

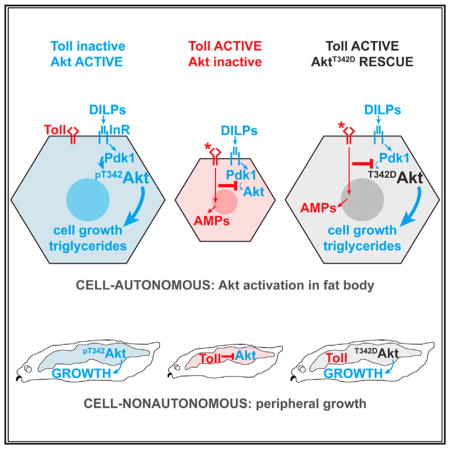
SUMMARY
In obese adipose tissue, Toll-like receptor signaling in macrophages leads to insulin resistance in adipocytes. Similarly, Toll signaling in the Drosophila larval fat body blocks insulin-dependent growth and nutrient storage. We find that Toll acts cell autonomously to block growth but not PI(3,4,5)P3 production in fat body cells expressing constitutively active PI3K. Fat body Toll signaling blocks whole-animal growth in rictor mutants lacking TORC2 activity, but not in larvae lacking Pdk1. Phosphorylation of Akt on the Pdk1 site, Thr342, is significantly reduced by Toll signaling, and expression of mutant AktT342D rescues cell and animal growth, nutrient storage, and viability in animals with active Toll signaling. Altogether, these data show that innate immune signaling blocks insulin signaling at a more distal level than previously appreciated, and they suggest that manipulations affecting the Pdk1 arm of the pathway may have profound effects on insulin sensitivity in inflamed tissues.
In Brief
Innate immune signaling through Toll family receptors induces insulin resistance. Roth et al. find that phosphorylation of Akt by Pdk1 is inhibited by Toll signaling in the Drosophila larval fat body. Toll-dependent impairments in fat body cell growth, nutrient storage, and peripheral growth are rescued by mimicking Akt activation loop phosphorylation.
INTRODUCTION
Throughout the animal kingdom, loss-of-function mutations in insulin signaling molecules such as the kinase Akt disrupt cell, organ, and animal growth and derail glucose homeostasis. In humans, insulin resistance and diabetes are rarely due to single-gene mutations. Rather, obesity is a major risk factor for diabetes, and one characteristic of obese adipose tissue is an increased number of macrophages (Weisberg et al., 2003; Xu et al., 2003). Macrophages can account for up to 40% of cells in obese fat, and most monocytes that infiltrate obese fat differentiate into macrophages that express proinflammatory cytokines. Toll-like receptor 4 (TLR4) signaling in these cells drives secretion of cytokines that interfere with insulin signaling in adipocytes (Lackey and Olefsky, 2016). Deletion of Tlr4 in macrophages and other bone marrow-derived cells or in hepatocytes or adipocytes reduces insulin resistance in high-fat diet-fed mice (Jia et al., 2014; Saberi et al., 2009; Tao et al., 2017), indicating that TLR4 expression is required in immune and metabolic tissues for diet-induced insulin resistance.
How innate immune signaling promotes insulin resistance is unknown. In mammals, inhibition of insulin signaling by cytokine and TLR signaling is associated with serine phosphorylation of insulin receptor substrate (IRS) proteins by kinases such as JNK and IKK (Boura-Halfon and Zick, 2009). Surprisingly, knockin mutations that prevent Irs1 serine phosphorylation lead to impaired rather than enhanced insulin sensitivity. This calls into question the role of these sites in the mechanism of insulin resistance and suggests that more distal steps of the pathway are subject to regulation (Copps et al., 2010, 2016; Hoehn et al., 2008).
The highly conserved insulin and Toll signaling pathways operate in the same cells of the fat body in the model organism Drosophila melanogaster. Insulin signaling in the fat body promotes nutrient storage, fat body cell growth, and whole-animal growth (Arrese and Soulages, 2010; Géminard et al., 2009). In response to infection, Toll signaling in the fat body drives secretion of antimicrobial peptides that kill invading microorganisms (Buchon et al., 2014). In flies, as in mammals, the insulin signaling pathway and innate immune system interact. Infection of adult flies with the bacterial pathogen Mycobacterium marinum leads to progressive wasting and reduced Akt phosphorylation (Dionne et al., 2006). Reduced IRS function in adult chico mutants improves survival in response to infection (Libert et al., 2008). Activation of Toll signaling in the larval fat body, either genetically or by infection, inhibits insulin signaling and whole-animal growth (DiAngelo et al., 2009). Here, we show that Akt phosphorylation by Pdk1 is strongly inhibited in response to Toll pathway activation in the Drosophila larval fat body.
