

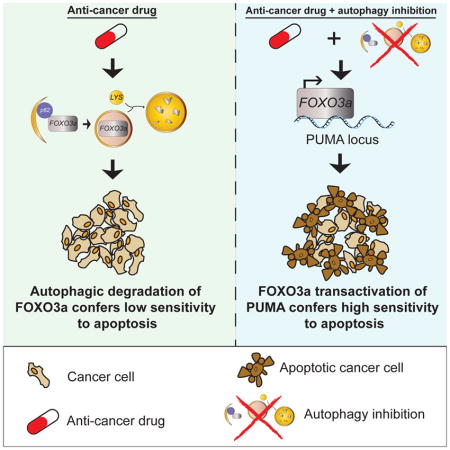
Summary
Macroautophagy (autophagy) is intimately linked with cell death and allows cells to evade apoptosis. This has prompted clinical trials to combine autophagy inhibitors with other drugs with the aim of increasing the likelihood of cancer cells dying. However, the molecular basis for such effects is unknown. Here, we describe a transcriptional mechanism that connects autophagy to apoptosis. The autophagy-regulating transcription factor, FOXO3a, is itself turned over by basal autophagy creating a potential feedback loop. Increased FOXO3a upon autophagy inhibition stimulates transcription of the pro-apoptotic BBC3/PUMA gene to cause apoptosis sensitization. This mechanism explains how autophagy inhibition can sensitize tumor cells to chemotherapy drugs and allows an autophagy inhibitor to change the action of an MDM2-targeted drug from growth inhibition to apoptosis, reducing tumor burden in vivo. Thus, a link between two processes mediated via a single transcription factor binding site in the genome can be leveraged to improve anti-cancer therapies.
Keywords: autophagy, apoptosis, FOXO3a, PUMA, Nutlin, p53, MDM2, homeostasis, CRISPR/Cas9, chloroquine
eTOC Blurb
Fitzwalter et al. uncover a link between autophagy and apoptosis, explaining how autophagy inhibitors can improve anticancer drugs by increasing sensitivity to apoptosis. The transcription factor FOXO3a, an autophagy regulator, is itself degraded by basal autophagy. Disruption of autophagy allows FOXO3a to upregulate BBC3/PUMA expression and thus cause apoptosis sensitization.
Introduction
Autophagy, the mechanism by which cellular material is delivered to lysosomes via double membrane vesicles called autophagosomes, has multiple and oftentimes competing roles in cancer (Amaravadi et al., 2016; White, 2012). However, it is well established that autophagy can protect cancer cells against apoptosis (Fitzwalter and Thorburn, 2015) and this has provided a basis for many pre-clinical and clinical studies where autophagy inhibitors are intended to increase tumor cell death when used in combination with other anti-cancer agents (Levy et al., 2017; Towers and Thorburn, 2016). Although autophagy’s ability to protect against apoptosis is well established, the molecular machinery that links autophagy and apoptosis to govern cell-fate decisions is poorly understood.
We previously reported (Thorburn et al., 2014) that the pro-apoptotic protein PUMA (p53-upregulated modulator of apoptosis, also known as BBC3) is increased upon autophagy inhibition. This increase is not sufficient to cause cells to die on their own but can sensitize them to an apoptosis inducer. Here, we investigated the underlying mechanism by which this occurs revealing a transcriptional feedback loop that links basal autophagy homeostasis to apoptosis sensitivity. This mechanism explains why autophagy inhibition can increase apoptosis in response to anti-cancer drugs. Moreover, this mechanism also allows autophagy inhibitors to promote activation of apoptosis by an inhibitor of MDM2, which can activate the p53 transcriptional program but often merely cause growth inhibition and fail to induce tumor cell apoptosis (Burgess et al., 2016). Thus, by capitalizing on a mechanism that connects autophagy to apoptosis, it is feasible to improve and even change the mode of action of an anti-cancer drug.
Results
Basal autophagy inhibition increases expression of PUMA
Previous work demonstrated that shRNA knockdown of essential autophagy regulators or pharmacological inhibition of autophagy caused higher PUMA protein levels (Thorburn et al., 2014). A simple explanation for this effect would be that PUMA protein is degraded by autophagy. However, inhibition of autophagy by knockdown of multiple autophagy regulators including, ATG7, PIK3C3/Vps34, and ULK1 (Unc-51-like Autophagy Activating Kinase 1) caused increased PUMA mRNA levels in HCT116 colorectal cancer cells (Figures 1A–1F) as well as other cancer cell lines including MCF7 and MCF10a (Figures S1A–S1D). An increase in PUMA mRNA levels was also observed when two essential autophagy regulators ATG7 or ATG5 were knocked out using CRISPR/Cas9 (Figure 1G). Treatment with bafilomycin A1, an inhibitor of vacuolar-type H+-ATPase that blocks autophagy by preventing lysosomal acidification also caused increases in PUMA mRNA (Figure 1H), and this was abolished following drug washout (Figure 1I). PUMA is a well-known target gene for p53 however, basal autophagy inhibition in HCT116 cells that lack p53 displayed similar increases in PUMA mRNA compared to HCT116 wild-type cells (Figure 1J). To test if autophagy inhibition causes increased PUMA gene transcription, chromatin immunoprecipitation (ChIP) was performed at the BBC3/PUMA locus using an antibody that recognizes elongating RNA Polymerase II (Komarnitsky et al., 2000). Autophagy inhibition with bafilomycin A1 (Figure 1K) or chloroquine (Figure S1E) caused enrichment of active RNA Pol II occupancy at the BBC3/PUMA locus that was comparable to other stimuli that activate PUMA transcription (Figure S1F). Taken together, these data indicate that basal autophagy inhibition leads to increased PUMA mRNA transcription in a p53-independent manner.
Figure 1. Autophagy inhibition activates PUMA transcription.

(A–F) HCT116 cells were transduced with lentiviral shRNAs targeting autophagy regulators (shATG7, shVps34, shULK1) or shCtrl and the resulting BBC3 (PUMA) mRNA levels were measured relative to 18s rRNA control. See also Figure S1.
(G) HCT116 cells were transduced with a lentiviral CRISPR/Cas9 plasmid targeting ATG5, ATG7, or a non-targeting control and BBC3 (PUMA) mRNA levels were measured relative to 18s rRNA control.
