

Abstract
Indigo is an ancient dye uniquely capable of producing the signature tones in blue denim; however, the dyeing process requires chemical steps that are environmentally damaging. We describe a sustainable dyeing strategy that not only circumvents the use of toxic reagents for indigo chemical synthesis but also removes the need for a reducing agent for dye solubilization. This strategy utilizes a glucose moiety as a biochemical protecting group to stabilize the reactive indigo precursor indoxyl to form indican, preventing spontaneous oxidation to crystalline indigo during microbial fermentation. Application of a β-glucosidase removes the protecting group from indican, resulting in indigo crystal formation in the cotton fibers. We identified the gene coding for the glucosyltransferase PtUGT1 from the indigo plant Polygonum tinctorium and solved the structure of PtUGT1. Heterologous expression of PtUGT1 in Escherichia coli supported high indican conversion, and biosynthesized indican was used to dye cotton swatches and a garment.
Indigo is a dye that has been prized since antiquity for its vibrancy and deep blue hue, with evidence of its use dating back 6,000 years1. For more than a century, its unique properties have been leveraged to produce the popular textile blue denim. Historically, indigo was extracted from dye-producing plants that were important cash crops on European farms. Production later expanded to plantations in India, the Spice Islands, Central America, and South Carolina2. This critical economy was supplanted at the turn of the 20th century when a chemical synthesis was developed.
Other blue dyes are now synthesized that can be used on a variety of textiles, but none have replaced indigo as a denim dye. Unlike most dyes, indigo does not covalently bind to cotton, but instead adsorbs to the fibers. The adsorbed indigo is robust to strong detergents used for laundering, yet it flakes off with persistent abrasion to expose the internal white yarn core, yielding the desired worn-in look that personalizes an individual’s pair of jeans. This unique combination of robustness to detergents and susceptibility to abrasion makes indigo irreplaceable as a denim dye and contributes to the enduring popularity of blue denim. As of 2011, 50,000 tons of indigo have been synthesized per year, of which 95% was used to dye the over 4 billion denim garments manufactured annually3,4.
The high demand for indigo presents a serious sustainability problem for two reasons. First, indigo is chemically synthesized on an industrial scale from aniline, a toxic compound derived from the petroleum product benzene. The synthesis involves hazardous chemicals including formaldehyde, hydrogen cyanide, sodamide, and strong bases (Fig. 1a)5,6. Second, indigo is insoluble in water, and it must be reduced to the water-soluble leuco form by an excess of reducing agent to be used as a dye. In the current industrial process, sodium dithionite is the preferred reducing agent because of its low cost and short reduction time6,7. However, sodium dithionite decomposes to form sulfate and sulfite, which can corrode equipment and pipes in dye mills and wastewater treatment facilities7. Many dye mills avoid the additional cost of wastewater treatment by dumping the spent dye materials into rivers, where they have negative ecological impacts.
Figure 1. A glucosyl protecting group enables control over the timing and location of indigo dyeing.
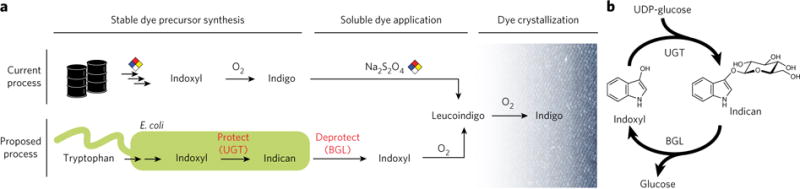
(a) The current industrial process involves chemically synthesizing indigo and adding a reducing agent (typically sodium dithionite) to the indigo vat for reduction to dye-competent, soluble leucoindigo. In the proposed microbial process, E. coli biosynthesizes indoxyl and glucosylates it at the C3 hydroxyl group before its spontaneous air oxidation to indigo. The glucoside, indican, is stable in air and can be stored. The glucosyl group is removed only at the point of dyeing, allowing the regenerated indoxyl to oxidize to indigo crystals in cotton fibers. No reducing agent is required when dyeing with indican. BGL, β-glucosidase; UGT, UDP-glycosyltransferase. (b) Glucose acts as a protecting group for indoxyl inside the production host and in the fermenter until it is removed by co-application with β-glucosidase to cotton.
To combat environmental concerns, microbial production of indigo has previously been targeted as a replacement for chemical synthesis8–11. These efforts offered considerable environmental improvements by removing the need for the harmful chemicals used in indigo synthesis; however, a reducing agent was still required to reduce the insoluble indigo to the soluble leucoindigo for dyeing. A number of other strategies (for example, bacterial fermentation12,13, catalytic hydrogenation14, and electrochemical reduction15) have been used to reduce indigo, but none are as fast-acting and cost-effective as sodium dithionite.
Here we present an alternative indigo dyeing process that mimics the natural biochemical protecting group strategy employed by the Japanese indigo plant P. tinctorium (Supplementary Fig. 1). In the leaf, indole is oxygenated at the C3 position to produce the highly reactive indigo precursor indoxyl. Instead of further oxidizing to form indigo, the hydroxyl group is glucosylated, protecting this reactive functional group and generating the colorless molecule indican. Only upon damage to the leaf tissue is blue coloration observed: the intracellular endomembranes of the vacuole (containing indican)16 and the chloroplast (containing β-glucosidase)17 are lysed, allowing their contents to mix. β-glucosidase hydrolyzes indican to indoxyl, which spontaneously oxidizes to form crystalline indigo through a leucoindigo intermediate. Thus, the glucose moiety serves as a biochemical protecting group, stabilizing the reactive indoxyl in the reduced state until treatment with a β-glucosidase. Accordingly, indican provides many desired attributes for a dye: in addition to water solubility and stability over a broad range of pH and temperatures, indican provides spatial and temporal control over indoxyl formation for direct dyeing of cotton fibers.
