

The first commercially viable hydrogen/air performance for a nonprecious metal catalyst–based PEMFC is demonstrated.
Abstract
Despite great progress in the development of nonprecious metal catalysts (NPMCs) over the past several decades, the performance and stability of these promising catalysts have not yet achieved commercial readiness for proton exchange membrane fuel cells (PEMFCs). Through rational design of the cathode catalyst layer (CCL), we demonstrate the highest reported performance for an NPMC-based membrane electrode assembly (MEA), achieving a peak power of 570 mW/cm2 under air. This record performance is achieved using a precommercial catalyst for which nearly all pores are <3 nm in diameter, challenging previous beliefs regarding the need for larger catalyst pores to achieve high current densities. This advance is achieved at industrially relevant scales (50 cm2 MEA) using a precommercial NPMC. In situ electrochemical analysis of the CCLs is also used to help gain insight into the degradation mechanism observed during galvanostatic testing. Overall, the performance of this NPMC-based MEA has achieved commercial readiness and will be introduced into an NPMC-based product for portable power applications.
INTRODUCTION
As part of a global effort to reduce our dependency on fossil fuels, many renewable energy sources are being actively developed (for example, wind and solar). However, it is widely recognized that these energy sources are highly intermittent, and thus, the ability to efficiently store energy will become critical in the coming years. Hydrogen represents one of the most promising forms of energy storage, because it is easier to store compared to electrons and can be generated from nearly any energy source through electrolysis of water. One of the most efficient methods of extracting the stored energy from hydrogen is through the use of proton exchange membrane fuel cells (PEMFCs), making it a key technology as society transitions toward cleaner forms of energy. Unfortunately, the electrodes in PEMFCs currently require significant amounts of platinum group metal (PGM) catalysts to drive the desired electrochemical reactions. This is particularly true at the cathode, which contains nearly 80% of the total PGM in the fuel cell due to the sluggish oxygen reduction reaction (ORR) (1).
Thus, it is necessary to reduce/replace PGM catalysts to make PEMFCs cost-competitive with conventional technologies (for example, internal combustion engines and batteries). This must be achieved while maintaining acceptable performance, stability, and durability. Although significant advances in PGM electrocatalysts have provided a clear pathway to achieving lower PGM loadings (2–4), recent advances in nonprecious metal catalysts (NPMCs) have given hope for achieving the “holy grail” of PEMFC cathode electrocatalysis: PGM-free catalyst layers. Since their original discovery in 1964 (5), the activity/performance of metal/nitrogen/carbon (M/N/C) NPMCs has been greatly improved (6–11). However, a review of recent literature demonstrates that meeting real-world commercial requirements remains a significant challenge for these materials. Specifically, although power densities of >0.9 W/cm2 have been reported under O2 (8, 11–13), values under air are rarely reported, and when they are, a far more modest value of 0.2 to 0.4 W/cm2 is achieved (13–16).
Here, we incorporate a precommercial NPMC catalyst into high-performing catalyst layers through optimization of ionomer and catalyst loading. Although higher catalyst loadings were found to lead to (expected) transport losses, through drawing on knowledge gained in previous work by our group (17), we are able to optimize the ionomer content of the higher catalyst loading design to achieve the highest power ever reported under air/H2 in a large-scale (50 cm2) NPMC-based membrane electrode assembly (MEA) (570 mW/cm2). These results represent an important step in the commercialization of NPMCs, because the reported performance is now sufficient to meet beginning-of-life requirements for certain (for example, backup power/portable power) applications. Finally, a stability test is performed to help uncover the most likely mechanism for degradation. Through this work, we identify carbon oxidation (attack by H2O2) as being a primary mechanism for the observed performance loss during galvanostatic experiments.
RESULTS AND DISCUSSION
Catalyst loadings of 1.0, 2.5, and 4.0 mg/cm2 were coated onto a gas diffusion layer (GDL) to form a gas diffusion electrode (GDE). Before performing polarization analysis, cyclic voltammetry was used to verify that the expected loadings were achieved (fig. S1 and table S1). Polarization analysis was performed on all three designs, under both air and O2 (Fig. 1, A and B). Under O2, the trend in kinetic overpotentials was 1.0 > 2.5 > 4.0 mg/cm2 (Fig. 1C). This trend was fully expected, because reducing the catalyst loading will decrease the number of available active sites, leading to increased overpotential.
Fig. 1. Polarization analysis obtained under air and oxygen for MEAs prepared from the three different catalyst loadings.
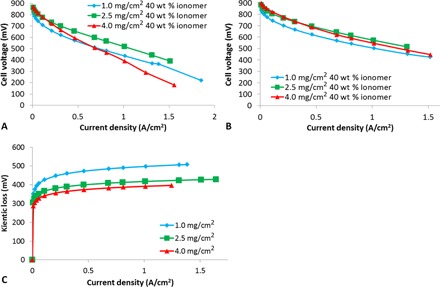
Polarization curves obtained under (A) air and (B) O2 at 100% relative humidity (RH) (anode/cathode). (C) Kinetic overpotential (assuming Butler-Volmer kinetics) for the three different catalyst loadings.
At higher current densities, the 1.0-mg/cm2 design does appear to show lower mass transport effects than the other designs in Fig. 1. However, despite this advantage in mass transport, the very low initial kinetic performance of this design renders it inferior to the 2.5-mg/cm2 design over all current densities. Also as expected, the 4.0-mg/cm2 design shows worse mass transport than the lower loaded designs (although the degree of the difference is perhaps larger than expected), and by moderate current densities of 0.6 A/cm2, it is already the worst-performing design when tested under air.