RESULTS
Toll Signaling Blocks Insulin Signaling in a Cell-Autonomous Manner
To identify the point of interaction between fat body Toll and insulin signaling pathways, we carried out genetic epistasis experiments using Akt phosphorylation, cell and animal growth, and triglyceride storage as readouts for insulin signaling. Following insulin receptor (InR) activation, phosphatidylinositol 3-kinase (PI3K) generates PI(3,4,5)P3, which recruits Pdk1, mTORC2, and Akt to the plasma membrane where Pdk1 and mTORC2 phosphorylate Akt on Thr342 and Ser505 (T308 and S473 in mAkt1), respectively. Expression of constitutively active Toll receptors (Toll10b) in larval fat body under control of r4-GAL4 led to reduced Akt S505 phosphorylation in this organ, in agreement with previous findings (Figure 1A) (DiAngelo et al., 2009). Using an antibody that we generated to recognize Akt phosphorylated on T342 (Figure S1A), we found that phosphorylation on this site was also decreased in fat bodies expressing Toll10b, while total Akt levels were unchanged (Figure 1A). Larvae expressing GFP or Toll10b in fat body had equivalent hemolymph levels of Drosophila insulin-like peptide 2 (Dilp2), an InR ligand secreted by insulin-producing cells in the brain (Figure 1B) (Brogiolo et al., 2001), suggesting that Toll signaling induces insulin resistance rather than insulin insufficiency. To test this, we asked whether Toll signaling could disrupt insulin signaling in animals with elevated Dilp2. Transgenic misexpression of Dilp2 in fat body led to increased Akt phosphorylation in whole larvae, and this was blocked when Toll10b was co-expressed (Figure 1C). Similarly, fat body Toll signaling blocked triglyceride storage in larvae misexpressing Dilp2 in fat body (Figure S2A).
Figure 1. Toll Signaling Blocks Insulin Signaling in a Cell-Autonomous Manner.

(A) Western blot analysis of phosphorylated (pT342 and pS505) and total Akt in fat bodies, n = 4/group (gp).
(B) Hemolymph Dilp2 levels in mid-third-instar larvae, n = 4/gp.
(C) Western blot analysis of pT342, pS505, and total Akt in whole larvae, n = 3–4/gp.
(D) Mosaic analysis using the FLP; Act5c > Cd2 > GAL4 system.
(E–J) (E), (G), and (I): micrographs of fat bodies containing WT, Cd2+, GAL4− cells (red) and transgene-expressing, Cd2−, GAL4+ cells (arrowheads). Nuclei are stained with DAPI (blue). Scale bar, 50 μm. (F), (H), and (J): nuclear area in Cd2+, GAL4− (red), and Cd2−, GAL4+ (gray) cells. ****p < 0.0001 versus Cd2+. UAS-transgenes expressed in Cd2−, GAL4+ cells are as follows: (E and F) left, no transgene (Cd2+, n = 169; Cd2−, n = 36), and right, Toll10b (Cd2+, n = 368; Cd2−, n = 108); (G and H) left, InRA1325D (Cd2+, n = 56; Cd2−, n = 22), and right, InRA1325D + Toll10b (Cd2+, n = 246; Cd2−, n = 45); (I and J) left, myrAkt (Cd2+, n = 100; Cd2−, n = 57), and right, myrAkt + Toll10b (Cd2+, n = 176; Cd2−, n = 81). Data are presented as mean ± SD. See also Table S1 and Figures S1 and S2.
Because fat body Toll signaling blocked Akt activation in animals with normal or elevated Dilp2 levels, we asked whether the Toll pathway acted cell autonomously to induce insulin resistance. We generated mosaic fat bodies containing clones of cells expressing Toll10b alone or with transgenes encoding active insulin signaling pathway components (Figure 1D). Endoreplication in fat body cells leads to tandem increases in nuclear and cell size; we therefore measured nuclear area as a proxy for cell growth (Britton et al., 2002). Nuclear area was equivalent in GAL4+, Cd2− cells lacking UAS transgenes and their wild-type (WT) GAL4−, Cd2+ neighbors. In contrast, GAL4+, Cd2-cells expressing Toll10b were small with significantly smaller nuclei than WT cells (Figures 1E and 1F). Expression of constitutively active InRA1325D drove an increase in nuclear area, and this was blocked by co-expression of Toll10b (Figures 1G and 1H). We noted that Toll signaling reduced whole-cell area in cells expressing InRA1325D, perhaps by blocking nutrient storage. Indeed, triglyceride levels were reduced when Toll10b was expressed alone or with InRA1325D (Figure S2B).