The reversible nature of biochemical protecting groups can be a powerful tool in metabolic engineering for controlling metabolite activity, solubility, and recognition by enzymes and transporters. Addition and removal of the protecting group are catalyzed by separate enzymes; thus, these processes can be independently regulated. Several biochemical groups (for example, sulfo, acetyl, methyl, malonyl, and glycosyl groups) can serve as protecting groups, each relating their own chemical properties, such as the charge of a sulfo group or the hydrophobicity of an acetyl group. Transferase enzymes (for example, sulfo-, methyl-, acetyl-, and glycosyltransferases) can site-selectively protect a functional group, and another set of enzymes (for example, sulfatases, demethylases, esterases, and glycosidases) can catalyze the removal of these protecting groups. An interesting example is the use of acetylation in the noscapine biosynthetic pathway in Papaver somniferum L., in which one hydroxyl group is selectively acetylated before hydroxylation at another carbon18. The acetyl protecting group prevents off-pathway reactivity and is subsequently hydrolyzed by an esterase. Similarly, biochemical protecting group strategies can be used by the metabolic engineer to separately control the properties of a metabolite inside the cell relative to those outside the bioreactor.
In our specific application, we sought to gain temporal control over the regeneration of the indoxyl intermediate to achieve indigo crystallization in the cotton fibers, eliminating the need for a reducing agent (Fig. 1a). We leveraged the work of previous groups to biosynthesize the indigo precursor indoxyl from tryptophan in Escherichia coli8–11. However, we also expressed a glucosyltransferase to stabilize indoxyl, preventing spontaneous dimerization by protecting the reactive hydroxyl with a glucosyl group. This glucoside, indican, is secreted from the cell into the fermentation broth and is sufficiently stable for long-term storage and concentration via gentle boiling. At the point of dyeing, indican can be enzymatically hydrolyzed (or deprotected) with a β-glucosidase to regenerate indoxyl (Fig. 1b). Indoxyl spontaneously oxidizes to leucoindigo, and, as in the chemical process, leucoindigo crystallizes to indigo directly in the cotton fibers of the textile, where it adsorbs and becomes trapped.
RESULTS
Identification of glucosyltransferase from P. tinctorium
The described biochemical protecting strategy requires a UDP-glucose:indoxyl glucosyltransferase (UGIG) to produce indican; however, none have previously been reported. Furthermore, when initiating our search, no genome or transcriptome sequence had been available for any indigo-producing plant. To our knowledge, though transcriptome sequencing data have now been reported for two indigo plants19–21, no UGIG amino acid sequence has been identified. We chose P. tinctorium for our enzyme discovery efforts because this Japanese indigo plant has been reported to be among the highest indican-yielding plants22,23 and there exists a published method for purifying its UGIG, which is active on indoxyl in vitro16. We adapted this protocol to purify the glucosyltransferase from P. tinctorium leaves. In parallel, we sequenced cDNA isolated from the leaves and assembled a transcriptome de novo using the Trinity algorithm24. The purified glucosyltransferase was then analyzed by fragmentation and MS, and fragments were matched to transcrip-tome-predicted sequences to identify the UGIG gene sequence.
Sequence analysis showed that the identified glucosyltransferase (UGT72B29, referred to here as PtUGT1) belongs to the UDP-glycosyltransferase (UGT) superfamily25 and the GT1 family of inverting glycosyltransferases in the Carbohydrate Active Enzymes database (CAZy)26. When amplifying the PtUGT1 gene sequence from the cDNA, we also identified a second isoform of the glucosyltransferase gene, encoding a protein (UGT72B30, or PtUGT2) that differs from PtUGT1 by four amino acid residues (V19M, G225A, E230Q, and A423D) (Fig. 2a and Supplementary Fig. 2). When expressed and purified from E. coli, PtUGT1 and PtUGT2 were both active on indoxyl and had similar kinetic parameters in vitro (PtUGT1 kcat = 9.1 ± 0.5 s−1; PtUGT2 kcat = 12 ± 0.8 s−1; Supplementary Fig. 3). PtUGT1 was used for the rest of this work because it was the predominant isoform in the transcriptome sequencing.
Figure 2. The crystal structure of PtUGT1 with bound indoxyl sulfate.
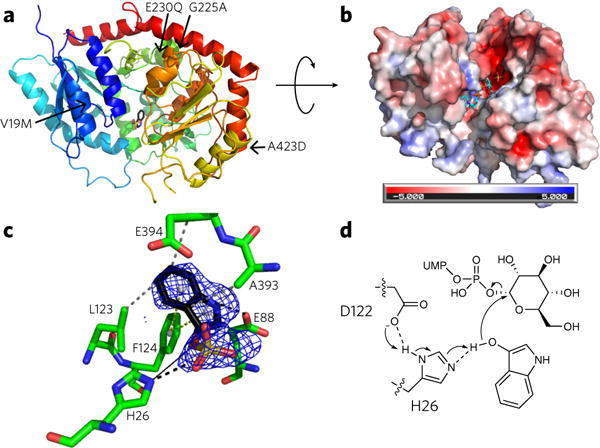
(a) Overall GT-B fold of PtUGT1 (PDB ID 5NLM). The N-terminal Rossmann domain (blue) consists of a seven-stranded parallel β-sheet surrounded by nine α-helical segments, and the C-terminal Rossmann domain (red) consists of a six-stranded parallel β-sheet and five α-helices. The indoxyl sulfate bound in the active site is shown in stick representation. The structure is annotated with the four amino acids differing between PtUGT1 and PtUGT2 (arrows). (b) Electrostatic surface potential of PtUGT1. Colors show the electrostatic potential, from −5 kT/e (red) to 5 kT/e (blue). The view is rotated ~90° around the x axis with respect to a. Bound indoxyl sulfate is depicted in stick representation (black) together with the donor substrate UDP-glucose superposed from the homologous AtUGT72B1 complex structure (cyan, PDB ID 2VCE). (c) The acceptor binding site with the bound indoxyl sulfate and interacting residues shown in stick representation. The indoxyl sulfate omit map is displayed as a blue grid contoured at 3.0 σ. Interactions are depicted as dashed lines (black, salt bridge/hydrogen bond; gray, hydrophobic interaction; yellow, π-stacking). A black solid line indicates the distance from the catalytic histidine to the glucose-accepting oxygen. (d) In the proposed catalytic mechanism based on homology to other characterized UGTs29,31, H26 deprotonates the indoxyl hydroxyl group, which then performs an SN2 attack on the anomeric carbon of glucose. The conserved D122 is believed to balance the charge on the catalytic histidine29. Consistent with this hypothesis, D122 forms a 2.6 Å hydrogen bond to H26 in the present structure.