To help understand the source of the mass transport limitations (proton and/or O2 transport), we measured the difference in performance under air versus O2 at 1 A/cm2 for each of the three designs. It is clear from this analysis (fig. S2) that the 4.0-mg/cm2 design [the thickest of the three cathode catalyst layers (CCLs)] shows the largest dependence on O2 concentration. Thus, it is reasonable to assume that higher performance could be achieved by increasing the porosity of this layer to facilitate O2 transport. One way to achieve this would be to decrease the ionomer content in the CCL (18, 19). However, doing so would result in a concomitant decrease in protonic conductivity, eventually leading to proton transport limitations. Fortunately, we previously developed an impedance method to determine the critical CCL conductance for NPMC-based CCLs (17). As shown in that work, provided the CCL conductance is beyond a critical value (100 S), protonic transport within the CCL is not a dominant mechanism of performance loss. In addition, we demonstrated that the CCL conductance varied linearly with ionomer loading, at least within a loading range of 30 to 40 weight % (wt %) (17). With this in mind, we decreased the ionomer content of the 4.0-mg/cm2 design from 40 to 35 wt %, because this would still put the CCL conductance above the critical value of 100 S (fig. S3) but should simultaneously increase CCL porosity. As expected, this did result in a clear performance gain, with peak power and resulting performance for the two designs shown in Table 1 and Fig. 2 (A and B).
Table 1. Peak power for the 35 and 40 wt % ionomer designs under air or O2 for the 4.0-mg/cm2 CA#1 loaded CCLs.
| Oxidant gas | 35 wt % ionomer | 40 wt % ionomer |
| Air | 570 mW/cm2 | 395 mW/cm2 |
| O2 | 750 mW/cm2 | 678 mW/cm2 |
Fig. 2. Polarization and performance curves obtained under air or O2.
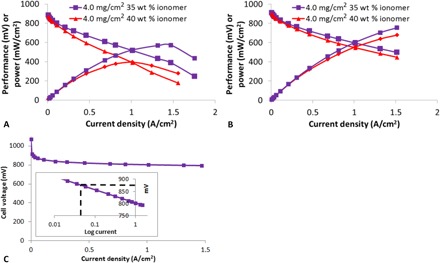
Performance obtained under (A) air and (B) O2 for the 4.0-mg/cm2 CCL design with 35 or 40 wt % ionomer. (C) Performance (corrected for ohmic losses) obtained under O2 (inset shows performance achieved at 0.044 A/cm2).
To the best of our knowledge, the results for the 35 wt % ionomer design (Table 1) represents the highest ever reported H2/air power density for an NPMC-based CCL at this current density (certainly for a 50-cm2 MEA).
Presently, the U.S. Department of Energy 2020 technical target for performance from an NPMC-based CCL is 900 mV at 0.044 A/cm2 under O2 (20). As shown in Fig. 2C, the 35 wt % ionomer design at 4.0 mg/cm2 is already achieving ~880 mV at 0.044 A/cm2. This is a highly promising result and is on par with the current state of the art (6, 11, 21, 22). This is also true for the H2/O2 performance data, which at 750 mW/cm2 is also comparable to the state of the art. However, it is evident from Table 1 that despite only matching the state of the art for activity under O2, the performance under air is the highest ever reported. The fact that such high performance under air was achieved using a catalyst that does not greatly exceed the state of the art demonstrates the greatly improved transport characteristics of the CCL design described here versus previously reported designs, and also highlights that other researchers may be able to leverage the CCL design strategies described here to achieve similar performance with their own catalysts. These results are now high enough to meet the beginning-of-life requirements for some portable power/backup power applications (23).
It is worth highlighting that this high H2/air performance (current densities of >1.5 A/cm2) was achieved using a catalyst that is >90% microporous based on t-plot analysis (table S3) (nearly 100% of pores are <3 nm; fig. S5). Because it is highly improbable that the ionomer is able to penetrate into these pores [based on the size of hydrophilic domains of the ionomer (24)], this has important implications for the understanding of proton transport to/within the catalyst. Many researchers in this field have been specifically focusing on increasing the mesoporosity of their catalysts (8, 10, 25). Our results indicate that through rational design of the catalyst layer, extremely high performance can actually be achieved with a catalyst that is largely microporous. Although a full discussion of this falls beyond the scope of the present work, one possibility could be rapid transport through water to active sites sufficiently close to the ionomer (26). This has previously been suggested by Herranz et al. (9) and Zhang et al. (21) and is supported by our current findings.
It has been widely reported that many NPMCs suffer from poor stability (performance loss during potentiostatic or galvanostatic experiments) (7, 8, 27), with the exact mechanism for this loss still a matter of investigation (21, 28–30). The following are the primary hypotheses to explain this instability: (i) demetalation of the NPMC (30–34); (ii) attack by H2O2 (35, 36) (and/or free radicals) (37); (iii) protonation of the active site (38) or protonation of N species neighboring the active site, followed by anion adsorption (9); and (iv) micropore flooding (12, 21, 39, 40). Considering these possible mechanisms, it is instructive to predict how each of them might be affected by a change in catalyst loading.
Mechanism 1 (demetalation) is likely not to be a significant contributor to current generations of NPMCs. This is because most NPMCs (including CA#1 used in this study) are now acid-washed before incorporating into the CCL, which should remove most of the leachable iron (41–43). In addition, recent work has convincingly demonstrated that although demetalation likely does occur, it has no correlation with performance/stability loss within the first several hours (where most of the instability is observed) (30, 34, 44). Although this hypothesis cannot be completely ruled out, it appears less likely than the three remaining hypotheses.