(H) HCT116 cells were treated with vehicle or bafilomycin A1 10nM, an autophagy inhibitor, for 24hrs and BBC3 (PUMA) mRNA levels were measured relative to 18s rRNA control.
(I) HCT116 cells treated with bafilomycin 10nM for 24hrs, then washed out for 16hrs and 24hrs. BBC3 (PUMA) mRNA levels were measured relative to 18s rRNA control.
(J) HCT116 cells or HCT116 p53 −/− cells were treated with vehicle or bafilomycin A1 10nM, an autophagy inhibitor, for 24hrs and BBC3 (PUMA) mRNA levels were measured relative to 18s rRNA control.
(K) Chromatin immunoprecipitation analysis in HCT116 cells of RNA polymerase II phospho serine 2 (indicative of active, elongating RNA pol II) occupancy at the BBC3 (PUMA) genomic locus relative to IgM control with vehicle or bafilomycin A1 10nM treatment, an autophagy inhibitor, for 24hrs. X-axis numbers indicate the probed position along the BBC3 (PUMA) locus relative to the transcriptional start site. PUMA locus schematic is not to scale; stars along the locus indicate the approximate position of each amplicon. See also Figure S1.
Statistical significance in B, D, F, H was determined by unpaired t-test, one-way ANOVA (Tukey post-hoc) in G, I, K, and two-way ANOVA (Tukey post-hoc) in J. *P<0.05; **P<0.01; ***P<0.001. Data represented as mean ± SEM.
Autophagy regulation of PUMA is mediated through FOXO3a
The basal rate of autophagy is regulated by several transcription factors (Feng et al., 2015; Füllgrabe et al., 2014) including FOXO3a, which is critical for maintenance of a gene expression program that allows autophagy to protect hematopoietic stem cells from cytokine deprivation (Warr et al., 2013). Additionally, FOXO3a can regulate PUMA expression (Amente et al., 2011; You et al., 2006) leading us to hypothesize that FOXO3a might be responsible for increased PUMA expression upon autophagy inhibition. In support of this idea, bafilomycin A1 failed to increase PUMA mRNA when FOXO3a was knocked down via shRNA (Figure 2A) or knocked out with CRISPR/Cas9 (Figures 2B and S2A). ChIP analysis revealed that FOXO3a displays a constitutive level of occupancy at the BBC3/PUMA locus in an intronic region containing a Forkhead Response Element (FHRE) +1,902 basepairs downstream of the transcriptional start site as was previously reported (Eijkelenboom et al., 2013). Upon basal autophagy inhibition with bafilomycin A1 or chloroquine, FOXO3a became enriched approximately 20-fold over IgG control at this site (Figures 2C and S2B). Mutation of the FOXO3a-binding FHRE in the BBC3/PUMA locus using CRISPR/Cas9 gene editing to create a set of three pooled HCT116 clones each with deletions in the FHRE (Figure 2D) was sufficient to block the increase in PUMA expression upon pharmacological (Figure 2E) or genetic (Figure S2C) autophagy inhibition, but did not affect the ability of p53 to activate PUMA via Nutlin treatment (Figure S2D). These data indicate that inhibition of basal autophagy results in increased FOXO3a-driven transcription of the PUMA gene through a single transcription factor binding site in the genome.
Figure 2. FOXO3a binding to the BBC3/PUMA locus is necessary for PUMA transcriptional activation.

(A) (left) HCT116 p53 −/− cells were transduced with two different lentiviral shRNAs targeting FOXO3a transcripts. After 24hrs of vehicle or bafilomycin A1 10nM treatment, PUMA mRNA levels were measured relative to 18s rRNA control. (right) western blot showing FOXO3a protein levels after treatment with two independent shRNAs targeting FOXO3a or shCtrl.
(B) Parental or FOXO3a−/− pooled HCT116 cells were treated with bafilomycin A1 10nM, an autophagy inhibitor, for 24hrs and BBC3 (PUMA) mRNA levels were measured relative to GAPDH control. See also Figure S2.
(C) Chromatin immunoprecipitation analysis in HCT116 cells of FOXO3a occupancy at the BBC3 (PUMA) genomic locus relative to IgG control with vehicle or bafilomycin A1 10nM, an autophagy inhibitor, treatment for 24hrs. X-axis indicates the probed position along the BBC3 locus relative to the transcriptional start site. PUMA locus schematic is not to scale; stars indicate the approximate position of each amplicon. See also Figure S2.
(D) Sequencing alignments showing ΔForkhead Response Element (ΔFHRE) deletions in HCT116 cells at the BBC3 locus at position +1,902 relative to the transcriptional start site using CRISPR/Cas9 targeting. Blue and Green base pairs indicate FHRE. Multiple clones were obtained with homozygous indels and were pooled to limit clonal variation.
(E) HCT116 parental or ΔForkhead Response Element (ΔFHRE) cells were treated for 24hrs with vehicle or bafilomycin A1 10nM, an autophagy inhibitor, and BBC3 (PUMA) mRNA levels were measured relative to 18s rRNA control. See also Figure S2.
Statistical significance in C was determined by one-way ANOVA (Tukey post-hoc) and two-way ANOVA (Tukey post-hoc) in A, B, E. **P<0.01; ***P<0.001; n.s.=not significant. Data represented as mean ± SEM.
FOXO3a also had a constitutive level of occupancy at the BIM locus that increased nearly two-fold with bafilomycin A1 treatment for 24hrs (Figure S2E), but FOXO3a occupancy at non-apoptotic proteins like SOD2 did not change with autophagy inhibition at that time point (Figure S2F). Pharmacological or genetic autophagy inhibition by CRISPR/Cas9 knockout of essential autophagy genes resulted in higher BIM mRNA levels (Figures S2G and S2H). Several other known FOXO3a target genes including non-apoptotic genes were also activated (albeit with varying kinetics) upon bafilomycin A1 treatment (Figure S2I–S2M). These data suggest that FOXO3a increases its transcriptional activity at multiple gene targets upon genetic or pharmacological autophagy inhibition.