Structural characterization of PtUGT1
To investigate the structural basis for indoxyl glucosylation by PtUGT1, we solved the PtUGT1 crystal structure at 2.14-Å resolution with indoxyl sulfate, a glycosylation-incompetent indoxyl analog, in the active site (PDB ID 5NLM; Fig. 2a). PtUGT1 displays the canonical GT-B fold27, consisting of two Rossmann domains connected with a linker. Of the four amino acids that differ between PtUGT1 and PtUGT2, the one closest to the active site (V19) is located in the core β-sheet of the N-terminal Rossmann domain, 12 Å from indoxyl sulfate (Fig. 2a). The remaining three substitutions are solvent-exposed residues. In line with our kinetic observations, this structural analysis suggests that none of the substitutions are likely to affect catalysis.
Indoxyl sulfate binds at one end of the catalytic cleft between the two domains, similarly to other UGT acceptors28 (Fig. 2b). This rather large cavity excessively accommodates indoxyl rather than fitting it snugly as in the classical lock-and-key model. Hence, structural determinants of indoxyl specificity can be speculated to be limited to those amino acids in direct contact with the indoxyl molecule (Fig. 2c). In addition to hydrophobic interactions with residues L123, A393, and E394 and perpendicular π-stacking with F124, indoxyl sulfate interacts with the enzyme through a hydrogen bond between the indoxyl sulfate ring nitrogen and the conserved E88, which could therefore play a major role in indoxyl specificity and turnover. By analogy to other UGTs, H26 is expected to be the catalytic Brønsted base29 (Fig. 2d). Indeed, the H26A mutant produces no detectable indican in an enzyme kinetics assay (Supplementary Fig. 3). This histidine is oriented toward the glucose-accepting oxygen at a distance of 3.9 Å. It is possibly displaced with respect to the catalytically competent conformation by the bulky sulfate group on the acceptor substrate mimic, with which it interacts through a salt bridge in the current structure.
A search for PtUGT1 structural homologs revealed substantial similarity (Z-scores above 30) to several plant UGTs belonging to the GT1 family. The closest structural homolog found is the AtUGT72B1 enzyme (Z-score 53.6; r.m.s. deviation = 1.6 Å for 361 aligned residues), which is a bifunctional N- and O-glucosyltransferase implicated in xenobiotic metabolism in Arabidopsis thaliana30. Its structure has been solved in complex with a donor analog, UDP-2F-glucose (PDB ID: 2VCE)31, revealing a donor binding mode very similar to that of other characterized UGTs. An extensive hydrogen bonding network is formed between all parts of the nucleotide sugar (i.e., the nucleobase, pentose, phosphates, and sugar moiety) and enzyme residues primarily from the C-terminal Rossmann domain. In AtUGT72B1, 21 residues participate in this network; 19 of these are conserved in PtUGT1 and align well (r.m.s deviation. = 0.6 for 131 aligned atoms) with the corresponding AtUGT72B1 residues (Supplementary Fig. 4). It is therefore likely that the donor binding sites in PtUGT1 and AtUGT72B1 are similar.
Production of indican in E. coli
We chose E. coli as the production host because of its short generation time, its ability to support high flux through engineered metabolic pathways, its use as an industrial production host for metabolic products32–34, and its ability to efficiently glucosylate small molecules35,36. Furthermore, indoxyl production in E. coli has previously been accomplished at high titer for indigo biosynthesis9–11.
We developed an appropriate E. coli background strain and growth conditions to test heterologous expression of PtUGT1 in vivo. Previously, the flavin-dependent monooxygenase (FMO) from Methylophaga aminisulfidivorans was shown to efficiently convert indole to indoxyl37, the precursor to indican and indigo (Fig. 3a). We expressed the FMO gene on a plasmid under the control of the constitutive bacterial promoter J23100 (ref. 38) to test indoxyl production under various carbon sources. Previous reports have shown that the expression and activity of TnaA, an E. coli enzyme required for conversion of tryptophan to indole, is repressed by glucose8,39, but not by glycerol40. Indeed, a glycerol-fed culture produced 400 mg/L indigo after 24 h growth in EZ Rich defined medium when an optimal concentration of tryptophan (over 15 mM or 3.1 g/L) was co-fed, whereas a culture grown with glucose instead of glycerol produced almost no indigo (Supplementary Fig. 5). Tryptophan supplementation is required for visible indigo production, as native tryptophan production is insufficient to sustain indoxyl production in the laboratory strain MG1655 grown in EZ Rich medium (containing only 0.1 mM tryptophan).
Figure 3. Heterologous expression of PtUGT1 stabilizes indoxyl before it dimerizes, producing indican.
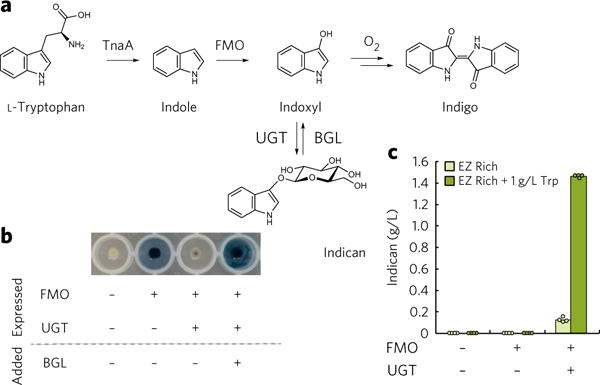
(a) Tryptophan is converted to indole by the native E. coli enzyme TnaA, oxygenated to indoxyl by FMO, and glucosylated by PtUGT1. A β-glucosidase hydrolyzes indican into indoxyl, which spontaneously oxidizes to indigo. (b) Expression of PtUGT1 reduces indigo formation in an FMO-expressing ΔbglA strain, but indigo precipitate forms upon addition of β-glucosidase. (c) Indican can be produced in a ΔbglA strain by heterologous expression of both FMO and PtUGT1, and titers improve with exogenous feeding of more tryptophan than the 20 mg/L present in EZ Rich media. Bars represent the mean of four biological replicates, and each replicate is represented by a circle.