Expression of Toll10b throughout the fat body impairs whole-animal growth, and this is rescued by co-expression of myristoylated Akt (myrAkt), a constitutively active Akt transgene (DiAngelo et al., 2009). Fat body cells expressing myrAkt were large with large nuclei. In contrast to results with InRA1325D, cells co-expressing Toll10b and myrAkt had significantly larger nuclei than their WT neighbors (Figures 1I and 1J). Expression of myr-Akt also rescued triglyceride storage in larvae with active Toll signaling (Figure S2C). Together, these data show that Toll acts cell autonomously to block cell growth and triglyceride storage downstream of InR and upstream of Akt.
Toll Acts Independently of TORC2 to Block Insulin Signaling
Clonal expression of Dp110CAAX, a constitutively active PI3K catalytic subunit, produced extremely large cells with massive nuclei. Toll10b co-expression blocked growth driven by Dp110CAAX (Figure 2A). We saw robust recruitment of the PI(3,4,5)P3 reporter tGPH (Britton et al., 2002) to the plasma membrane of cells expressing Dp110CAAX alone or with Toll10b, suggesting that the Toll pathway acts downstream of PI(3,4,5)P3 to block insulin signaling (Figure 2B). Similarly, PI3K activation led to elevated Akt phospho-S505 signal in cells expressing Dp110CAAX alone or with Toll10b compared with surrounding WT cells (Figure 2C).
Figure 2. Toll Signaling Acts Independently of TORC2 to Block Insulin Signaling.

(A–C) Micrographs of fat bodies containing Cd2+ WT cells (red, nuclear area, left: 240.0 ± 80.2 μm2, n = 101; right: 245.6 ± 99.0 μm2, n = 118) and Cd2−, GAL4+ cells (arrowheads) expressing Dp110CAAX (left, nuclear area: 648.7 ± 193.0 μm2, n = 31, p < 0.0001 versus Cd2+) or Dp110CAAX + Toll10b (right, nuclear area: 334.3 ± 119.5 μm2, n = 33, p < 0.0001 versus Cd2+). Nuclei are stained with DAPI (blue). Scale bars, 50 μm. (A) Immunocytochemistry for Cd2 (red). (B) Plasma membrane localization of the ubiquitously expressed PI(3,4,5)P3 reporter tGPH (green) in cells expressing Dp110CAAX alone or with Toll10b. (C) Immunocytochemistry for Akt pS505 (green).
(D–G) r4-GAL4 was used to drive GFP or Toll10b in fat bodies of larvae heterozygous or hemizygous for rictorΔ2. (D) Western blot analysis of Akt (pS505 and total) in whole-larval lysates, n = 2–4/gp. (E) Whole-animal triglycerides, n = 8/gp. *p = 0.0326 and **p = 0.0011 versus GFP. (F) Images of pupae. Scale bar, 2 mm. (G) Pupal body weights, n = 14–16/gp except rictorΔ2/Y; r4-GAL4 > Toll10b, n = 6. **p = 0.0020 and ****p < 0.0001 versus GFP. Data are presented as mean ± SD. See also Table S1.
We formally tested whether mTORC2 mediates the ability of the Toll pathway to inhibit insulin signaling by asking whether Toll10b could reduce growth and nutrient storage in larvae lacking the essential mTORC2 component rictor. In Drosophila, null mutations in rictor abolish mTORC2 activity, block Akt S505 phosphorylation, and lead to a minor whole-animal growth defect (Hietakangas and Cohen, 2007; Lee and Chung, 2007). Akt S505 phosphorylation was undetectable in lysates of larvae hemizygous for the null allele rictorΔ2 (Figure 2D). Nonetheless, fat body expression of Toll10b reduced triglyceride storage (Figure 2E) and whole-animal growth (Figures 2F and 2G) in rictorΔ2 heterozygotes and hemizygotes.