Basal FOXO3a turnover by autophagy
FOXO3a is degraded by the proteasome in response to various signaling events (Huang and Tindall, 2011) but it is not known if autophagy also regulates FOXO3a. To test this and determine if autophagy-mediated turnover of FOXO3a explains how autophagy inhibition can enhance FOXO3a transcriptional activity, multiple cell types were treated with bafilomycin A1 (Figures 3A and S3A), or chloroquine (Figures S3B and S3C) resulting in a time-dependent increase in the amount of FOXO3a protein indicating that basal FOXO3a turnover can occur via the lysosome. Consistent with this turnover being due to (macro)autophagy, HCT116 cells that lack the essential autophagy genes ATG5 or ATG7 by CRISPR/Cas9-mediated knockout showed no changes in FOXO3a mRNA (Figure S3D), but showed a robust increase in FOXO3a protein compared to control cells (Figure 3B). Similarly, ATG7 shRNA knockdown also caused a robust increase in FOXO3a protein compared to shCtrl cells (Figure S3E). Additionally, shRNA knockdown targeting p62/SQSTM1, a protein that recruits substrates to the autophagosome, also increased FOXO3a protein in multiple cells lines (Figures 3C and S3F–G) without increased FOXO3a mRNA (Figure S3H). Importantly, shRNA targeting of another autophagy receptor that recruits cargo to the autophagosome, NBR1, did not cause an increase in FOXO3a protein (Figure S3I). Exogenously expressed FOXO3a protein also increased when cells were treated with bafilomycin A1 (Figure 3D) or p62/SQSTM1 shRNA (Figure 3E).
Figure 3. FOXO3a is a substrate for basal autophagy.

(A) HCT116 cells treated with bafilomycin A1 10nM, an autophagy inhibitor, for indicated duration. Whole-cell lysates were probed for FOXO3a protein levels. See also Figure S3.
(B) HCT116 cells were transduced with lentiviral constructs containing Cas9 and gRNA targeting ATG5, ATG7, or a non-targeting control and whole-cell lysates were probed for indicated proteins. See also Figure S3
(C) HCT116 cells were transduced with lentiviral shRNAs targeting p62/SQSTM1 or shCtrl and probed for indicated proteins. See also Figure S3.
(D) Western blot analysis on whole-cell lysates of HCT116 cells expressing mCherry-GFP-FOXO3a treated with bafilomycin A1 10nM, an autophagy inhibitor, for 8hrs.
(E) Western blot analysis of whole-cell lysates of HCT116 cells expressing mCherry-GFP-FOXO3a transduced with shCtrl or shp62/SQSTM1 lentivirus.
(F) Schematic representation of expression construct and how it was used to understand the environment of FOXO3a localization.
(G) Confocal images of HCT116 cells expressing mCherry-GFP-FOXO3a and treated with a lysosomal dye. See also Figure S4.
(H) Confocal images of HCT116 cells expressing mCherry-GFP-FOXO3a transduced with shRNA targeting ATG5.
(I) Confocal images of HCT116 cells expressing mCherry-GFP-FOXO3a transduced with shRNA targeting ATG7.
(J) Confocal images of HCT116 cells expressing mCherry-GFP-FOXO3a treated with bafilomycin A1 10nM, an autophagy inhibitor, for 8hrs. Scale bars= 10μm.
If FOXO3a is a substrate for autophagy, it should localize to autophagosomes and lysosomes. To test this, we expressed a construct expressing mCherry-GFP-FOXO3a (Figure 3F). GFP fluorescence, but not mCherry, is sensitive to decreased pH and this provides a basis to monitor autophagosome and autolysosome targeting of autophagy markers such as LC3B (Gump et al., 2014; Kimura et al., 2007) and autophagy cargos (Dou et al., 2015). If FOXO3a is in a pH-neutral environment, such as the cytoplasm or nucleus, both GFP and mCherry should fluoresce resulting in a diffuse yellow signal from the overlayed mCherry and GFP. If FOXO3a is concentrated into a small pH neutral environment, like an autophagosome, we expect a punctate yellow signal and, if FOXO3a resides in an acidic environment, such as an autolysosome, we should see punctate red signals from the overlayed mCherry and GFP fluorescent signals (Figure 3F). Cells expressing the mCherry-GFP-FOXO3a protein under basal conditions showed a significant amount of FOXO3a localized to acidic compartments that were also positively stained by a lysosomal marker (Figures 3G and S4A–D). Upon shRNA knockdown of ATG5 or ATG7 (Figures 3H and 3I) or blocking lysosomal acidification with bafilomycin A1 (Figure 3J), the predominance of punctate red signal was reverted to yellow and FOXO3a accumulated in the nucleus consistent with the data above showing that FOXO3a binds to and transactivates the BBC3/PUMA locus and causes new transcription upon autophagy inhibition. These data indicate that FOXO3a is targeted to autolysosomes allowing FOXO3a levels to be controlled by basal autophagy in addition to the proteasome as previously identified (Huang and Tindall, 2011). These data support a model whereby a transcription factor that controls autophagy is itself degraded by autophagy and, when this is blocked, the elevated FOXO3a sensitizes cells to apoptosis by increasing PUMA transcription. This creates a mechanism whereby a homeostatic loop that potentially controls autophagy also ensures that autophagy deficiency sensitizes to apoptosis.
Autophagy regulation of PUMA expression via FOXO3a can be leveraged to improve anti-cancer drugs
Autophagy inhibition using chloroquine or hydroxychloroquine is being tested in clinical studies in combination with many different anti-cancer drugs based on the idea that this will enhance tumor cell apoptosis (Levy et al., 2017; Towers and Thorburn, 2016). We therefore tested if the FOXO3a mediated PUMA activation mechanism is important for sensitizing cells to anti-cancer drugs upon autophagy inhibition. Consistent with numerous previous studies with anti-cancer drugs, etoposide (Figure 4A–B) or doxorubicin (Figure 4C–D) caused greater caspase 3/7 activity when combined with autophagy inhibition by chloroquine compared to the individual agents. However, this ability of autophagy inhibition to sensitize parental HCT116 tumor cells to drug-induced apoptosis was abolished in isogenic cells that either lack PUMA (PUMA−/−) or merely lack the ability to bind FOXO3a at the single site in the PUMA locus (ΔFHRE) (Figures 4E–H). These data suggest that the ability of autophagy inhibitors like chloroquine to sensitize tumor cells to apoptosis by standard chemotherapy is via the FOXO3a-dependent mechanism described above. Moreover, although autophagy inhibition can lead to increased transcription of other FOXO3a target genes (Figure S2I–S2M), including other pro-apoptotic genes including BIM (Figures S2G and S2H) and BNIP3 (Figure S2L), the increased drug-induced apoptosis that we observe when autophagy is blocked is mediated largely through a single binding site in the PUMA gene.