Mechanism 2 (attack by H2O2 or associated radicals): Very early on in the development of NPMCs, it was hypothesized that H2O2, produced at the NPMC surface through an undesirable 2e− reduction of O2, could oxidize the catalyst/active sites, leading to reduction in overall ORR activity (35–37). This oxidative attack could be exacerbated by the trace iron in these NPMCs, which could lead to a Fenton’s reaction forming aggressive hydroxyl radicals (45). It has previously been suggested that, for PGM-based CCLs, the majority of the current is generated at the membrane/CCL interface (46, 47). This is particularly true at high current densities, where proton transport limitations in the CCL become more significant. Recently, a similar conclusion was reached for NPMC-based CCLs (16). In this case, it could be predicted that thinner (lower loading) CCLs would suffer more from H2O2 attack than thicker (higher loading) CCLs. This is because the thicker CCLs would have a higher percentage of catalyst (close to the GDL interface) not generating significant current and subsequently generating less H2O2 than for the thinner CCLs. This would also be in-line with Pt-based MEAs, where the most severe degradation is observed at the membrane/CCL interface (48). This is shown schematically in fig. S4.
Mechanism 3 (active-site protonation/anion binding): This mechanism was proposed by several research groups (9, 38) and, if true, could represent a major hurdle for NPMCs because it would predict that these catalysts will be inherently unstable in the presence of protons/anions (both ubiquitous in the CCL of a PEMFC). However, it does seem clear that this mechanism would predict no difference in stability for CCLs of varying thickness/loading, provided that the ionomer content was kept constant (as it was in the present work). Furthermore, there would be no reason to expect any carbon oxidation during the stability testing, and certainly no reason to predict differences in the relative rate of carbon oxidation as loading was varied.
Mechanism 4 (micropore flooding): This mechanism was proposed relatively recently to help explain the rapid initial (<20 hours) decay in performance (12, 21, 39, 40). Briefly, it is believed that as current is generated, water can start to fill the micropores of the NPMC. Because it is believed that for some NPMCs the active sites are housed in micropores (49), flooding of the micropores should result in substantial mass transport limitations. However, recent work by our group has provided contrary evidence (29), suggesting that micropore flooding may not be a primary factor behind the observed instability. Unlike mechanism 3, this mechanism appears to predict higher instability with lower catalyst loading, because the volumetric water production would be increased, thus conceivably leading to more rapid performance loss. However, unlike mechanism 2, no increase in carbon oxidation would be anticipated for the lower catalyst loadings.
Because each mechanism has a unique predicted outcome, a stability study of the three catalyst loadings should help to isolate the primary mechanism. Briefly, the predictions are summarized in Table 2.
Table 2. Mechanisms and corresponding predictions.
| Mechanism |
Stability affected by loading |
Carbon oxidation affected by loading |
| Mechanism 2 (attack by H2O2) | Expected | Expected |
| Mechanism 3 (active-site protonation/anion binding) |
Not expected | Not expected |
| Mechanism 4 (micropore flooding) |
Expected | Not expected |
The results of the stability test are shown in Fig. 3, with full polarization curves available in fig. S8. Note that both potentiostatic and galvanostatic stability tests have previously been reported in the literature, with potentiostatic being the most common (39). For this work, we chose the galvanostatic mode, because this most closely represents the operating mode in a commercial portable/backup power fuel cell product.
Fig. 3. Stability of the three catalyst loadings during galvanostatic experiments at 0.5 A/cm2.
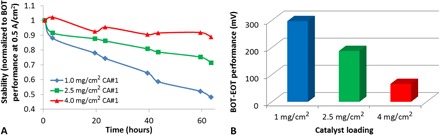
The stability test was performed under air/H2, 100% RH (cathode/anode). (A) The performance at 0.5 A/cm2 is normalized to the beginning-of-test (BOT) performance at 0.5 A/cm2. (B) Absolute differences in BOT versus end-of-test (EOT) performances.
It is clear that the higher loading design resulted in improved stability. The 4.0-mg/cm2 design appears to show very high stability, losing ~10% of its initial activity after 60 hours versus ~55% for the 1.0-mg/cm2 design (Fig. 3). Although further improvements would be required for most commercial applications, this result does demonstrate that CCL strategies can be used to improve the stability/lifetime of NPMC-based MEAs. It should also be noted that although going to higher loadings could result in further improvements in stability, this strategy could lead to additional challenges, including (i) poor CCL quality (for example, cracking/flaking) and (ii) greatly increased MEA thickness, making drop-in replacement of Pt-based MEAs difficult for many commercial stacks. Thus, we chose to limit this study to 4.0 mg/cm2.
Overall, the fact that higher loadings result in improved stability seems sufficient to rule out mechanism 3 as being the primary mechanism, because this mechanism does not predict such a trend to exist (Table 2).