Toll Signaling Acts at or Downstream of Pdk1 to Block Growth
Larvae homozygous for the strong hypomorphic Pdk1Δ33 allele (Cheng et al., 2011) lack detectable phosphorylation of Akt T342 (Figure S1B) and weigh 60% less than heterozygotes. Expression of Toll10b in fat body reduced growth in Pdk1Δ33 heterozygotes, but not homozygotes (Figures 3A and 3B). Fat bodies expressing Toll10b had elevated Pdk1 mRNA levels compared with controls, a finding that was echoed for other insulin signaling intermediates (Figure S3). Toll10b expression had no effect on endogenous Pdk1 protein levels in fat body (Figure 3C), as measured using a Pdk1 allele with a C-terminal myc tag, inserted using CRISPR/Cas9 (Figure S4).
Figure 3. Toll Signaling Acts at or Downstream of Pdk1 to Block Growth.

(A) Images of Pdk1Δ33 mutant pupae expressing GFP or Toll10b in fat body. Scale bar, 2 mm.
(B) Pupal body weights, n = 19–22/gp for Pdk1Δ33/+, n = 6–10/gp for Pdk1Δ33/Pdk1Δ33. ***p = 0.0002 versus GFP.
(C) Western blot analysis of endogenous Pdk1 with a C-terminal myc tag (Pdk1myc) in fat bodies expressing GFP or Toll10b, n = 2/gp.
(D) Micrographs of fat bodies containing Cd2−, GAL4+ cells (arrowheads) expressing Pdk1 (left) or Pdk1 + Toll10b (right). WT cells are Cd2+ (red); nuclei are stained with DAPI (blue). Scale bar, 50 μm.
(E) Nuclear area in Cd2+, GAL4− cells (red, Pdk1, n = 262; Pdk1+Toll10b, n = 379) and Cd2−, GAL4+ cells (gray, Pdk1, n = 50; Pdk1+Toll10b, n = 55). ****p < 0.0001 versus Cd2+.
(F) Images of pupae. Scale bar, 2 mm.
(G) Pupal body weights, n = 14–15/gp. ****p = 0.0001 versus RFP + GFP.
(H) Western blot analysis of pT342, pS505, and total Akt, transgenically expressed Pdk1 (myc-tagged) and Histone H3 (HisH3) in fat bodies, n = 3–4/gp. Data are presented as mean ± SD. See also Table S1 and Figures S1, S3, and S4.
The resistance of Pdk1Δ33 homozygotes to the growth-reducing effects of fat body Toll signaling suggested that the Toll pathway targets Pdk1 to block insulin signaling. To test this hypothesis, we asked whether elevated Pdk1 expression could rescue Toll10b-dependent phenotypes. GAL4-driven expression of Pdk1 in fat body clones led to increased cell and nuclear size. However, as with the InRA1325D and Dp110CAAX transgenes, this growth advantage was completely suppressed by co-expression of Toll10b (Figures 3D and 3E). Animals expressing Toll10b throughout the fat body, with or without Pdk1 co-expression, were smaller than controls (Figures 3F and 3G). Forced expression of Pdk1 drove Akt T342 phosphorylation relative to control fat bodies with no effect on phospho-S505 or total Akt levels. Toll signaling considerably reduced Pdk1-driven T342 phosphorylation without affecting Pdk1 trans-gene expression (Figure 3H).
Mimicking Akt Thr342 Phosphorylation Rescues Toll-Dependent Phenotypes
Toll signaling does not further reduce growth in Pdk1 mutants, and yet elevated Pdk1 is insufficient to rescue growth in animals expressing Toll10b in fat body. These results suggest that Toll signaling may inhibit Pdk1 activity or activate an Akt T342 phosphatase. Consequently, we asked whether expression of a phospho-mimicking T342D Akt mutant would rescue Toll10b-dependent growth and nutrient storage phenotypes. Unlike InRA1325D, Dp110CAAX, Pdk1, and myrAkt, AktT342D did not increase nuclear area when expressed in fat body cells. However, AktT342D expression substantially rescued growth in fat body cells co-expressing Toll10b (Figures 4A and 4B). Co-expression of Toll10b with AktT342D, but not with WT Akt, rescued impaired triglyceride storage caused by innate immune signaling (Figures 4C and 4D). Expression of the AktT342D trans-gene completely rescued the whole-animal growth defect caused by fat body Toll signaling (Figures 4E and 4F). The majority of flies that express Toll10b in fat body die just before eclosion. This reduced viability can be rescued by loss of the Toll pathway transcription factor Dif or by expression of AktT342D, but not by transgenes encoding WT, S505D, T342A, or S505A Akt (Figure 4G). Together, these data suggest that the reduction in insulin signaling, growth, and nutrient storage in cells with active Toll signaling is due to inhibition of Akt T342 phosphorylation.