Before biosynthesizing indican, we also examined whether this product would be stable in our E. coli production host. Indican has previously been shown to be hydrolyzed by a number of β-glucosidases41–43, and because E. coli contains several native β-glucosidases, indican could be susceptible to hydrolysis after production. To measure background hydrolysis, 1 g/L indican was fed to MG1655 E. coli, and the loss of indican was quantified over 4 d (Supplementary Fig. 6). We did observe background hydrolysis and hypothesized that it was a result of one or more endogenous E. coli β-glucosidases. A panel of single-gene glucosidase knockouts from the Keio collection (background strain BW25113)44 was tested for stability of fed indican (Supplementary Fig. 7). The only knockout that noticeably reduced hydrolysis was ΔbglA, and we confirmed its effect in our bacterial host, MG1655. There was considerably less indican degradation in the ΔbglA strain than in the wild-type strain, and even after 5 d of incubation at 37 °C there was no visible indigo formation in the ΔbglA strain (Supplementary Fig. 6).
The PtUGT1 gene and the FMO gene were then co-expressed in MG1655ΔbglA for in vivo indican production (Fig. 3a and Supplementary Fig. 8). The two-gene operon was driven by the T7 promoter (PT7) for high gene expression and integrated into the chromosome to minimize copy number variability (strain TMH011). In addition to producing indican, the strain also showed dramatically lower indigo production compared to the MG1655ΔbglA background strain expressing only FMO (strain TMH003; Fig. 3b), consistent with the prediction that glucosylation prevents indoxyl dimerization and oxidation into indigo crystals. Indican production, as quantified by LC–MS, was extremely efficient, with 97% of tryptophan converted into indican on a molar basis (Fig. 3c). Addition of purified β-glucosidase BglA from Bacillus circulans45 to the extracellular medium resulted in visible indigo precipitation (Fig. 3b). Thus, indican is both produced and secreted from our engineered E. coli strain TMH011.
The efficiency of PtUGT1 can also be seen when comparing titers of indican in TMH011 (9.8 mM or 2.9 g/L) to titers of indigo in TMH003 (5.3 mM or 1.4 g/L) when grown for 24 h in media with excess (26 mM or 5.4 g/L) fed tryptophan (Fig. 4a,b). Because indigo is an indoxyl dimer, and thus requires twice as many moles of indoxyl substrate as indican, these titers are approximately equivalent on an indoxyl molarity basis, with a 38% molar conversion of tryptophan to indican and a 41% molar conversion to indigo. Thus, indoxyl glucosylation appears to be competitively more efficient than indoxyl oxidation up to the titers achieved, which is consistent with the observation that only low amounts of indigo byproduct are made when expressing PtUGT1 (Fig. 4b) whereas tryptophan is fully used (Fig. 4c). Under the current production regime, the PT7-driven expression of FMO and PtUGT1, as well as their metabolic products, do not appear to cause toxicity to the cells (Fig. 4d).
Figure 4. Production and growth curves for indican and indigo production.
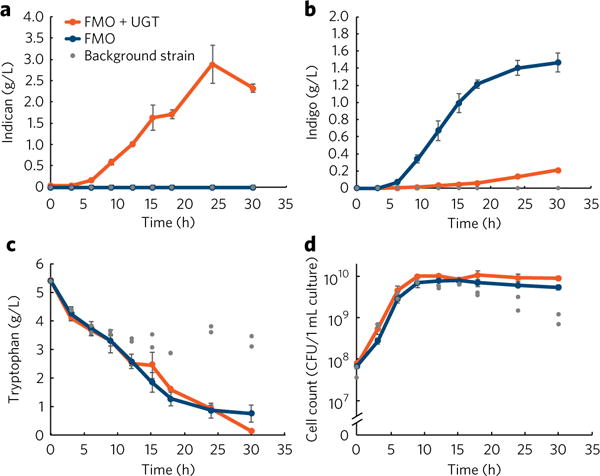
(a–d) Titers of indican (a), indigo (b), and tryptophan (c), and cell count (d; measured as colony-forming units (CFU), see “Determination of cell count” in Online Methods) over a 37 h growth period. Cells expressing FMO and PtUGT1 are represented by orange; cells expressing only FMO are represented by blue; cells expressing neither gene are represented by gray. Error bars represent the mean ± s.d. of six (FMO + UGT) or four (FMO) independently grown colonies; circles represent individual independently grown colonies.
Use of biosynthesized indican as a textile dye
The current indigo dye mill process consists of repeatedly dipping ropes of cotton yarn into multiple dye baths containing an aqueous solution of leucoindigo, sodium hydroxide, and sodium dithionite. Exposure to air between dips allows spontaneous oxidation of the leucoindigo, depositing indigo in the fibers6. We propose a dyeing process in which an indican solution and a β-glucosidase solution are alternately applied to the textile, resulting in indican hydrolysis. The resulting indoxyl spontaneously oxidizes and dimerizes in air to form leucoindigo and, subsequently, indigo. In our methodology, we apply indican as a spray to prevent contamination of the indican solution with β-glucosidase, which would result in premature hydrolysis (Fig. 1).
Our dyeing process requires the biosynthesis of indican, as well as a β-glucosidase capable of hydrolyzing indican in cotton fibers. The gene encoding the B. circulans β-glucosidase BglA was expressed under the control of PT7 in the Rosetta BL21(DE3) E. coli strain to produce lysate with an indican hydrolysis activity of 1.8 ± 0.8 μmol/min/mg. In parallel, to produce a sufficient amount of indican to test the dyeing process, we used a 14-L fermenter to grow 5 L of strain TMH011. The fermentation broth was used to dye fabric without the need for indican purification. As higher dye concentrations are correlated with darker shades of indigo, the broth was gently boiled to increase the indican concentration by 2.9-fold from 1.1 g/L to 3.2 g/L.
An unexpected orange color was produced in the media when biosynthesizing indican. This orange color was dependent on FMO expression and was more intense with co-expression of PtUGT1 (Supplementary Fig. 9a). However, this color was water soluble, as it was efficiently removed in both water and detergent washes (Supplementary Fig. 9b) and, accordingly, did not affect the final dyed product (Fig. 5).
Figure 5. Bio-indican can be used as an effective, reductant-free cotton textile dye.