This leaves mechanisms 2 and 4 as possibilities (again, assuming that mechanism 1 is a minor contributor). Looking again at Table 2, it should now be possible to differentiate between these two mechanisms by considering the degree of carbon corrosion observed. This was achieved by comparing the cyclic voltammograms (CVs) before and after the stability test (Fig. 4A). Comparing between mechanisms 2 and 4, both would appear to predict an increase in double-layer charge following the stability test due to either carbon oxidation (mechanism 2) or increased electrochemical surface area (mechanism 4). This was in fact observed for each of the CCL designs (Fig. 4A). In addition, both would conceivably predict a larger increase in the double-layer charge for the lower loaded designs due to either increased production of H2O2/carbon oxidation (mechanism 2) or increased volumetric water production, leading to more rapid micropore flooding (mechanism 4). Again, this was observed (Fig. 4B). However, only carbon surface oxidation would be expected to lead to the formation of new pseudocapacitive peaks as oxygen-containing surface groups are formed. In the case of micropore flooding, only a uniform increase in double-layer charge would be predicted. Examining the plot in Fig. 4A, it is clear that the 1.0-mg/cm2 design shows nonuniform growth in the double-layer charging currents, with the appearance of new pseudocapacitive peaks at 0.6 V [typically attributed to quinone/hydroquinone (50, 51)] as well as 0.8 V. Thus, micropore flooding alone appears insufficient to explain the stability results, because some degree of surface oxidation is occurring. Although there is no conclusive evidence that this oxidation is from H2O2 (or associated radicals), at these potentials, carbon is not expected to oxidize (52) and, thus, some oxidizing agent must be present (most likely H2O2/associated radicals). At a loading of 2.5 mg/cm2, these peaks are far less evident, and at 4.0 mg/cm2, hardly any change in the double layer is observed.
Fig. 4. Comparison of the change in double layer charge for each MEA between beginning of test (BOT) and end of test (EOT).
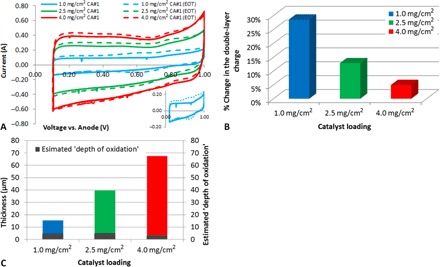
(A) CVs obtained before and after the stability test. The inset highlights the pseudocapacitive peaks for the 1.0-mg/cm2 design. (B) Percent increase in double-layer charge following the stability test for each of the three loadings. (C) Measured (scanning electron microscopy cross section) CCL thickness and estimated depth of oxidation into the CCL.
The results in Fig. 4B show that a higher percentage of the catalyst is oxidized as CCL loading is decreased. This could be explained either by degradation occurring uniformly, but to a decreasing degree, as the CCL loading is increased or, as previously described, by the membrane/CCL interface degrading at a higher rate than the rest of the CCL (fig. S4). However, on the basis of the facts that the majority of the current is generated at the membrane/CCL interface (16, 46, 47) and that degradation is known to be more severe at the membrane/CCL interface for Pt-based MEAs (48), it appears most likely that the interpretation shown in fig. S4 is correct (rapid degradation at the membrane/CCL interface). To further examine this result, the percentage of oxidation shown in Fig. 4B was used to estimate the “depth of oxidation” into the CCL by multiplying the percent change in double-layer charge by the measured CCL thickness (Fig. 4C).
Figure 4C shows that, when the percentage of oxidation is multiplied by CCL thickness, a similar depth of oxidation is observed. This does appear to support the hypothesis that the majority of the reaction (and degradation) occurs close to the membrane/CCL interface and that this reaction penetration is somewhat independent of overall CCL thickness. It could also help to explain why stability increases with CCL loading. As catalyst oxidation occurs, the primary reaction zone will be pushed deeper into the CCL (away from the membrane). For the higher loading CCLs, additional (nonoxidized) catalyst is available, and so, only an additional ohmic overpotential would be expected (because the protons now have to travel further to react). For the thinner CCLs, both ohmic and kinetic overpotentials would be expected, because not only do the protons have to transport further but also fewer catalyst sites are available to buffer the losses occurring at the membrane/CCL interface (fig. S4).
Although these results most strongly support mechanism 2 (carbon oxidation by H2O2) as the primary culprit for the instability of this particular NPMC, it should be highlighted that these results do not preclude the possibility that other mechanisms are also occurring. Rather, the results suggest that mechanism 3 and/or 4 on their own cannot properly explain the observations, and if either is occurring, it must be occurring in conjunction with mechanism 2 (the primary mechanism). Finally, it must be reiterated that our assumption of a minor contribution from mechanism 1 draws on recently reported work that suggests demetalation is not a critical concern (30, 34, 44), but in the present work, we cannot fully rule it out.
CONCLUSION
Overall, the results reported here show, for the first time, an NPMC-based MEA capable of meeting commercial performance requirements for a subset of portable power applications. High current densities (up to 1.5 A/cm2) were achieved using a catalyst that was >90% microporous (nearly all pores <3 nm), highlighting how rational catalyst layer design can be leveraged to overcome catalyst-level transport problems. These results have propelled NPMCs into a new realm of maturation. With each new advance in performance, stability, and durability, new markets can be penetrated, but the most difficult step (achieving commercial performance requirements) has now been achieved and has ushered in a new paradigm for NPMC-based PEMFCs.
MATERIALS AND METHODS
Catalyst synthesis (CA#1)
The synthesis of CA#1 is outlined in our patent (53). Briefly, 1.0 g of a polyacrylonitrile-polymethacrylic acid copolymer was dissolved in 15 g of dimethylformamide (solution a). Next, 1.0 g of 2-methylimidazole and 5.78 g of zinc chloride (ZnCl2) were dissolved in 15 g of dimethylformamide (solution b). Next, solution a and solution b were mixed, and 0.187 g of iron powder was added with stirring. Afterward, the obtained mixed solution was subjected to vacuum drying at 60°C for 24 hours. The mixture obtained by the abovementioned vacuum drying was then heated in air to be made infusible. Specifically, the abovementioned mixture was heated from room temperature to 150°C over 30 min and then increased in temperature from 150° to 220°C over 2 hours. Following this, the mixture was kept at 220°C for 3 hours.