Figure 4. Mimicking Akt Thr342 Phosphorylation Rescues Toll-Dependent Phenotypes.

(A) Micrographs of fat bodies containing Cd2−, GAL4+ cells (arrowheads) expressing AktT342D (left) or AktT342D + Toll10b (right). WT cells are Cd2+ (red); nuclei are stained with DAPI (blue). Scale bar, 50 μm.
(B) Nuclear area in Cd2+, GAL4− cells (red, AktT342D, n = 138; AktT342D+Toll10b, n = 260), and Cd2−, GAL4+ cells (gray, AktT342D, n = 42;AktT342D+Toll10b, n = 40). ****p < 0.0001 versus Cd2+.
(C and D) Whole-animal triglycerides in animals expressing Toll10b in fat body with AktT342D (C) or WT Akt (D), n = 11–13/gp. ***p = 0.0005 and ****p < 0.0001 versus RFP + GFP.
(E) Images of pupae. Scale bar, 2 mm.
(F) Pupal body weights, n = 9–14/gp. ***p = 0.0001 versus RFP + GFP.
(G) Percent recovery of viable adult r4-GAL4, UAS-Toll10b flies co-expressing indicated trans-genes in fat body, n = 78–146 adults/cross. Data in (B), (C), (D), and (F) are presented as mean ± SD. See also Table S1.
(H) Model for inhibition of T342 phosphorylation by Toll signaling.
DISCUSSION
Using a genetic approach, we show that Toll signaling acts in a cell-autonomous manner to inhibit insulin signaling downstream of IRS/chico and PI3K. Constitutive PI3K activity drives PI(3,4,5)P3 accumulation and mTORC2 activity in cells expressing Toll10b, but these two signaling events are uncoupled from cell growth when the Toll pathway is active. Our data indicate that Akt phosphorylation on the Pdk1 site, T342, is the critical step regulated by Toll signaling. Our data fit the consensus in the literature that phosphorylation on the activation loop (T342) rather than on the hydrophobic motif (S505) is the more critical step for insulin signaling. First, we find that elevated Pdk1 expression robustly drives cell growth and T342 phosphorylation without affecting S505 phosphorylation or total Akt levels. Second, loss of rictor leads to minor growth defects compared with loss of Pdk1 (this study; Cheng et al., 2011; Hietakangas and Cohen, 2007; Lee and Chung, 2007). Third, loss of mTORC2 components in mice blocks mAkt S473 phosphorylation, but this affects only a subset of Akt targets (Guertin et al., 2006; Jacinto et al., 2006; Kumar et al., 2010). Finally, mAkt T308 phosphorylation promotes full Akt activity via activation of mTORC2 and subsequent S473 phosphorylation (Yang et al., 2015).
Toll signaling does not reduce Pdk1 or Akt levels, and future work will be required to determine whether regulation of T342 phosphorylation occurs through impaired interaction between Pdk1 and Akt, reduced Pdk1 activity, or increased phosphatase activity toward T342 (Figure 4H). Mammalian Akt is subject to post-translational modifications that regulate translocation to the plasma membrane and, consequently, interaction with Pdk1 (Risso et al., 2015). Pdk1 activity is regulated by subcellular localization, activating tyrosine phosphorylation, and auto-phosphorylation that reduces homodimerization and increases activity toward Akt (Calleja et al., 2014). The phosphatases PP2A and Phlpp dephosphorylate mAkt1 (Gao et al., 2005; Kuo et al., 2008), but knockdown of Phlpp fails to rescue growth in fat body cells expressing Toll10b (data not shown).