(a) Top row, Pure indican with no β-glucosidase (BGL); pure indican with β-glucosidase; bio-indican with β-glucosidase. Bottom row, Indigo, nonreduced; indigo, reduced with sodium dithionite. All swatches are dyed on white cotton denim. Scale bar, 1 cm. (b) Scarf (100% white cotton gauze fabric) dyed with indican. All samples were photographed after numerous, vigorous water washes. Scale bar, 5 cm.
To demonstrate the indican dye strategy and compare it to the current, chemically reduced indigo dyeing process, we dyed swatches of white cotton denim cloth using concentrated fermentation broth containing 3.2 g/L indican (Fig. 5a). A solution of 3.2 g/L purchased indican standard was also used to examine the effect of indican purity on dyeing. The swatches were dyed by spraying them with indican, dipping them into β-glucosidase cell lysate, and oxidizing them in air. An equimolar chemically reduced indigo dye bath of 1.4 g/L indigo (taking into account that two indoxyl molecules are required to make one leucoindigo molecule) was used to mimic the conventional industrial process for comparison. An unreduced indigo vat did not visibly dye cloth, but the reduced-indigo dye was effective, as were the indican dyes after deprotection. Although the indican-dyed cloths did not dye as darkly as the conventionally dyed cloths, they remained blue even after a water wash (Fig. 5a). The difference in color intensity is likely attributable to the higher pH used for the chemical dyeing process, as indigo dyeing is more efficient under alkaline conditions than at the neutral pH used for the indican dyeing.
The concentrated bio-indican media and β-glucosidase lysate were used to dye a garment, a cotton gauze fabric scarf, as a proof-of-concept application. A total of three dye cycles were applied to each side of the fabric to achieve a deep indigo color, wherein one cycle is defined as an indican broth spray application followed by a β-glucosidase lysate spray application. Color lasted even after the fabric was vigorously rinsed in water until the runoff water was clear (Fig. 5b). The scarf was subsequently laundered in a commercial laundry machine with detergent, and although some intensity was lost, ample color was maintained (Fig. 5b for pre-machine wash; Supplementary Fig. 10 for post-machine wash).
DISCUSSION
The indigo dyeing process has undergone minimal changes over the last hundred years, yet demand for the dye is higher than ever before, making its ecological consequences unsustainable. Here, we report a promising new strategy for dyeing that not only removes the need for the hazardous chemicals required for indigo synthesis, as previously addressed with indoxyl production in E. coli9–11, but also eliminates the need for the environmentally unfriendly reducing agent used for solubilization by instead using a glucosyl biochemical protecting group. We identified the gene sequence of a novel glucosyltransferase from P. tinctorium (PtUGT1) with activity on indoxyl, characterized the enzyme structurally, and successfully co-expressed it with the oxygenase FMO in E. coli to produce up to 2.9 g/L indican in vivo from fed tryptophan. This bio-indican was shown to be an effective fabric dye when hydrolyzed by a β-glucosidase.
Addition of the glucosyl protecting group onto indoxyl confers several favorable properties. First, indican is stable in air until incubated with a β-glucosidase, providing control over when and where the indoxyl molecule will be deprotected for dyeing. Second, the hydrophilic glucosyl group keeps indican soluble in water, which is the preferred solvent of dye mills. Third, the stability of the gluco-side bond allows not only long-term storage but also concentration via gentle boiling of the crude fermentation broth. Thus, indican can be produced by batch, or eventually continuous, fermentation. Product can be purified or even used directly as concentrated spent media for dyeing cotton. A conventional leucoindigo bath is typically around 4 g/L (ref. 6), so a fermentation producing an equivalent concentration, based on indoxyl molarity, of ~9 g/L indican would not require additional concentration.
Glucosylation has previously been employed as a metabolic engineering tool to change the chemical and biological properties of the aglycone. For example, vanillin titers in Schizosaccharomyces pombe have been improved through glucosylation. Vanillin gluco-side has improved solubility and lower toxicity; consequently, it can be produced at considerably higher titers in S. pombe without causing growth defects46. In fact, several glucosides have been shown to be less toxic than their aglycone forms46,47. The glucose moiety can increase the water solubility of many metabolites; in addition, this modification may block either spontaneous or enzyme-catalyzed reactions (by obstructing substrate recognition) that produce toxic metabolites. Thus, glucosylation provides the metabolic engineer with a valuable tool for achieving desired biochemical properties of a molecule in the cell, similarly to how an organic chemist utilizes protecting groups to modify specific functional groups in an appropriate sequential manner. The ability to hydrolyze the glycosidic bond with a β-glucosidase enables reversible control, as the glucosyl group can be removed enzymatically when and where desired.
Incorporation of indican into a dye mill process poses some practical challenges that must be addressed. Although indican’s stability is advantageous for storing and shipping, it requires an enzyme or strong acid for hydrolysis. A strong acid is challenging to incorporate into the mill process, as the dyeing properties of leucoindigo are optimal at a basic pH, conventionally around pH 11 (refs. 7,48). Although the use of β-glucosidase to hydrolyze the glucosyl protecting group provides many advantages of control, there are a couple of challenges in adapting it into an industrial process. First, use of an enzyme increases the cost and process time of the dyeing procedure. Second, the use of a six-carbon glucosyl moiety as the protecting group means that a large portion of the feedstock carbon will not go into the final dye molecule, instead comprising a moiety that will ultimately be removed. Commercialization will require further improvements; however, it is reasonable to expect that these will be achievable. First, a more active and cheaply produced β-glucosidase must be obtained. As β-glucosidases are mass produced by filamentous fungi hosts such as Aspergillus niger for biofuel production from cellulosic material49,50, inexpensive industrial production of an engineered β-glucosidase seems feasible. Furthermore, engineering a high activity β-glucosidase for faster hydrolysis and activity under higher pH conditions should be possible by using the formation of visible indigo color for high-throughput screens. Similar screens can be employed for screening strain modifications increasing flux to indoxyl directly from glucose rather than relying on bioconversion from tryptophan.
The use of enzymes to add biochemical protecting groups site specifically to small molecules in the cell, with subsequent removal either inside or outside the cell, could prove as powerful to synthetic biologists as chemical protecting groups are for synthetic organic chemists. Indigo provides a motivating case study where nature has provided a blueprint. Following this model has provided a promising new dyeing strategy offering a much-needed update to the historic, but unsustainable, indigo dyeing process.