The raw material obtained by the abovementioned process was then ball-milled using silicon nitride balls having a diameter of 10 mm in a planetary ball mill (P-7; Fritsch Japan Co. Ltd.). This raw material was then carbonize-heated to 1100°C in an image furnace under a nitrogen atmosphere and was kept in this state for 1 hour. The carbonized material obtained by the abovementioned procedure was ball-milled using the previously mentioned balling procedure. Further ball milling was then performed using zirconia beads having a diameter of 0.3 mm and methanol in a different ball-milling apparatus (Imex Co. Ltd.).
Metal removal treatment was performed by acid washing. Specifically, 20 ml of concentrated hydrochloric acid was added to 1.0 g of the carbonized material, and this mixture was then stirred for 30 min. The carbonized material was precipitated, and the solution was removed. This treatment was repeated several times. Distilled water was then added, followed by additional stirring. The solution containing the carbonized material was filtered with a filtration film and washed with distilled water until the filtrate became neutral. The collected carbonized material was then vacuum-dried. The dried carbonized material was pulverized with a mortar and pestle.
Next, further heat treatment was performed. Specifically, the carbonized material, which had already been subjected to the metal removal treatment as described above, was placed in a quartz tube. This material was then heated to 700°C in an image furnace under a nitrogen atmosphere and kept in this state for 1 hour, followed by planetary ball milling, resulting in the final catalyst (CA#1).
MEA preparation and evaluation
A precommercial NPMC (CA#1) was supplied by Nisshinbo Holdings. MEAs were prepared using 0.1-mg/cm2 Pt at the anode, with Nafion NR211 as the membrane. The cathodes were prepared by coating 1.0, 2.5, or 4.0 mg/cm2 of CA#1 onto a GDL to form a GDE. An ionomer with equivalent weight (EW) of 700 (SS700C/25, Asahi Kasei) was used, typically at 40 wt % (I/C ratio of 0.67:1). MEAs (50 cm2) were evaluated under air or O2 at 173 kPa, 75°C, and 100% RH. Fuel and oxidant stoichiometries were both 40. All CVs were obtained from 0.1 to 1.0 V versus anode with CorrWare software using a PAR Model 263A potentiostat. During CV measurements, hydrogen and nitrogen were fed to the anode and cathode, respectively, with the anode thus acting as a dynamic hydrogen electrode (DHE) during CV measurements. In situ electrochemical impedance spectroscopy (EIS) was also performed using a Solartron SI 1287 potentiostat, Solartron SI 1255 frequency response analyzer, ZPlot/ZView software, and a transmission-line equivalent circuit. The EIS measurements were carried out at 0.45 V versus DHE using ac amplitude of 10 mV and a frequency range from 50,000 to 0.5 Hz. The EIS analysis was performed at 100% RH.
Physical characterization
X-ray photoelectron spectroscopy (XPS): XPS analysis followed the same procedure outlined in our recent work (17). Briefly, XPS measurements were obtained using an x-ray photoemission spectroscopy device (AXIS Nova, Kratos Analytical). An Al Kα ray (10 mA; 15 kV; pass energy, 40 eV) was used as an x-ray source. Narrow scans were performed to C1s (10 scans), N1s (10 scans), O1s (45 scans), Fe2p (90 scans), and Zn2p (45 scans) core levels using a pass energy of 40 eV with a dwell time of 120 ms. The resultant spectra were corrected for the binding energy based on a C1s spectrum peak of 284.5 eV. From peak areas and detection sensitivity coefficients of the spectra, elemental concentrations (%) of nitrogen atoms and carbon atoms were determined. The atomic ratio of the nitrogen atoms to the carbon atoms was evaluated as an “N/C” ratio. The results are summarized in table S2.
Specific surface area measurement: The specific surface area was determined by using a BELSORP-max (available from BEL Japan Inc.) instrument. The BET (Brunauer-Emmett-Teller) method was used to measure the surface area, and pore size distribution was determined using the MP (Mikhail and Brunauer) method. The presence of mesopores was evaluated using the BJH (Barrett-Joyner-Halenda) method, and the contribution from micropores was quantified using t-plot analysis. The results are summarized in fig. S5 and table S3.
X-ray diffraction measurement (XRD) was carried out using an x-ray diffractometer (Rigaku RINT2000/PC, Rigaku Corporation). The voltage and current applied to the x-ray vacuum tube were 50 kV and 300 mA, respectively. Measurements were performed at a sampling interval of 0.1°, a scanning speed of 1°/min, and a measurement angle range (2θ) of 5° to 90°. CuKα was used as an incident x-ray. The catalyst sample was placed on a concave portion of a glass sample plate (2 cm × 2 cm × 0.2 mm thick) and pressed with a glass slide, and the concave portion was uniformly filled with the sample so that the surface corresponded to a reference level. The glass sample plate was then fixed on a wide-angle XRD stage so that the sample filling the concave portion was not deformed.