Figure 4. The combination effect of autophagy inhibition and anti-cancer agents requires FOXO3a binding the Forkhead Response Element to induce apoptosis.

(A) HCT116 cells were treated with vehicle, chloroquine 40μM, etoposide 2.5μM, or etoposide + chloroquine. Caspase 3/7 activation (an indicator of apoptosis execution) was measured and normalized to cell number per mm2.
(B) Representative images of caspase 3/7 activation from panel A treated with vehicle, chloroquine 40μM, etoposide 2.5μM, or etoposide + chloroquine.
(C) HCT116 cells were treated with vehicle, chloroquine 40μM, doxorubicin 2.5μM, or doxorubicin + chloroquine. Caspase 3/7 activation (an indicator of apoptosis execution) was measured and normalized to cell number per mm2.
(D) Representative images of caspase 3/7 activation from panel C treated with vehicle, chloroquine 40μM, doxorubicin 2.5μM, or doxorubicin + chloroquine.
(E) HCT116 parental cells, PUMA−/− cells, or ΔFHRE cells lacking the endogenous Forkhead Response Element in the BBC3/PUMA locus were treated with etoposide 2.5μM + chloroquine 40μM. Caspase 3/7 activation was measured and normalized to cell number per mm2.
(F) Representative images of caspase 3/7 activation from panel E in HCT116 parental cells, PUMA−/− cells, or ΔFHRE cells lacking the Forkhead Response Element in the endogenous BBC3 locus treated with etoposide 2.5μM + chloroquine 40μM.
(G) HCT116 parental cells, PUMA−/− cells, or ΔFHRE cells lacking the endogenous Forkhead Response Element in the BBC3/PUMA locus were treated with doxorubicin 2.5μM + chloroquine 40μM. Caspase 3/7 activation was measured and normalized to cell number per mm2.
(H) Representative images of caspase 3/7 activation from panel G in HCT116 parental cells, PUMA−/− cells, or ΔFHRE cells lacking the Forkhead Response Element in the endogenous BBC3/PUMA locus treated with doxorubicin 2.5μM + chloroquine 40μM. Statistical significance in A, C, E, G was determined by repeated measures two-way ANOVA (Tukey post-hoc). ***P<0.001. Data represented as mean ± SEM.
We next asked if this idea could be taken a step further and change the mode of action of a drug. Nutlin is an inhibitor of MDM2 that activates the p53 transcriptional program including PUMA but only results in growth inhibition rather than tumor cell death in most cancer cell types (Huang et al., 2009; Paris et al., 2008; Tovar et al., 2006). We used publicly available datasets (Garnett et al., 2012) to identify determinants of Nutlin response and found that PUMA levels, but not other BH3-only proteins, is positively correlated with Nutlin sensitivity (IC50 < 5μM) in different cell types (Figures 5A and S5A–C). This led us to hypothesize that by combining MDM2 inhibition with autophagy inhibition, both of which increase PUMA levels but through different transcription factors (i.e. p53 and FOXO3a, respectively), it might be possible to enhance PUMA transcription more than with MDM2 inhibition alone and thus cause cancer cells that would otherwise only undergo growth inhibition with Nutlin to commit to apoptosis instead.
Figure 5. FOXO3a mediated upregulation of PUMA changes the mode of action of an anti-cancer drug.

(A) Publicly available gene expression data set of cancer cell lines from multiple tumor types via Oncomine shows that cell lines sensitive to Nutlin (IC50 < 5 μM) have increased expression of BBC3/PUMA. Values were Log2 transformed and the median values were scaled to zero according to Garnett et al. 2012. See also Figure S5.
(B) HCT116 cells were treated with vehicle, bafilomycin A1 10nM, chloroquine 40μM, Nutlin 20μM—an MDM2 inhibitor that activates p53, Nutlin + bafilomycin A1, or Nutlin + chloroquine for 24hrs and the resulting BBC3/PUMA mRNA levels were measured relative to 18s rRNA control.
(C) Clonogenic survival assay using HCT116 cells treated for 72hrs with vehicle, chloroquine 40μM, Nutlin 20μM, or Nutlin + chloroquine. Cells were then allowed to grow back for 7 days in full media.
(D) HCT116 cells were treated with vehicle, chloroquine 40μM, Nutlin 20μM, or Nutlin + chloroquine. Cell permeability by Sytox green (an indicator of cell death) was measured and normalized to cell number per mm2.
(E) Representative images from panel D of cell permeability by Sytox green in HCT116 parental cells treated with vehicle, chloroquine 40μM, Nutlin 20μM, or Nutlin + chloroquine.
(F) HCT116 cells were treated with vehicle, chloroquine 40μM, Nutlin 20μM, or Nutlin + chloroquine. Caspase 3/7 activation (an indicator of apoptosis execution) was measured and normalized to cell number per mm2. See also Figure S5.
(G) Representative images of caspase 3/7 activation from panel F in HCT116 parental cells treated with vehicle, chloroquine 40μM, Nutlin 20μM, or Nutlin + chloroquine.
(H) HCT116 parental cells or ΔFHRE cells lacking the endogenous Forkhead Response Element in the BBC3/PUMA locus were treated with Nutlin 20μM + chloroquine 40μM. Caspase 3/7 activation was measured and normalized to cell number per mm2. See also Figure S5.
(I) Representative images of caspase 3/7 activation from panel H in HCT116 parental cells or ΔFHRE cells lacking the Forkhead Response Element in the endogenous BBC3/PUMA locus treated with Nutlin 20μM + chloroquine 40μM.
(J) Athymic nude mice given subcutaneous parental or ΔFHRE HCT116 tumors were treated with vehicle, Nutlin, chloroquine, or Nutlin + chloroquine and PUMA mRNA levels in the tumors were measured relative to 18s rRNA control.