Elevated Pdk1 expression drives Akt T342 phosphorylation, and Toll signaling impairs this, albeit incompletely. Nonetheless, fat body Toll signaling blocks growth in animals co-expressing the Pdk1 transgene, but not in those co-expressing AktT342D. Several possibilities could account for these results. First, there may be discordance between experimentally observable Akt phosphorylation and the activity of its growth-promoting substrates such that maximal Akt activity, achieved in the absence of Toll signaling, is needed for InRA1325D, Dp110CAAX, and Pdk1 transgenes to drive growth. Indeed, our results with these trans-genes indicate a considerable reserve capacity for Akt activity and cell growth in vivo. Second, the Toll and insulin signaling pathways may operate in parallel to regulate a downstream target critical for growth, and the AktT342D transgene may more efficiently correct downstream signaling compared with the Pdk1 transgene. Third, fat bodies that co-express Pdk1 and Toll10b might maintain relatively elevated Akt T342 phosphorylation because of the kinetics of induction of negative regulators of Pdk1 and/or T342 phosphorylation. Future work will be required to distinguish among these possibilities.
Toll signaling in the fat body acts not only cell autonomously to disrupt insulin signaling, but also cell nonautonomously to reduce whole-animal growth. Hemolymph Dilp2 levels are not changed by Toll signaling, suggesting that reduced whole-animal growth caused by fat body Toll signaling may be due to peripheral insulin resistance and not impaired Dilp secretion from insulin-producing cells. Similarly, in mice, loss of insulin signaling in adipose tissue leads to insulin resistance in liver and muscle (Abel et al., 2001; Kumar et al., 2010; Shearin et al., 2016; Softic et al., 2016). A major question arising from these studies and ours is the following: what mechanisms communicate a reduction in insulin signaling in one organ to the rest of the animal? Genetic screens in model organisms such as Drosophila may pave the way for addressing this outstanding matter.
In conclusion, our findings demonstrate that Toll signaling disrupts insulin signaling downstream of IRS, PI3K, and Pdk1 by interfering with Akt phosphorylation on its activation loop site. Our results echo findings in mammalian cells that many treatments that provoke insulin resistance do so independently of IRS and through a variety of mechanisms (Hoehn et al., 2008). Furthermore, our results define the point in the insulin signaling pathway that is blocked by Toll signaling and provide a genetic model with robust phenotypes that can be used to identify the cell-autonomous and cell-nonautonomous mechanisms employed by the Toll pathway to disrupt insulin signaling.
EXPERIMENTAL PROCEDURES
Drosophila Stocks and Husbandry
Flies were raised on standard food at 25°C, and 6- to 8-hr egg lays were conducted to prevent overcrowding. Experiments were performed using mid-third-instar larvae (96–108 hr after egg lay) or white prepupae (0–5 hr after puparium formation). Stocks used in this study, the UAS-AktT342D and UAS-Pdk1 transgenes, and the CRISPR/Cas9 strategy used to generate a myc-tagged Pdk1 allele are described in Supplemental Experimental Procedures. Full genotypes are listed in Table S1.
Antibodies and Western Blotting
Sources and dilutions of primary and secondary antibodies used in this study are listed in Supplemental Experimental Procedures. The rabbit anti-phosphoThr342-Akt antibody was generated against the phosphopeptide YGRTTK(Tp)FCGTPE. Peptide synthesis, immunization, and antibody purification were performed at Alpha Diagnostics International (San Antonio, TX). Western blotting was performed using larval or fat body lysates as described in Supplemental Experimental Procedures.
Mosaic Analysis of Cell Size
Act5c > Cd2 > GAL4 flies were crossed to hsFLP flies carrying UAS transgenes. After overnight egg lays, embryos were heat shocked (37°C, 45 min) to induce FLP recombinase. Larval fat bodies were subjected to immunostaining for Cd2 as described in Supplemental Experimental Procedures. Z-stack images (2 μm interval) were collected on a Perkin Elmer UltraVIEW High Speed Confocal Imaging microscope with Volocity software or a Zeiss LSM 700 confocal microscope with ZEN imaging software. Maximum intensity projection images were colorized in Adobe Photoshop. For analysis of nuclear area, images of DAPI-stained nuclei were imported into ImageJ and subjected to thresholding, and nuclear area was calculated in Cd2+ and Cd2− cells.
Dilp2, Triglyceride, and Animal Weight Measurements
Hemolymph was collected on ice from 10 to 12 larvae, and 1 μL of pooled hemolymph was used for Dilp2 measurement by ELISA (Park et al., 2014). Triglycerides were measured as described (Bland et al., 2010). Pupae were weighed in groups of 4–6 following removal of food from the cuticle.