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
ONLINE METHODS
cDNA library construction
P. tinctorium plants (Companion Plants, Athens, Ohio) were grown in a laboratory environment. Samples of leaf tissue were taken from live plants at several points during the day and frozen in liquid nitrogen. These samples, a total of 300 mg, were mixed, kept frozen with liquid nitrogen, and crushed using 5 mm diameter steel beads in a beadbeater at 30 Hz for 2 min. Total RNA was extracted from 100 mg of powdered frozen leaf tissue using the Qiagen RNeasy Plant Mini kit obtained from Qiagen GMBH (Hilden, Germany). The UC Berkeley Functional Genomics Laboratory completed the rest of the cDNA preparation as follows: The mRNA was extracted from the total RNA using magnetic beads coated with oligo (dT)25. The mRNA was then sheared to approximately 550 base pairs in length using a Covaris S2 ultrasonicator from Covaris, Inc. (Woburn, MA). A cDNA library was generated using the Apollo 324 Next-Gen Library Prep System from WaferGen Biosystems, Inc. (Fremont, CA) using the manufacturer-supplied PrepX RNA-Seq Library Preparation Kit. The cDNA library was then clustered using the cBot from Illumina Inc. (San Diego, CA), and the clustered sample was loaded onto an Illumina HiSeq 2500 courtesy of the UC Berkeley Vincent J. Coates Genomics Sequencing Laboratory and sequenced using the Rapid Run reagent kit for 150 base, paired-end reads.
Transcriptomics
Paired-end reads received from the Illumina HiSeq 2500 sequencer were first trimmed to remove low-quality reads using the Trimmomatic software package51 in paired-end mode to remove Illumina adaptor sequences and using a sliding quality window of 30 or greater, whereby reads with under 36 acceptable bases are dropped. Overlapping paired-end reads were then merged using the FLASH software package52 with a minimum overlap size of 15 bases and an expected fragment length of 350 bases. The remaining merged and unmerged reads were pooled and digitally normalized to remove redundant data using the khmer software package53 with options set to paired-end, k-mer size of 19, culling count of 20, and 4 hash tables of 4 GiB each. Following digital normalization, reads were assembled into transcript scaffolds using the Trinity RNA-seq assembly package24 in paired-end mode or Oases using k-mer sizes between 17 and 31 (ref. 54). Scaffolds were then annotated using BLASTX against a library of known plant glycosyltransferases retrieved from UniProt with an E-value threshold of 1e-60 to identify glucosyltransferase candidates. These candidates were then translated into peptides using the Trinity package, Transdecoder, using the default settings24. This work was done on the XSEDE Blacklight system55.
UDP-glucose:indoxyl glucosyltransferase (UGIG) purification
The UGIG purification protocol was adapted from Minami et al.16. 200 g leaves were harvested from P. tinctorium plants grown in a greenhouse, flash frozen in liquid nitrogen, and ground to a fine powder. 400 mL extraction buffer (100 mM potassium phosphate pH 7.0, 2 mM EDTA, 20 mM β-mercaptoethanol (BME), 1 tablet cOmplete protease inhibitor (obtained from Roche)) was added to the ground leaves, and the mixture was thawed at 4 °C for 30 min. The slurry was then centrifuged twice at 24,000 × g for 30 min at 4 °C. Three parts PEG solution (50% (w/v) PEG 6000, 100 mM potassium phosphate pH 7.0, 2 mM EDTA, 20 mM BME) was added to two parts supernatant, and the mixture was centrifuged at 143,000 × g. The supernatant was dialyzed against equilibration buffer (50 mM HEPES pH 7.0, 1 mM EDTA, 10 mM BME).
The supernatant was run through DEAE Sepharose Fast Flow beads (GE Healthcare) and eluted with a solution of 50 mM HEPES pH 7.0, 100 mM NaCl, 1 mM EDTA, and 10 mM BME. Fractions were assayed for the presence of a UGIG (see “Glucosyltransferase assay for purification”), as they were for all subsequent chromatography steps. The desired fractions were then run through hydroxyapatite beads (Sigma), eluting with buffer containing 10 mM potassium phosphate pH 7.0, 50 mM HEPES-NaOH pH 7.0, 1 mM EDTA, 5 mM DTT, and 10% v/v glycerol. The fractions showing glucosyltransferase activity were then applied to a Mono Q 5/50 GL column (GE Healthcare) and eluted with a NaCl gradient from 0 to 200 mM. Lastly, the protein was concentrated using a 10,000 MWCO spin concentrator and run through a Superdex 200 10/300 GL column (GE Healthcare), eluting with a buffer made of 50 mM HEPES pH 7.0 and 5 mM DTT.
The protein preparation was electrophoresed on a NuPAGE Novex 4–12% Bis–Tris Protein Gel (Life Technologies) at 140 V for 75 min. The sequential rounds of chromatography yielded a highly enriched protein around 52 kDa in weight, similar to the previous report16. The band on the gel corresponding to the glucosyltransferase was extracted for trypsin digestion and HPLC separation followed by tandem mass spectrometry, carried out by the UC Berkeley Vincent J. Coates Proteomics/Mass Spectrometry Laboratory, to identify protein fragments. These protein fragments were correlated to the Transdecoder-predicted, BLASTX-limited sequences (see “Transcriptomics”) using SEQUEST and DTAselect56. The sequences with the most complete coverage of matching peptide fragments were selected for further study.
Glucosyltransferase assay for purification
After each purification step, a glucosyltransferase assay was performed using a protocol adapted from that of Minami et al.16. 100 μL protein sample was incubated in 100 mM glycine-NaOH pH 9, with 2 mM UDP-glucose, 2 mM indoxyl phosphate, 0.1 mg/mL potato acid phosphatase, 100 mM DTT, and 10 mM ascorbic acid, in a total volume of 200 μL. The mixture was incubated at 37 °C for 3 h. The reaction was terminated by adding 140 μL cold 40% (w/v) trichloroacetic acid, centrifuging at max speed for 5 min, and then adding 200 μL 500 mM NaOH. The sample was assayed for the presence of indican by LC–MS (see “Determination of metabolite concentrations”).