(K) Kaplan-Meier survival curves based on tumor burden of athymic nude mice given subcutaneous parental or ΔFHRE HCT116 tumors and treated with vehicle, Nutlin, chloroquine, or Nutlin + chloroquine over indicated duration.
(L) Schematic representation of global model when autophagy is perturbed. Upon genetic or pharmacological basal autophagy inhibition, autophagy homeostasis is perturbed and FOXO3a becomes transcriptionally active. Both autophagy genes and pro-apoptotic proteins like PUMA are transcriptionally activated to compensate for the autophagy perturbation. If homeostasis is not re-established, cells undergo apoptosis. Statistical significance in A was determined by unpaired t-test, one-way ANOVA (Tukey post-hoc) in B, a repeated measures two-way ANOVA (Tukey post-hoc) in D, F, H, a two-way ANOVA in J, and five individual comparisons using Log-rank (Mantel-Cox) test in K. Due to five individual comparisons in K, p=0.05 ÷ 5= 0.01 is the corrected threshold for statistical significance. *P<0.01 for KM survival curves; n.s.=not significant. *P<0.05; **P<0.01; ***P<0.001 for all others. Data represented as mean ± SEM
Consistent with this hypothesis, Nutlin plus bafilomycin A1 or chloroquine caused a greater increase in PUMA mRNA levels compared to either compound alone (Figure 5B). Previous reports (Paris et al., 2008; Tovar et al., 2006) indicate that HCT116 cells undergo growth arrest upon treatment with Nutlin, but no apoptosis. Consistent with this, the combination of Nutlin and chloroquine was much more effective at reducing colony formation than either drug alone (Figure 5C). As expected, Nutlin alone caused a robust cytostatic effect, however, Nutlin plus chloroquine resulted in a reduction in tumor cell number (Figure S5D) and resulted in more cell death compared to each agent alone as measured by a cell permeable dye (Figures 5D and 5E). Only the combination of Nutlin and either pharmacological or genetic autophagy inhibition was able to increase caspase 3/7 activity (Figures 5F, 5G, and S5E–S5G). Caspase activation was greatly reduced in HCT116 cells lacking either PUMA or containing deletions in the single FOXO3a-binding FHRE in the PUMA gene locus (Figures 5H–I, S5H, and SMovie1). However, cells that lack the FOXO3a binding element were still able to activate caspases similar to parental cells when treated with the broad apoptotic stimulus, staurosporine (Figure S5I). Taken together, these data indicate that the mechanism whereby autophagy inhibition promotes FOXO3a-driven PUMA expression is necessary for the combination effect of Nutlin and autophagy inhibitor that leads to tumor cell apoptosis rather than growth arrest.
A similar combinatorial effect of Nutlin and autophagy inhibition on gene activation of BIM, or other BH3 proteins, was not observed (Figure S5J). And although HCT116 cells had low apoptosis sensitivity upon BIM shRNA knockdown (Figure S5K), this occurred independent of BIM transcriptional regulation by Nutlin and autophagy inhibition. Thus, the primary mechanism by which the drug combination causes the switch to apoptosis seems to be mediated through the PUMA gene suggesting that the ability to activate two transcription factors (p53 and FOXO3a) that target the same gene (i.e. PUMA) is important for mediating apoptosis ability by the p53-activating drug upon autophagy inhibition.
Consistent with the in vitro studies, xenografted tumors grown from HCT116 parental cells responded to the combination of Nutlin and chloroquine by enhancing PUMA mRNA levels better than either drug alone (Figure 5J). However, tumors from HCT116 cells lacking the endogenous Forkhead Response Element (ΔFHRE) treated with the combination of Nutlin and chloroquine did not show this effect. As expected for a compound that activates p53, other p53 target genes, including BAX and p21, were also upregulated (Figure S5L). Importantly, mice with tumors grown from the parental HCT116 cells had markedly improved survival due to tumor burden by the drug combination compared with either drug alone. However, mice with tumors that lacked the single FHRE in the PUMA locus displayed no better survival due to tumor burden with the combination treatment than was seen with Nutlin alone in the HCT116 parental cell tumors (Figure 5K). Tumor growth rates were slowest with the drug combination in parental HCT116 tumors while tumor growth rate with the drug combination in tumors lacking the single FHRE in the PUMA locus were similar to that seen with Nutlin alone in the HCT116 parental tumors (Figure S5M). In contrast to the improvement in survival due to tumor burden (Figure 5K), the robust cytostatic effect of Nutlin alone meant that the study did not achieve statistical significance (p=0.16) when comparing tumor volume of Nutlin treated mice to Nutlin and chloroquine treated mice (Figure S5M). Additionally, we noted increased toxicity (e.g. as determined by weight loss, Figure S5N) in animals treated with the drug combination that led some animals to be removed from the study irrespective of their tumor burden. This is consistent with recent clinical studies where it has been found that autophagy inhibition with chloroquine or hydroxychloroquine can increase the toxicities that are known to be caused by the other drug that is used in combination (Levy et al., 2017).
Discussion
FOXO3a can orchestrate a gene transcription program that maintains autophagy homeostasis (Warr et al., 2013). In this study, we expand on this idea by showing that disturbance in the autophagy balance (i.e. inhibiting autophagy) causes cells to increase FOXO3a activity presumably with the goal of reestablishing homeostasis. This is achieved because FOXO3a is itself turned over by basal autophagy. Moreover, this mechanism of FOXO3a regulation also creates a link between autophagy and apoptosis that causes autophagy-inhibited cells to simultaneously be sensitized to apoptosis by upregulating BBC3/PUMA. Our data therefore fit with a model whereby FOXO3a is part of a homeostatic feedback surveillance loop to maintain autophagy homeostasis and ensure that in the event of autophagy inhibition, cells that fail to re-establish proper levels of autophagy are more likely to undergo apoptosis (Figure 5L). Although other pro-apoptotic FOXO3a targets (e.g. BIM) were also activated, our results indicate that the primary apoptosis sensitizing effect is achieved through the PUMA gene via a single FOXO3a binding site. However, different types of cancer cells may depend to a greater extent on different BH3-only proteins for apoptosis.