Supplementary Material
Acknowledgments
We would like to acknowledge our many colleagues who contributed to the very useful discussions and insights into this work, particularly R. Bashyam, A. Young, P. He, and S. Knights from Ballard Power Systems, and C. Fujii, Y. Kubota, and Y. Oshima from Nisshinbo Holdings Inc. From Ballard Power Systems, we would like to acknowledge E. Marquez and J. Song for assistance with the coating and the failure analysis of the catalyst layers. Funding: We acknowledge funding from both Nisshinbo Holdings Inc. and Ballard Power Systems. Author contributions: D.B., T.K., and S.Y. planned and designed the experiments. D.B. and S.Y. were primarily responsible for designing the catalyst layers. Y.Z. and K.B. prepared the catalyst layers and performed the majority of the testing. T.K. and T.S. were responsible for developing the catalyst. J.-i.O. and Y.I. provided overall supervision of the project, particularly with respect to catalyst development. D.B. and S.Y. were primarily responsible for drafting the manuscript, and every author helped with the final review and edits. Competing interests: Y.I., T.K., T.S., and J.-i.O. are inventors on a patent related to this work held by Nisshinbo Holdings Inc. and Gunma University (serial no. WO 2016/088716 A1; 9 June 2016). J.-i.O. is also an inventor on an additional patent held by Nisshinbo Holdings Inc. and Gunma University (serial no. WO 2017/209244 A1; 9 December 2016). T.K., D.B., S.Y., and K.B. are inventors on a third patent related to this work held by Nisshinbo Holdings Inc. and Ballard Power Systems (serial no. WO 2017/203980 A1; 9 November 2016). All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/3/eaar7180/DC1
Supplementary Text
fig. S1. In situ CVs obtained for three different loadings of CA#1.
fig. S2. Difference in performance under air versus O2 at 1 A/cm2 for each of the three designs.
fig. S3. CCL conductance versus ionomer EW.
fig. S4. Schematic depiction of how CCL degradation may be expected to proceed in the case of attack by H2O2.
fig. S5. N2 gas sorption analysis of CA#1.
fig. S6. Transmission electron microscopy images of CA#1 obtained at various magnifications.
fig. S7. X-ray photoelectron spectroscopy characterization of CA#1.
fig. S8. Polarization curves at beginning of test (BOT) (solid lines) and end of test (EOT) (dashed lines) for MEAs prepared from the three different catalyst loadings.
table S1. Double-layer charge normalized to the charge obtained for the 1.0 mg/cm2 design.
table S2. XPS characterization of CA#1.
table S3. Gas sorption analysis.
table S4. Fe and Zn content based on inductively coupled plasma measurements of CA#1.
REFERENCES AND NOTES
- 1.F. Barbir, PEM Fuel Cells: Theory and Practice (Elsevier Academic Press, 2005). [Google Scholar]
- 2.Banham D., Ye S., Current status and future development of catalyst materials and catalyst layers for PEMFCs: An industrial perspective. ACS Energy Lett. 2, 629–638 (2017). [Google Scholar]
- 3.Kongkanand A., Subramanian N. P., Yu Y., Liu Z., Igarashi H., Muller D. A., Achieving high-power PEM fuel cell performance with an ultralow-Pt-content core–shell catalyst. ACS Catal. 6, 1578–1583 (2016). [Google Scholar]
- 4.Kongkanand A., Mathias M., The priority and challenge of high-power performance of low-platinum proton-exchange membrane fuel cells. J. Phys. Chem. Lett. 7, 1127–1137 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Jasinski R., A new fuel cell cathode catalyst. Nature 201, 1212–1213 (1964). [Google Scholar]
- 6.Wu G., More K. L., Johnston C. M., Zelenay P., High-performance electrocatalysts for oxygen reduction derived from polyaniline, iron, and cobalt. Science 332, 443–447 (2011). [DOI] [PubMed] [Google Scholar]
- 7.Lefèvre M., Proietti E., Jaouen F., Dodelet J.-P., Iron-based catalysts with improved oxygen reduction activity in polymer electrolyte fuel cells. Science 324, 71–74 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Proietti E., Jaouen F., Lefèvre M., Larouche N., Tian J., Herranz J., Dodelet J.-P., Iron-based cathode catalyst with enhanced power density in polymer electrolyte membrane fuel cells. Nat. Commun. 2, 416 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Herranz J., Jaouen F., Lefèvre M., Kramm U. I., Proietti E., Dodelet J.-P., Bogdanoff P., Fiechter S., Abs-Wurmbach I., Bertrand P., Arruda T. M., Mukerjee S., Unveiling N-protonation and anion-binding effects on Fe/N/C Catalysts for O2 reduction in proton-exchange-membrane fuel cells. J. Phys. Chem. C 115, 16087–16097 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serov A., Artyushkova K., Niangar E., Wang C., Dale N., Jaouen F., Sougrati M.-T., Jia Q., Mukerjee S., Atanassov P., Nano-structured non-platinum catalysts for automotive fuel cell application. Nano Energy 16, 293–300 (2015). [Google Scholar]
- 11.Shui J., Chen C., Grabstanowicz L., Zhao D., Liu D.-J., Highly efficient nonprecious metal catalyst prepared with metal–organic framework in a continuous carbon nanofibrous network. Proc. Natl. Acad. Sci. U.S.A. 112, 10629–10634 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y.-C., Lai Y.-J., Song L., Zhou Z.-Y., Liu J.-G., Wang Q., Yang X.-D., Chen C., Shi W., Zheng Y.-P., Rauf M., Sun S.-G., S-doping of an Fe/N/C ORR catalyst for polymer electrolyte membrane fuel cells with high power density. Angew. Chem. Int. Ed. 54, 9907–9910 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Chung H. T., Cullen D. A., Higgins D., Sneed B. T., Holby E. F., More K. L., Zelenay P., Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst. Science 357, 479–484 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Larouche N., Chenitz R., Lefèvre M., Proietti E., Dodelet J.-P., Activity and stability in proton exchange membrane fuel cells of iron-based cathode catalysts synthesized with addition of carbon fibers. Electrochim. Acta 115, 170–182 (2014). [Google Scholar]
- 15.Serov A., Workman M. J., Artyushkova K., Atanassov P., McCool G., McKinney S., Romero H., Halevi B., Stephenson T., Highly stable precious metal-free cathode catalyst for fuel cell application. J. Power Sources 327, 557–564 (2016). [Google Scholar]
- 16.P. Zelenay, “Hydrogen and fuel cells program,” 2017 Annual Merit Review and Peer Evaluation Meeting (AMR) (2017).