Plasmids, strains, and growth media
Plasmids were constructed using a MoClo Golden Gate Assembly57 and propagated using E. coli strain TG1 (Lucigen). Strains for plasmid construction were grown in Luria Broth (LB) selected on 34 mg/L chloramphenicol, 100 mg/L ampicillin, and/or 25 mg/L kanamycin.
Unless otherwise noted, final production strains are based on E. coli strain MG1655 containing the DE3 prophage (F− ilvG− rfb-50 rph-1 λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5])), provided by the M. Chang laboratory (University of California, Berkeley). Genomic integrations to introduce heterologous genes were done by λ red recombination58 immediately downstream of base 44344920 in the E. coli genome. Oligos 1 and 2 (Supplementary Table 1) were used to amplify the gene of interest for integration. bglA was knocked out of the genome by replacing the open reading frame with a KanR antibiotic marker. KanR, flanked by FRT sites, was amplified from the ΔbglA Keio knockout strain (background: BW25113)44 using Oligos 3 and 4. This PCR product was integrated into MG1655(DE3) and selected on LB with kanamycin. The KanR gene was subsequently removed using pFLP2 (ref. 59), and the plasmid was cured from the strain with 5% sucrose. All strains and plasmids used in this work are listed in Supplementary Tables 2 and 3.
UGT crystallographic structure solution
Expression and purification of PtUGT1 for crystallization
Rosetta(DE3) cells (Novagen) were transformed with pTMH307 and selected on LB agar plates containing chloramphenicol and ampicillin. Overnight cultures were diluted into 4 L Terrific Broth (Fisher) with ampicillin selection, grown at 37 °C in an Innova 44 shaker (New Brunswick Scientific) at 200 r.p.m., and induced with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at OD600 ~3. Cultures were then grown at 18 °C for 21 h for protein expression, and the cells were harvested by centrifugation. The cell pellets were resuspended in 50 mM HEPES pH 7.0, 300 mM NaCl, 25 mM imidazole pH 8.0, and 1 mM DTT. The cell suspension was lysed by freeze/thaw and sonication. Lysate was purified using Ni-NTA agarose beads (Qiagen), and the protein was dialyzed against 25 mM HEPES pH 7.0, 50 mM NaCl, and 1 mM DTT. The N-terminal 6×His tag was TEV-cleaved, and the final purified protein was concentrated to 15 mg/mL using a 30,000 MWCO Amicon Ultra-15 Centrifugal Filter Unit (EMD Millipore). Protein concentration was determined using the Bradford assay in triplicate (Bio-Rad).
Crystallographic structure determination and analysis
PtUGT1 was co- crystallized with indoxyl sulfate (Sigma-Aldrich I3875) in sitting drops consisting of 0.2 μL protein solution (15 mg/mL in 25 mM HEPES pH 7.0, 50 mM NaCl, 1 mM DTT, 1 mM indoxyl sulfate) and 0.2 μL crystallization buffer (MCSG-1 screen (Anatrace) solution 9: 0.2 M MgCl2●6H2O, 0.1 M HEPES pH 7.5, 25% PEG 3350). The drops were set up with a Phoenix crystallization robot in 3-drop Intelli-Plate 96-well crystallization plates (Art Robbins Instruments). Crystals appeared after 2 d and grew to their final size after ~4 d. These crystals were cryoprotected in 10% glycerol and mounted in a nylon cryoloop (Hampton Research). 270° of data were collected at 100 K, 1.0000 nm, at the Berkeley Center for Structural Biology beamline 5.0.3 of the Advanced Light Source in Berkeley, California with a 1° oscillation and 5 s exposure time and a ADSC Q315r CCD detector. The data were processed with Xia2 (ref. 60) and XDS61. The structure was solved by molecular replacement using PDB ID 2VCE31 as a search model and Phaser62 from the Phenix software package63. The structure was rebuilt and refined using phenix.refine64 and Coot65 to a final R-factor of 22.5 and an Rfree of 25.2, with 94.74% of the residues in the favored region of the Ramachandran plot, 5.04% in the allowed region, and 0.22% outliers. The final structure was validated with MolProbity66 and deposited in the Protein Data Bank with PDB ID 5NLM. Data collection and refinement statistics are reported in Supplementary Table 4. Figures were prepared with the MacPyMOL Molecular Graphics System, Version 1.0 Schrödinger, LLC. PtUGT1–indoxyl sulfate interactions were analyzed with the Protein–Ligand Interaction Profiler (PLIP)67 using default distance cut-offs. Structural homology searches were performed with DaliLite v.3 (ref. 68).
Glucosyltransferase assay for enzymology
Rosetta (DE3) cells were transformed with pTMH307, pTMH308, pTMH634, or pTMH635, and selected on LB agar plates containing chloramphenicol and ampicillin. PtUGT1 and PtUGT2 were subsequently expressed and purified as described in “Expression and purification of PtUGT1 for crystallization” (Supplementary Fig. 11 for SDS–PAGE gel). The enzymes were stored in the final buffer with 10% glycerol and flash frozen in liquid nitrogen for storage.
The activity assay consisted of two parts: first, indoxyl was produced and quantified; second, the glucosyltransferase’s activity on indoxyl was quantified. To produce and quantify indoxyl, 100 mM indoxyl phosphate and 10 mg/mL potato acid phosphatase (both obtained from Sigma) were mixed 2:1 in an anaerobic chamber and allowed to react for 75 min at 30 °C. Two-fold serial dilutions of the indoxyl mixture were then made, and 50 μL of each indoxyl dilution was taken in triplicate to run a malachite green assay69, an assay that determines the concentration of free phosphate, and thus, free indoxyl. The indoxyl dilutions were first diluted by powers of 10 in water as necessary to keep the malachite green signal within the linear range of the assay. The Glycosyltransferase Activity Kit (R&D Systems), based on the malachite green phosphate assay, was used: 30 μL malachite green reagent A (containing ammonium molybdate in 3 M sulfuric acid), 100 μL water, and 30 μL malachite green reagent B (containing malachite green oxalate and polyvinyl alcohol) were sequentially added to 50 μL of each indoxyl dilution. The reactions were incubated for 20 min, and absorbance at 620 nm was measured on an Infinite M1000 PRO microplate reader (Tecan) and converted to indoxyl concentration by comparison against a six-point standard curve of two-fold dilutions of potassium phosphate monobasic (range: 3.1–100 μM).