Importantly, it is possible to leverage this mechanism to improve anti-cancer drugs. There are many ongoing clinical trials to inhibit autophagy for cancer therapy (Levy et al., 2017; Towers and Thorburn, 2016). The underlying idea is to enhance tumor cell apoptosis and thus make other drugs work more effectively but the molecular mechanism by which this occurs has been unclear making it difficult to determine which drugs might benefit from autophagy inhibition. The regulatory mechanism identified here begins to solve these problems and our data indicate that the ability of an autophagy inhibitor to increase tumor cell apoptosis by standard chemotherapy drugs like etoposide or doxorubicin requires that FOXO3a be able to increase PUMA gene expression. Moreover, we also show that it is possible to exploit the mechanistic link between autophagy homeostasis and apoptosis sensitivity to improve the therapeutic efficacy of a cancer drug like Nutlin that often only causes growth inhibition of cancer cells (Huang et al., 2009; Paris et al., 2008; Tovar et al., 2006) into one that kills cancer cells. Thus, pharmacological manipulation of the autophagic turnover of a transcription factor can work through a single genomic binding site in one gene to improve the ability of cancer drugs to cause tumor cell apoptosis and can even change the mode of action of a highly specific anti-cancer drug. Since there are several MDM2 inhibitors in clinical development (Burgess et al., 2016), our work suggests that they could be good candidates for combination with autophagy inhibition in the clinic (Sullivan et al., 2015). However, the increased general toxicity that we observed with the drug combination means that as with other attempts to widen the therapeutic window for a given cancer drug by inhibiting autophagy, it will be important to optimize doses of both the MDM2 inhibitor and the autophagy inhibitor in clinical studies.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Thorburn (Andrew.Thorburn@ucdenver.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines were maintained at 37 °C and 5 % CO2. HCT116 (male) cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) with 10 % fetal bovine serum. MCF7 (female) cells were maintained in Minimum Essential Media (MEM) with 10 % fetal bovine serum. MCF10a (female) cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) F12 with 5 % horse serum, 20 ng/mL epidermal growth factor, 0.5 mg/mL hydrocortisone, 100 ng/mL cholera toxin, 10 μg/mL insulin. HeLa (female) cells were maintained in RPMI with 10% FBS. The above cells lines were authenticated at the Barbara Davis Center at University of Colorado—Anschutz Medical Campus. Six week old athymic nude mice were purchased and housed according to IACUC guidelines.
METHOD DETAILS
Clonogenic assay
HCT116 cells were plated at 2000 cells per well of a 12-well plate. The next day, cells were treated with Nutlin-3a 20μM and/or chloroquine 40μM for 48 hours. These reagents were replenished after 24hrs. After 48 hours of exposure to the above reagents, full media was used to allow grow back for 7 days, replenishing media every 72 hours. Cells were then fixed and stained with crystal violet.
Chromatin Immunoprecipitation
HCT116 cells were plated at 40,000 cells/cm2 of a 150mm plate—40,000 × 152 cm2= 6,080,000 per plate (3 plates per condition). The next day, cells were treated with bafilomycin 10nM, Nutlin-3a 20μM, or chloroquine 40μM for 24 hours. Cells were then washed with PBS, fixed with a 1% formaldehyde/1× PBS solution for 15 minutes at room temperature. The fixing reaction was stopped by the addition of 1 mL of 2.5 M glycine (0.125 M final). Cells were then washed 2× with cold PBS and harvested with RIPA buffer (150 mM NaCl, 1% [v/v] Nonidet P-40, 0.5% [w/v] deoxycholate, 0.1% [w/v] SDS, 50mM EDTA, protease inhibitor cocktail, phosphatase inhibitor. Samples were then sonicated to generate <500 base pair DNA fragments. One mL of 1 mg/mL of protein lysate was precleared for 2 hours with 30μL of Protein A/G beads FOXO3a CST 75D8 (10 μL per IP) and RNA Polymerase II Clone H5 (20μL per IP) were used with 60μL of Protein A/G beads, and inverted overnight at 4°C. Beads were pre-washed twice with PBS, then twice with RIPA buffer from above. For phosphorylated CTD of polymerase, immunocomplexes were recovered using anti-mouse IgM/protein A/G beads. Beads were washed twice with RIPA buffer, 4 times with IP buffer (100mM Tris HCl pH 8.5, 500mM LiCl, 1% [v/v] Nonidet P-40, 1% [w/v] deoxycholic acid), twice again with RIPA buffer, then twice with TE. Immunocomplexes were eluted at 65°C for 10 minutes with 1% SDS. Reverse crosslinking was performed by adjusting to 200mM NaCl and incubated for 5hrs at 65°C. DNA was purified and then used in QPCR reaction. PCR primers used in these reactions are listed in the attached table.
RT-QPCR
RNA was isolated using Qiagen RNeasy kit following the manufacturer’s instructions. Reverse transcription reaction was performed using the Qiagen Quantitect RT kit according to manufacturer’s instructions. SYBR green CFX from Applied Biosystems was used in QPCR reactions with a standard curve ranging from 0.04 ng to 50ng of cDNA. Quantities were calculated relative to this standard curve, and normalized to 18s rRNA or GAPDH as housekeep gene control. Primers are listed in the Key Resources Table.