- 17.Banham D., Kishimoto T., Sato T., Kobayashi Y., Narizuka K., Ozaki J.-i., Zhou Y., Marquez E., Bai K., Ye S., New insights into non-precious metal catalyst layer designs for proton exchange membrane fuel cells: Improving performance and stability. J. Power Sources 344, 39–45 (2017). [Google Scholar]
- 18.Antolini E., Giorgi L., Pozio A., Passalacqua E., Influence of Nafion loading in the catalyst layer of gas-diffusion electrodes for PEFC. J. Power Sources 77, 136–142 (1999). [Google Scholar]
- 19.Lee S. J., Mukerjee S., McBreen J., Rho Y. W., Kho Y. T., Lee T. H., Effects of Nafion impregnation on performances of PEMFC electrodes. Electrochim. Acta 43, 3693–3701 (1998). [Google Scholar]
- 20.Fuel Cell Technologies Office, http://energy.gov/sites/prod/files/2016/10/f33/fcto_myrdd_fuel_cells.pdf (2016).
- 21.Zhang G., Chenitz R., Lefèvre M., Sun S., Dodelet J.-P., Is iron involved in the lack of stability of Fe/N/C electrocatalysts used to reduce oxygen at the cathode of PEM fuel cells? Nano Energy 29, 111–125 (2016). [Google Scholar]
- 22.Stariha S., Artyushkova K., Serov A., Atanassov P., Non-PGM membrane electrode assemblies: Optimization for performance. Int. J. Hydrogen Energy 40, 14676–14682 (2015). [Google Scholar]
- 23.Ballard, www.ballard.com/about-ballard/newsroom/news-releases/2017/09/13/ballard-to-offer-world-s-first-pem-fuel-cell-product-using-non-precious-metal-catalyst (2017).
- 24.Spry D. B., Goun A., Glusac K., Moilanen D. E., Fayer M. D., Proton transport and the water environment in nafion fuel cell membranes and AOT reverse micelles. J. Am. Chem. Soc. 129, 8122–8130 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Stariha S., Artyushkova K., Workman M. J., Serov A., Mckinney S., Halevi B., Atanassov P., PGM-free Fe-N-C catalysts for oxygen reduction reaction: Catalyst layer design. J. Power Sources 326, 43–49 (2016). [Google Scholar]
- 26.Wang Q. P., Eikerling M., Song D. T., Liu Z. S., Structure and performance of different types of agglomerates in cathode catalyst layers of PEM fuel cells. J. Electroanal. Chem. 573, 61–69 (2004). [Google Scholar]
- 27.Maruyama J., Abe I., Formation of platinum-free fuel cell cathode catalyst with highly developed nanospace by carbonizing catalase. Chem. Mater. 17, 4660–4667 (2005). [Google Scholar]
- 28.Banham D., Ye S., Pei K., Ozaki J.-i., Kishimoto T., Imashiro Y., A review of the stability and durability of non-precious metal catalysts for the oxygen reduction reaction in proton exchange membrane fuel cells. J. Power Sources 285, 334–348 (2015). [Google Scholar]
- 29.Choi J.-Y., Yang L., Kishimoto T., Fu X., Ye S., Chen Z., Banham D., Is the rapid initial performance loss of Fe/N/C non precious metal catalysts due to micropore flooding? Energ. Environ. Sci. 10, 296–305 (2017). [Google Scholar]
- 30.Hyuck Choi C., Baldizzone C., Polymeros G., Pizzutilo E., Kasian O., Schuppert A. K., Sahraie N. R., Sougrati M.-T., Mayrhofer K. J. J., Jaouen F., Minimizing operando demetallation of Fe-N-C electrocatalysts in acidic medium. ACS Catal. 6, 3136–3146 (2016). [Google Scholar]
- 31.Wang B., Recent development of non-platinum catalysts for oxygen reduction reaction. J. Power Sources 152, 1–15 (2005). [Google Scholar]
- 32.Deng D., Yu L., Chen X., Wang G., Jin L., Pan X., Deng J., Sun G., Bao X., Iron encapsulated within pod-like carbon nanotubes for oxygen reduction reaction. Angew. Chem. Int. Ed. 52, 371–375 (2013). [DOI] [PubMed] [Google Scholar]
- 33.Varnell J. A., Tse E. C. M., Schulz C. E., Fister T. T., Haasch R. T., Timoshenko J., Frenkel A. I., Gewirth A. A., Identification of carbon-encapsulated iron nanoparticles as active species in non-precious metal oxygen reduction catalysts. Nat. Commun. 7, 12582 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Choi C. H., Baldizzone C., Grote J.-P., Schuppert A. K., Jaouen F., Mayrhofer K. J. J., Stability of Fe-N-C catalysts in acidic medium studied by operando spectroscopy. Angew. Chem. Int. Ed. Engl. 54, 12753–12757 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Schulenburg H., Stankov S., Schünemann V., Radnik J., Dorbandt I., Fiechter S., Bogdanoff P., Tributsch H., Catalysts for the oxygen reduction from heat-treated iron(III) tetramethoxyphenylporphyrin chloride: Structure and stability of active sites. J. Phys. Chem. B 107, 9034–9041 (2003). [Google Scholar]
- 36.Goellner V., Armel V., Zitolo A., Fonda E., Jaouen F., Degradation by hydrogen peroxide of metal-nitrogen-carbon catalysts for oxygen reduction. J. Electrochem. Soc. 162, H403–H414 (2015). [Google Scholar]