Statistics
Statistical analyses were performed using GraphPad Prism 7. Sample sizes (referring to biological replicates of cells or animals) are listed in the figure legends. Unpaired two-tailed t tests were used to assess differences in nuclear area between Cd2+, GAL4−, and Cd2−, GAL4+ cells and between rictorΔ2 and Pdk1Δ33 heterozygotes or homo/hemizygotes expressing GFP or Toll10b in fat body. For other triglyceride and body weight experiments, one-way ANOVA was used to determine differences between groups.
Supplementary Material
Highlights.
Toll acts cell autonomously to induce insulin resistance in the Drosophila fat body
Fat body Toll signaling reduces growth and triglyceride storage without altering circulating Dilp2 levels
Toll signaling acts at or downstream of the kinase Pdk1 to reduce Akt phosphorylation
Mimicking Akt activation loop phosphorylation rescues Toll-dependent phenotypes
Acknowledgments
We thank Tony Ip (University of Massachusetts, Worcester) and Graeme Davis (UCSF) for fly stocks, Mazhar Adli (University of Virginia) for advice on the CRISPR/Cas9 strategy, Young Jun Lee and Miyuki Suzawa for technical assistance, members of the Bland lab for discussions, and the Bloomington Drosophila Stock Center and the Drosophila Genomics Research Center for reagents. This work was supported by NIH Grants R21DK089391 to M.J.B. and R01DK099601 to M.L.B.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.033.
DECLARATION OF INTERESTS
M.J.B. is a full-time employee of Pfizer, Inc.
AUTHOR CONTRIBUTIONS
Conceptualization, M.J.B. and M.L.B.; Methodology, M.L.B.; Investigation, S.W.R., M.D.B., and M.L.B.; Writing, M.L.B.; Funding Acquisition, M.J.B. and M.L.B.
References
- Abel ED, Peroni O, Kim JK, Kim YB, Boss O, Hadro E, Minnemann T, Shulman GI, Kahn BB. Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature. 2001;409:729–733. doi: 10.1038/35055575. [DOI] [PubMed] [Google Scholar]
- Arrese EL, Soulages JL. Insect fat body: energy, metabolism, and regulation. Annu Rev Entomol. 2010;55:207–225. doi: 10.1146/annurev-ento-112408-085356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bland ML, Lee RJ, Magallanes JM, Foskett JK, Birnbaum MJ. AMPK supports growth in Drosophila by regulating muscle activity and nutrient uptake in the gut. Dev Biol. 2010;344:293–303. doi: 10.1016/j.ydbio.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boura-Halfon S, Zick Y. Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab. 2009;296:E581–E591. doi: 10.1152/ajpendo.90437.2008. [DOI] [PubMed] [Google Scholar]
- Britton JS, Lockwood WK, Li L, Cohen SM, Edgar BA. Drosophila’s insulin/PI3-kinase pathway coordinates cellular metabolism with nutritional conditions. Dev Cell. 2002;2:239–249. doi: 10.1016/s1534-5807(02)00117-x. [DOI] [PubMed] [Google Scholar]
- Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr Biol. 2001;11:213–221. doi: 10.1016/s0960-9822(01)00068-9. [DOI] [PubMed] [Google Scholar]
- Buchon N, Silverman N, Cherry S. Immunity in Drosophila melanogaster—from microbial recognition to whole-organism physiology. Nat Rev Immunol. 2014;14:796–810. doi: 10.1038/nri3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calleja V, Laguerre M, de Las Heras-Martinez G, Parker PJ, Requejo-Isidro J, Larijani B. Acute regulation of PDK1 by a complex interplay of molecular switches. Biochem Soc Trans. 2014;42:1435–1440. doi: 10.1042/BST20140222. [DOI] [PubMed] [Google Scholar]
- Cheng L, Locke C, Davis GW. S6 kinase localizes to the pre-synaptic active zone and functions with PDK1 to control synapse development. J Cell Biol. 2011;194:921–935. doi: 10.1083/jcb.201101042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copps KD, Hançer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab. 2010;11:84–92. doi: 10.1016/j.cmet.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copps KD, Hançer NJ, Qiu W, White MF. Serine 302 phosphorylation of mouse insulin receptor substrate 1 (IRS1) is dispensable for normal insulin signaling and feedback regulation by hepatic S6 kinase. J Biol Chem. 2016;291:8602–8617. doi: 10.1074/jbc.M116.714915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAngelo JR, Bland ML, Bambina S, Cherry S, Birnbaum MJ. The immune response attenuates growth and nutrient storage in Drosophila by reducing insulin signaling. Proc Natl Acad Sci USA. 2009;106:20853–20858. doi: 10.1073/pnas.0906749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionne MS, Pham LN, Shirasu-Hiza M, Schneider DS. Akt and FOXO dysregulation contribute to infection-induced wasting in Drosophila. Curr Biol. 2006;16:1977–1985. doi: 10.1016/j.cub.2006.08.052. [DOI] [PubMed] [Google Scholar]