Table 1.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal FOXO3a | Cell Signaling Technology | Cat# 3938, RRID:AB_2106669 |
| Rabbit monoclonal ATG7 | Cell Signaling Technology | Cat# 8558, RRID:AB_10831194 |
| Rabbit monoclonal ATG5 | Cell Signaling Technology | Cat# 9980S, RRID:AB_10829153 |
| Rabbit polyclonal PUMA/BBC3 | Cell Signaling Technology | Cat# 4976, RRID:AB_2064551) |
| Rabbit polyclonal ULK1 | Cell Signaling Technology | Cat# 4773S, RRID:AB_2288252 |
| Rabbit polyclonal PI3 Kinase Class III | Cell Signaling Technology | Cat# 3811S, RRID:AB_2062856 |
| Rabbit polyclonal LC3 | Novus | Cat# NB100-2220, RRID:AB_10003146 |
| Mouse monoclonal beta-Actin | Sigma-Aldrich | Cat# A5441, RRID:AB_476744 |
| Mouse monoclonal p62/SQSTM1 | Novus | Cat# H00008878-M01, RRID:AB_548364 |
| Mouse monoclonal Purified anti-RNA Polymerase II RPB1 antibody Clone H5 | Biolegend | Cat# 920204, RRID:AB_2616695 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bafilomycin A1 | Sigma-Aldrich | B1792; CAS RN: 88899-55-2 |
| Chloroquine | MP Biomedicals | 93919; CAS RN: 50-63-5 |
| Nutlin-3a | Cayman Chemicals | 10004372; CAS RN: 548472-68-0 |
| Protease inhibitor cocktail | Roche | 11836153001 |
| Phosphatase inhibitor | Sigma-Aldrich | P5726 |
| Protein A/G Plus beads | Santa Cruz | SC-2003 |
| Critical Commercial Assays | ||
| Qiagen RNeasy RNA isolation kit | Qiagen | Cat# 74104 |
| QuantiTect Reverse Transcription Kit | Qiagen | Cat# 205311 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| HCT116 | Authenticated | N/A |
| MCF7 | Authenticated | N/A |
| MCF10a | Authenticated | N/A |
| HeLa | N/A | |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| See Table S1 for ChIP primers | This paper | N/A |
| See Table S1 for QPCR primers | This paper | N/A |
| See Table S1 for gRNAs | This paper | N/A |
| Recombinant DNA | ||
| pBABE puro mCherry-GFP-HA-FOXO3a | This paper and Addgene x 2 | #1787 and #1764 |
| Software and Algorithms | ||
| Other | ||
| SYBR Green CFX for QPCR | Applied Biosystems | 4472942 |
| Mirus Transit LT1 transfection reagent | Mirus | MIR2304 |
| CRISPR Max | ThermoFisher | CMAX00001 |
| Cell Event Caspase 3/7 | Invitrogen | C10423 |
| MatTek 35mm dishes for living imaging | MatTek | P35GC-1.5-14-C |
| LysoTracker Blue DND-22 | ThermoFisher | Cat# L7525 |
| Incucyte ZOOM imaging system | Essen BioScience | N/A |
Expression constructs, shRNAs, and transduction
Protein depletion by shRNA was achieved by using the pLKO.1 system. These plasmids were obtained from the University of Colorado Functional Genomics Core. Lentiviruses were obtained by cotransfection of pLKO.1 shRNA plasmids with the lentiviral plasmids pMD2G, pRRE, and pRSV into HEK293FT using Mirus Transit LT1 transfection reagent. Media was harvested at 48 and 72hours post-transfection and stored at −80 °C. Cells were plated at 30,000 cells per well of a 6-well plate and 1mL of virus used. Puromycin was used for selection (1 μg/mL). Sequences are listed in the Key Resources Table.
CRISPR/Cas9 mediated knockout
For CRISPR/Cas9 knockout of ATG5 and ATG7, lentiviral constructs containing gRNAs targeting ATG5 and ATG7 (Table S1) were transduced into HCT116 cells as previously published (O’Prey et al., 2017). To avoid complications caused by clonal variation, ATG5 or ATG7 knockout cells from polyclonal populations were assayed by Western Blot for complete loss of protein expression and the polyclonal population used for functional tests. For CRISPR/Cas9 mediated knockout of the forkhead response element (FHRE), ribonuclear protein complexes (RNPs) were transfected into HCT116 cells with Lipofectamine CRISPR Max based on previously published methods (Liang et al., 2015). Single cell clones were isolated and genomic DNA analyzed to determine whether the FHRE was mutated, then to minimize clonal variation, clones with 6 different mutated FHRE sequences (Figure 2D) were pooled and used for functional tests. Sequences of gRNAs are listed in Table S1.
Immunoblots
Western blotting was performed using standard methods with antibodies in 5% milk or 5% bovine serum albumin fraction V in TBST (0.1% Tween-20). Proteins were separated on SDS-PAGE 1.0mm mini gels and transferred to nitrocellulose membranes. Blots were then probed with antibodies diluted as above. Semi-dry transfer apparatus was run at 15V for 70 minutes. Antibodies are listed in the Key Resources Table.
Fluorescence Microscopy
Lentiviral plasmid pBABE puro mCherry-eGFP-HA-FOXO3a was cloned by digesting pBABE puro mCherry-eGFP-LC3 with MfeI and SalI, and ligating FOXO3a cDNA generated from AddGene plasmid #1787 (HA-FOXO3a). Sequences are listed in the Key Resources Table. Lentiviral transduction of this construct into MCF7, MCF10a, and HCT116 was performed, expressed for 72hrs to reach steady-state, and imaged. Live cells were plated at 2,000 cells and imaged in MatTek 35mm glass bottom culture dishes using a confocal laser scanning Olympus FV1000 with a 60× objective. LysoTracker was used at concentration of 0.5μM.
Incucyte Cell Imaging
Cells were plated in 96-well plates at 3,000 cells/well. After cell attachment (48hrs), cells were treated with indicated concentrations of Nutlin-3a, chloroquine, bafilomycin A1, or staurosporine. CellEvent Green Caspase-3/7 reagent was from Invitrogen C10423 (used at 5μM final concentration). Sytox Green (Invitrogen S7020) cell dye reagent was used at 50nM. Images were taken using the Incucyte Zoom system every 4hrs using a 4× objective. Results are displayed normalized to the cell number and the initial time point (time point zero).
