943-015-0455-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Zhang S, Zhao P, Li G, Wu L, He J. Report of incidence and mortality in China cancer registries, 2009. Chin J Cancer Res. 2013;25:10–21. doi: 10.3978/j.issn.1000-9604.2012.12.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 4.Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, Horii A, Koyama K, Utsunomiya J, Baba S, Hedge P. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 5.Binefa G, Rodríguez-Moranta F, Teule A, Medina-Hayas M. Colorectal cancer: From prevention to personalized medicine. World J Gastroenterol. 2014;20:6786–6808. doi: 10.3748/wjg.v20.i22.6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu HP, Gao ZH, Cui SX, Sun DF, Wang Y, Zhao CR, Lou HX, Qu XJ. Inhibition of intestinal adenoma formation in APC mice by riccardin d, a natural product derived from liverwort plant Dumortiera hirsuta. PLoS One. 2012;7:e33243. doi: 10.1371/journal.pone.0033243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiurillo MA. Role of the Wnt/β-catenin pathway in gastric cancer: An in-depth literature review. World J Exp Med. 2015;5:84–102. doi: 10.5493/wjem.v5.i2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larriba MJ, Ordóñez-Morán P, Chicote I, Martín-Fernández G, Puig I, Muñoz A, Pálmer HG. Vitamin D receptor deficiency enhances Wnt/β-catenin signaling and tumor burden in colon cancer. PLoS One. 2011;6:e23524. doi: 10.1371/journal.pone.0023524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carothers AM, Davids JS, Damas BC, Bertagnolli MM. Persistent cyclooxygenase-2 inhibition downregulates NF-{kappa}B, resulting in chronic intestinal inflammation in the min/+ mouse model of colon tumorigenesis. Cancer Res. 2010;70:4433–4442. doi: 10.1158/0008-5472.CAN-09-4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McClellan JL, Davis JM, Steiner JL, Day SD, Steck SE, Carmichael MD, Murphy EA. Intestinal inflammatory cytokine response in relation to tumorigenesis in the Apc (Min/+) mouse. Cytokine. 2012;57:113–119. doi: 10.1016/j.cyto.2011.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murphy EA, Davis JM, McClellan JL, Gordon BT, Carmichael MD. Curcumin's effect on intestinal inflammation and tumorigenesis in the ApcMin/+ mouse. J Interferon Cytokine Res. 2011;31:219–226. doi: 10.1089/jir.2010.0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coghill AE, Newcomb PA, Campbell PT, Burnett-Hartman AN, Adams SV, Poole EM, Potter JD, Ulrich CM. Prediagnostic non-steroidal anti-inflammatory drug use and survival after diagnosis of colorectal cancer. Gut. 2011;60:491–498. doi: 10.1136/gut.2010.221143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robbins D, Zhao Y. Imaging NF-κB signaling in mice for screening anticancer drugs. Methods Mol Biol. 2011;716:169–177. doi: 10.1007/978-1-61779-012-6_10. [DOI] [PubMed] [Google Scholar]
- 14.Wang S, Liu Z, Wang L, Zhang X. NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell Mol Immunol. 2009;6:327–334. doi: 10.1038/cmi.2009.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiu W, Wang X, Leibowitz B, Liu H, Barker N, Okada H, Oue N, Yasui W, Clevers H, Schoen RE. Chemoprevention by nonsteroidal anti-inflammatory drugs eliminates oncogenic intestinal stem cells via SMAC-dependent apoptosis; Proc Natl Acad Sci USA; 2010; pp. 20027–20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB, Whelan J. Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in Apc(Min/+) mice. Cancer Res. 2002;62:403–408. [PubMed] [Google Scholar]
- 17.Klampfer L. Cytokines, inflammation and colon cancer. Curr Cancer Drug Targets. 2011;11:451–464. doi: 10.2174/156800911795538066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu ZQ, Fan PH, Ji M, Lou HX. Terpenoids and bisbibenzyls from Chinese liverworts conocephalum conicum and Dumortiera hirsute. J Asian Nat Prod Res. 2006;8:187–192. doi: 10.1080/1028602042000325537. [DOI] [PubMed] [Google Scholar]
- 19.Rosenfeld RJ, Goodsell DS, Musah RA, Morris GM, Goodin DB, Olson AJ. Automated docking of ligands to an artificial active site: Augmenting crystallographic analysis with computer modeling. J Comput Aided Mol Des. 2003;17:525–536. doi: 10.1023/B:JCAM.0000004604.87558.02. [DOI] [PubMed] [Google Scholar]
- 20.Mariani F, Sena P, Roncucci L. Inflammatory pathways in the early steps of colorectal cancer development. World J Gastroenterol. 2014;20:9716–9731. doi: 10.3748/wjg.v20.i29.9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samowitz WS, Slattery ML, Sweeney C, Herrick J, Wolff RK, Albertsen H. APC mutations and other genetic and epigenetic changes in colon cancer. Mol Cancer Res. 2007;5:165–170. doi: 10.1158/1541-7786.MCR-06-0398. [DOI] [PubMed] [Google Scholar]