- 37.Lefèvre M., Dodelet J.-P., Fe-based catalysts for the reduction of oxygen in polymer electrolyte membrane fuel cell conditions: Determination of the amount of peroxide released during electroreduction and its influence on the stability of the catalysts. Electrochim. Acta 48, 2749–2760 (2003). [Google Scholar]
- 38.Liu G., Li X., Popov B., Stability study of nitrogen-modified carbon composite catalysts for oxygen reduction reaction in polymer electrolyte membrane fuel cells. ECS Trans. 25, 1251–1259 (2009). [Google Scholar]
- 39.Shao M., Chang Q., Dodelet J.-P., Chenitz R., Recent advances in electrocatalysts for oxygen reduction reaction. Chem. Rev. 116, 3594–3657 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Yang L., Larouche N., Chenitz R., Zhang G., Lefèvre M., Dodelet J.-P., Activity, performance, and durability for the reduction of oxygen in PEM Fuel Cells, of Fe/N/C electrocatalysts obtained from the pyrolysis of metal-organic-framework and iron porphyrin precursors. Electrochim. Acta 159, 184–197 (2015). [Google Scholar]
- 41.Ishii T., Maie T., Hamano M., Kishimoto T., Mizushiri M., Imashiro Y., Ozaki J.-i., Synergistically enhanced oxygen reduction activity of iron-based nanoshell carbons by copper incorporation. Carbon 116, 591–598 (2017). [Google Scholar]
- 42.Sougrati M. T., Goellner V., Schuppert A. K., Stievano L., Jaouen F., Probing active sites in iron-based catalysts for oxygen electro-reduction: A temperature-dependent 57Fe Mössbauer spectroscopy study. Catal. Today 262, 110–120 (2016). [Google Scholar]
- 43.Chung H. T., Won J. H., Zelenay P., Active and stable carbon nanotube/nanoparticle composite electrocatalyst for oxygen reduction. Nat. Commun. 4, 1922 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrandon M., Wang X., Kropf A. J., Myers D. J., Wu G., Johnston C. M., Zelenay P., Stability of iron species in heat-treated polyaniline–iron–carbon polymer electrolyte fuel cell cathode catalysts. Electrochim. Acta 110, 282–291 (2013). [Google Scholar]
- 45.J. P. Dodelet, Oxygen reduction in PEM fuel cell conditions: heattreated nonprecious metal-N4 macrocycles and beyond, in N4-Macrocyclic Metal Complexes: Oxygen Reduction in PEM Fuel Cell Conditions, J. H. Zagal, F. Bedioui, J. P. Dodelet, Eds. (Springer Science+Business Media, Inc., 2006). [Google Scholar]
- 46.Cruz-Manzo S., Chen R., Rama P., Study of current distribution and oxygen diffusion in the fuel cell cathode catalyst layer through electrochemical impedance spectroscopy. Int. J. Hydrogen Energy 38, 1702–1713 (2013). [Google Scholar]
- 47.Young A. P., Stumper J., Knights S., Gyenge E., Ionomer degradation in polymer electrolyte membrane fuel cells. J. Electrochem. Soc. 157, B425–B436 (2010). [Google Scholar]
- 48.Rasouli S., Ortiz Godoy R. A., Yang Z., Gummalla M., Ball S. C., Myers D., Ferreira P. J., Surface area loss mechanisms of Pt3Co nanocatalysts in proton exchange membrane fuel cells. J. Power Sources 343, 571–579 (2017). [Google Scholar]
- 49.Jaouen F., Lefèvre M., Dodelet J.-P., Cai M., Heat-treated Fe/N/C catalysts for O2 electroreduction: Are active sites hosted in micropores? J. Phys. Chem. B 110, 5553–5558 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Frackowiak E., Béguin F., Carbon materials for the electrochemical storage of energy in capacitors. Carbon 39, 937–950 (2001). [Google Scholar]
- 51.Banham D., Feng F., Burt J., Alsrayheen E., Birss V., Bimodal, templated mesoporous carbons for capacitor applications. Carbon 48, 1056–1063 (2010). [Google Scholar]
- 52.Roen L. M., Paik C. H., Jarvi T. D., Electrocatalytic corrosion of carbon support in PEMFC cathodes. Electrochem. Solid-State Lett. 7, A19–A22 (2004). [Google Scholar]
- 53.Y. Imashiro, T. Kishimoto, T. Sato, J.-i. Ozaki, T. Maie, S. Kusadokoro, US20170194653 (2017).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/3/eaar7180/DC1
Supplementary Text
fig. S1. In situ CVs obtained for three different loadings of CA#1.
fig. S2. Difference in performance under air versus O2 at 1 A/cm2 for each of the three designs.
fig. S3. CCL conductance versus ionomer EW.
fig. S4. Schematic depiction of how CCL degradation may be expected to proceed in the case of attack by H2O2.
fig. S5. N2 gas sorption analysis of CA#1.
fig. S6. Transmission electron microscopy images of CA#1 obtained at various magnifications.
fig. S7. X-ray photoelectron spectroscopy characterization of CA#1.
fig. S8. Polarization curves at beginning of test (BOT) (solid lines) and end of test (EOT) (dashed lines) for MEAs prepared from the three different catalyst loadings.
table S1. Double-layer charge normalized to the charge obtained for the 1.0 mg/cm2 design.
table S2. XPS characterization of CA#1.
table S3. Gas sorption analysis.
table S4. Fe and Zn content based on inductively coupled plasma measurements of CA#1.