In vivo tumor studies
Six week old athymic nude mice were purchased and housed according to IACUC guidelines. Two million HCT116 parental or ΔFHRE cells were counted using the Vi-cell (Beckman Coulter) resuspended, on ice, in 100μL of 0.2% matrigel:PBS (1:1) and injected into each flank using 28g needles. Mice were anesthetized using isoflurane. Dosing of Nutlin-3a and chloroquine began when 80% of the mice had tumors that were ≥100mm3. Mice were given oral gavage (28g needles, 38mm long) twice daily of Nutlin-3a (Selleckchem—S1061) at 200 mg/kg—a 20 mg/mL stock was formulated in 2% Klucel (Hydroxypropyl cellulose—AlfaAesar) and 0.5% Tween 80 (Fischer BP338–500) in PBS. Mice were also given intraperitoneal injections once daily of chloroquine diphosphate (MPBio—193919) at 60 mg/kg—a 6 mg/mL stock was formulated in PBS. Tumor volume was measured and calculated regularly (roughly every other day) using a Sylvac-Fowler Bluetooth S_cal EVO caliper. Mice were sacrificed when tumor volume reached 200mm3, or a combined tumor burden per mouse of 300mm3. Mice were sacrificed by CO2 followed by cervical dislocation according to IACUC protocol and tumors were harvested for RNA and protein. Tumor samples were homogenized and processed for RNA using above methods for RT-QPCR analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as means ± standard errors of the mean (SEMs) as indicated within each figure legend. Independently prepared samples from at least two experiments, displayed as circles, squares or triangles, are displayed on column graphs. One-way analyses of variance (ANOVA), two-way analyses of variance, or unpaired Student’s t-tests were performed where indicated in figure legends using Prism/Graphpad. *P < 0.05; **P < 0.01; ***P < 0.001.
Supplementary Material
Table S1. Primers, gRNAs, and shRNA target sequences, related to STAR Methods.
ΔFHRE cells (right) lacking the endogenous Forkhead Response Element in BBC3 locus show less caspase 3/7 activation compared to parental cells (left).
Highlights.
Basal autophagy regulates apoptosis via turnover of the FOXO3a transcription factor
FOXO3a regulates PUMA to provide a mechanistic link between autophagy and apoptosis
Autophagy inhibitors can change the mode of action of a cancer drug via FOXO3a
A single FOXO3a binding site in the genome mediates these effects
Acknowledgments
This work was supported by National Institutes of Health grants RO1CA150925 (AT), RO1CA190170 (AT) and RO1CA117907 (JE) and shared resources supported by the University of Colorado Cancer Center P30CA046934. We would like to thank Michal Mokry who kindly provided processed FOXO3a ChIP-seq data.
Footnotes
Authorship contributions
BF and AT conceived the project, designed experiments and wrote the paper. BF performed the experiments. CT, KS, ZA, MH, and ML helped design or carry out experiments. CT performed quantitative analysis of cell death data. JO and KR contributed new reagents. JE and MM contributed to experimental design and interpretation of data. AT oversaw the project. All authors discussed the results and commented on the paper.
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913–1930. doi: 10.1101/gad.287524.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amente S, Zhang J, Lavadera ML, Lania L, Avvedimento EV, Majello B. Myc and PI3K/AKT signaling cooperatively repress FOXO3a-dependent PUMA and GADD45a gene expression. Nucleic Acids Res. 2011;39:9498–9507. doi: 10.1093/nar/gkr638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical Overview of MDM2/X-Targeted Therapies. Front Oncol. 2016;6:7. doi: 10.3389/fonc.2016.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP, et al. Autophagy mediates degradation of nuclear lamina. Nature. 2015;527:105–109. doi: 10.1038/nature15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelenboom A, Mokry M, de Wit E, Smits LM, Polderman PE, van Triest MH, van Boxtel R, Schulze A, de Laat W, Cuppen E, et al. Genome-wide analysis of FOXO3 mediated transcription regulation through RNA polymerase II profiling. Mol Syst Biol. 2013;9:638. doi: 10.1038/msb.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends in cell biology. 2015;25:354–363. doi: 10.1016/j.tcb.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzwalter BE, Thorburn A. Recent insights into cell death and autophagy. The FEBS journal. 2015;282:4279–4288. doi: 10.1111/febs.13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füllgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol. 2014;15:65–74. doi: 10.1038/nrm3716. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DWH, Thorburn A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol. 2014;16:47–54. doi: 10.1038/ncb2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Deo D, Xia M, Vassilev LT. Pharmacologic p53 activation blocks cell cycle progression but fails to induce senescence in epithelial cancer cells. Mol Cancer Res. 2009;7:1497–1509. doi: 10.1158/1541-7786.MCR-09-0144. [DOI] [PubMed] [Google Scholar]
- Huang H, Tindall DJ. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim Biophys Acta. 2011;1813:1961–1964. doi: 10.1016/j.bbamcr.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Komarnitsky P, Cho EJ, Buratowski S. Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 2000;14:2452–2460. doi: 10.1101/gad.824700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nature Reviews Cancer. 2017;17:528–542. doi: 10.1038/nrc.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, Carte J, Chen W, Roark N, Ranganathan S, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol. 2015;208:44–53. doi: 10.1016/j.jbiotec.2015.04.024. [DOI] [PubMed] [Google Scholar]
- O’Prey J, Sakamaki J, Baudot AD, New M, Van Acker T, Tooze SA, Long JS, Ryan KM. Application of CRISPR/Cas9 to Autophagy Research. Methods Enzymol. 2017;588:79–108. doi: 10.1016/bs.mie.2016.09.076. [DOI] [PubMed] [Google Scholar]
- Paris R, Henry RE, Stephens SJ, McBryde M, Espinosa JM. Multiple p53-independent gene silencing mechanisms define the cellular response to p53 activation. Cell Cycle. 2008;7:2427–2433. doi: 10.4161/cc.6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Palaniappan VV, Espinosa JM. ATM regulates cell fate choice upon p53 activation by modulating mitochondrial turnover and ROS levels. Cell Cycle. 2015;14:56–63. doi: 10.4161/15384101.2014.973330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorburn J, Andrysik Z, Staskiewicz L, Gump J, Maycotte P, Oberst A, Green DR, Espinosa JM, Thorburn A. Autophagy controls the kinetics and extent of mitochondrial apoptosis by regulating PUMA levels. Cell Rep. 2014;7:45–52. doi: 10.1016/j.celrep.2014.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, et al. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: implications for therapy. Proc Natl Acad Sci U S A. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers CG, Thorburn A. Therapeutic Targeting of Autophagy. EBioMedicine. 2016;14:15–23. doi: 10.1016/j.ebiom.2016.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegué E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013 doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nature reviews Cancer. 2012;12:401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J Exp Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers, gRNAs, and shRNA target sequences, related to STAR Methods.
ΔFHRE cells (right) lacking the endogenous Forkhead Response Element in BBC3 locus show less caspase 3/7 activation compared to parental cells (left).