- 22.Abdullah M, Rani AA, Sudoyo AW, Makmun D, Handjari DR, Hernowo BS. Expression of NF-kB and COX2 in colorectal cancer among native Indonesians: The role of inflammation in colorectal carcinogenesis. Acta Med Indones. 2013;45:187–192. [PubMed] [Google Scholar]
- 23.Phelps RA, Broadbent TJ, Stafforini DM, Jones DA. New perspectives on APC control of cell fate and proliferation in colorectal cancer. Cell Cycle. 2009;8:2549–2556. doi: 10.4161/cc.8.16.9278. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto K, Arakawa T, Ueda N, Yamamoto S. Transcriptional roles of nuclear factor kappa B and nuclear factor-interleukin-6 in the tumor necrosis factor alpha-dependent induction of cyclooxygenase-2 in MC3T3-E1 cells. J Biol Chem. 1995;270:31315–31320. doi: 10.1074/jbc.270.52.31315. [DOI] [PubMed] [Google Scholar]
- 25.Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. NF-kappaB function in growth control: Regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol. 1999;19:2690–2698. doi: 10.1128/MCB.19.4.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Esteve PO, Chicoine E, Robledo O, Aoudjit F, Descoteaux A, Potworowski EF, St-Pierre Y. Protein kinase C-zeta regulates transcription of the matrix metalloproteinase-9 gene induced by IL-1 and TNF-alpha in glioma cells via NF-kappa B. J Biol Chem. 2002;277:35150–35155. doi: 10.1074/jbc.M108600200. [DOI] [PubMed] [Google Scholar]
- 27.Vaiopoulos AG, Athanasoula KCh, Papavassiliou AG. NF-κB in colorectal cancer. J Mol Med (Berl) 2013;91:1029–1037. doi: 10.1007/s00109-013-1045-x. [DOI] [PubMed] [Google Scholar]
- 28.Sakamoto K, Maeda S, Hikiba Y, Nakagawa H, Hayakawa Y, Shibata W, Yanai A, Ogura K, Omata M. Constitutive NF-kappaB activation in colorectal carcinoma plays a key role in angiogenesis, promoting tumor growth. Clin Cancer Res. 2009;15:2248–2258. doi: 10.1158/1078-0432.CCR-08-1383. [DOI] [PubMed] [Google Scholar]
- 29.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 30.Shaked H, Hofseth LJ, Chumanevich A, Chumanevich AA, Wang J, Wang Y, Taniguchi K, Guma M, Shenouda S, Clevers H, et al. Chronic epithelial NF-κB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation; Proc Natl Acad Sci; 2012; pp. 14007–14012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Temraz S, Mukherji D, Shamseddine A. Potential targets for colorectal cancer prevention. Int J Mol Sci. 2013;14:17279–17303. doi: 10.3390/ijms140917279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kucharczak J, Simmons MJ, Fan Y, Gelinas C. To be, or not to be: NF-κB is the answer-role of Rel/NF-κB in the regulation of apoptosis. Oncogene. 2003;22:8961–8982. doi: 10.1038/sj.onc.1207230. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-kappaB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107:135–142. doi: 10.1172/JCI11914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim SM, Lee SY, Yuk DY, Moon DC, Choi SS, Kim Y, Han SB, Oh KW, Hong JT. Inhibition of NF-kappaB by ginsenoside Rg3 enhances the susceptibility of colon cancer cells to docetaxel. Arch Pharm Res. 2009;32:755–765. doi: 10.1007/s12272-009-1515-4. [DOI] [PubMed] [Google Scholar]
- 35.Takada Y, Kobayashi Y, Aggarwal BB. Evodiamine abolishes constitutive and inducible NF-kappaB activation by inhibiting IkappaBalpha kinase activation, there by suppressing NF-kappaB-regulated antiapoptoticand metastatic gene expression, up-regulating apoptosis and inhibiting invasion. J Biol Chem. 2005;280:17203–17212. doi: 10.1074/jbc.M500077200. [DOI] [PubMed] [Google Scholar]
- 36.Elbaz M, Yanay N, Laban S, Rabie M, Mitrani-Rosenbaum S, Nevo Y. Life or death by NF-κB, Losartan promotes survival in dy2J/dy2J mouse of MDC1A. Cell Death Dis. 2015;6:e1690. doi: 10.1038/cddis.2015.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun CC, Xu HM, Yuan Y, Gao ZH, Lou HX, Qu XJ. Riccardin D, a macrocyclicbisbibenzy, inhibits human breast cancer growth through the suppression of telomerase activity. Basic Clin Pharmacol Toxicol. 2014;115:488–498. doi: 10.1111/bcpt.12267. [DOI] [PubMed] [Google Scholar]
- 38.Teo H, Ghosh S, Luesch H, Ghosh A, Wong ET, Malik N, Orth A, de Jesus P, Perry AS, Oliver JD, et al. Telomere-independent Rap1 is an IKK adaptor and regulates NF-kappaB-dependent gene expression. Nat Cell Biol. 2010;12:758–767. doi: 10.1038/ncb2080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All of the materials used in the present study are commercially available and all data included in the present study were obtained by the co-authors.