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18:13–24. doi: 10.1016/j.molcel.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Géminard C, Rulifson EJ, Léopold P. Remote control of insulin secretion by fat cells in Drosophila. Cell Metab. 2009;10:199–207. doi: 10.1016/j.cmet.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Hietakangas V, Cohen SM. Re-evaluating AKT regulation: role of TOR complex 2 in tissue growth. Genes Dev. 2007;21:632–637. doi: 10.1101/gad.416307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoehn KL, Hohnen-Behrens C, Cederberg A, Wu LE, Turner N, Yuasa T, Ebina Y, James DE. IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 2008;7:421–433. doi: 10.1016/j.cmet.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Jia L, Vianna CR, Fukuda M, Berglund ED, Liu C, Tao C, Sun K, Liu T, Harper MJ, Lee CE, et al. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat Commun. 2014;5:3878. doi: 10.1038/ncomms4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Lawrence JC, Jr, Jung DY, Ko HJ, Keller SR, Kim JK, Magnuson MA, Harris TE. Fat cell-specific ablation of rictor in mice impairs insulin-regulated fat cell and whole-body glucose and lipid metabolism. Diabetes. 2010;59:1397–1406. doi: 10.2337/db09-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem. 2008;283:1882–1892. doi: 10.1074/jbc.M709585200. [DOI] [PubMed] [Google Scholar]
- Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol. 2016;12:15–28. doi: 10.1038/nrendo.2015.189. [DOI] [PubMed] [Google Scholar]
- Lee G, Chung J. Discrete functions of rictor and raptor in cell growth regulation in Drosophila. Biochem Biophys Res Commun. 2007;357:1154–1159. doi: 10.1016/j.bbrc.2007.04.086. [DOI] [PubMed] [Google Scholar]
- Libert S, Chao Y, Zwiener J, Pletcher SD. Realized immune response is enhanced in long-lived puc and chico mutants but is unaffected by dietary restriction. Mol Immunol. 2008;45:810–817. doi: 10.1016/j.molimm.2007.06.353. [DOI] [PubMed] [Google Scholar]
- Park S, Alfa RW, Topper SM, Kim GES, Kockel L, Kim SK. A genetic strategy to measure circulating Drosophila insulin reveals genes regulating insulin production and secretion. PLoS Genet. 2014;10:e1004555. doi: 10.1371/journal.pgen.1004555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso G, Blaustein M, Pozzi B, Mammi P, Srebrow A. Akt/PKB: one kinase, many modifications. Biochem J. 2015;468:203–214. doi: 10.1042/BJ20150041. [DOI] [PubMed] [Google Scholar]
- Saberi M, Woods NB, de Luca C, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM. Hematopoietic cell-specific deletion of Toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009;10:419–429. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearin AL, Monks BR, Seale P, Birnbaum MJ. Lack of AKT in adipocytes causes severe lipodystrophy. Mol Metab. 2016;5:472–479. doi: 10.1016/j.molmet.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic S, Boucher J, Solheim MH, Fujisaka S, Haering MF, Homan EP, Winnay J, Perez-Atayde AR, Kahn CR. Lipodystrophy due to adipose tissue-specific insulin receptor knockout results in progressive NAFLD. Diabetes. 2016;65:2187–2200. doi: 10.2337/db16-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao C, Holland WL, Wang QA, Shao M, Jia L, Sun K, Lin X, Kuo YC, Johnson JA, Gordillo R, et al. Short-term versus long-term effects of adipocyte Toll-like receptor 4 activation on insulin resistance in male mice. Endocrinology. 2017;158:1260–1270. doi: 10.1210/en.2017-00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Murashige DS, Humphrey SJ, James DE. A positive feedback loop between Akt and mTORC2 via SIN1 phosphorylation. Cell Rep. 2015;12:937–943. doi: 10.1016/j.celrep.2015.07.016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.