The rest of the indoxyl dilutions were used in the indoxyl glucosyltransferase activity assay, taking place in an anaerobic chamber. A 100-μL reaction mixture was prepared in triplicate per dilution, consisting of 50 mM HEPES pH 7.8, 50 mM NaCl, 10 mM DTT, 5 mM UDP-glucose, and 20 nM PtUGT1 or PtUGT2 enzyme, and including 15 μL of the indoxyl solution. The reactions were carried out for 10 min at 30 °C, and they were quenched with 100 μL 100 mM NaOH.
The reactions were spun down and run on a 6520 Accurate-Mass Q-TOF LC–MS (Agilent). Indican was detected by LC–MS as described in “LC–MS detection and quantification of indican,” and indican concentration was determined by comparison against a six-point indican standard curve (Sigma, ≥97% pure) ranging from 8.3 to 265 μM. Michaelis–Menten graphs and turnover numbers were generated by Prism (GraphPad Software, Inc.). The KM for the two PtUGT enzymes cannot be determined with accuracy because it is not possible to quantify with certainty the concentration of the indoxyl substrate because of low levels of oxidation, even under anaerobic conditions.
Small-scale assays for indican and indigo
Cultures were grown to saturation in MOPS EZ Rich Defined Medium (Teknova, ingredients listed in Supplementary Table 5), with kanamycin selection and 2% glycerol in place of the typical 0.2% glucose as the carbon source. Cells were diluted to OD600 0.1 in fresh media containing 1 mM IPTG (if strain contains T7 promoter), and indican or tryptophan as described in the text. They were grown at 37 °C in a Multitron shaker (INFORS HT) at 750 r.p.m. for 24 h (or as described in the text, if otherwise).
β-glucosidase purification
Rosetta (DE3) cells were transformed with pRLP121 and selected on LB agar plates containing chloramphenicol and ampicillin. BglA was subsequently expressed and purified as described in “Expression and purification of PtUGT1 for crystallization.” The enzyme was stored in the final buffer with 10% glycerol at a concentration of 2 mg/mL and flash frozen in liquid nitrogen for storage at −80 °C.
Shake-flask time course fermentations
Cultures were grown to saturation in MOPS EZ Rich Defined Medium (Teknova), with kanamycin selection and 2% glycerol in place of the typical 0.2% glucose as the carbon source. Cells were diluted to OD600 0.05 in 50 mL fresh media containing 26 mM tryptophan and 1 mM IPTG. They were grown for 37 h at 37 °C in 250-mL baffled shake flasks, in an Innova 44 shaker (New Brunswick Scientific) set to 200 r.p.m. Aliquots were taken at time points, to determine metabolite concentration or cell count.
Determination of metabolite concentrations
LC-MS detection and quantification of indican
Indican was detected by LC–MS/MS using a 6520 Accurate-Mass Q-TOF LC-MS (Agilent). Cultures were filtered through a 0.2 μm filter and diluted 10-fold in water. Five microliters of sample were injected onto a ZORBAX Eclipse Plus C18 4.6 × 100 mm 3.5 μm column (Agilent) using a flow rate of 0.5 mL/min. The solvents were water with 0.05% ammonium hydroxide and acetonitrile with 0.05% ammonium hydroxide. The column was flushed with 95% water/5% acetonitrile plus 0.05% ammonium hydroxide for 3 min, before an elution with a linear gradient to 2% water/98% acetonitrile plus 0.05% ammonium hydroxide over 11 min. Indican (m/z 294.098 [M-H]−, retention time (Rt) 10.0 min) was ionized by electrospray ionization in negative mode using a fragmentor voltage of 100 V, and for MS/MS, fragmented again using collision-induced dissociation (CID) at 10 V. The LC-MS trace was integrated in MZmine2 (http://mzmine.github.io). For quantification of indican, traces were compared against a seven-point indican standard curve (Sigma, ≥97%) ranging from 0 to 2.5 g/L.
HPLC quantification of indican and tryptophan
Indican and tryptophan concentrations were quantified by absorbance at 280 nm via diode array detector on a 1260 Infinity LC System (Agilent). Five microliters of sample were injected onto the column as described in “LC-MS detection and quantification of indican”, using the same solvents and gradient as before (tryptophan Rt 5 min, indican Rt 9.7 min). Integrations were done in ChemStation (Agilent). Indican content in the samples were compared against a 10-point standard curve made of twofold dilutions of indican (Sigma, ≥97%), ranging from 0.01 to 5 g/L. Tryptophan content in the samples were compared against a ten-point standard curve made of two-fold dilutions of tryptophan (Sigma, ≥99%), ranging from 0.05 to 25 mM.
Extraction and quantification of indigo
Cultures were spun down at max speed and the supernatant removed. The pellet was resuspended in 100% DMSO (Sigma) and pelleted again. Supernatant absorbance was measured at 620 nm on an Infinite M1000 PRO microplate reader (Tecan) and converted to indigo concentration by an 8-point standard curve of commercially available indigo (Spectrum Chemical) dissolved in DMSO, ranging from 2 to 250 μM.
Determination of cell count
Indigo absorbs light at 600 nm, so OD600 was not an appropriate method to determine cell density. Instead, 10 μL of culture underwent repeated tenfold dilutions in LB broth, and 5 μL of each dilution was spotted onto an LB agar plate with kanamycin selection. The most densely populated spot with distinct colonies was counted, and the colony forming units per 1 mL original culture was back-calculated.
Production of dye reagents
β-glucosidase lysate production
Rosetta(DE3) cells transformed with pRLP121 were grown in 4 L Terrific Broth and induced with 1 mM IPTG as described in “Expression and purification of PtUGT1 for crystallization.” The culture was spun down, and the cell pellet was resuspended in 250 mL 25 mM HEPES pH 7.8, 50 mM NaCl, 1 mM DTT, and 5 tablets cOmplete protease inhibitor (Roche). The cells were lysed by freeze/thaw and sonication. Lysate was spun down at 4 °C at 4,800g, and the crude soluble protein was directly used for dyeing.
Indican production in bioreactor
TMH011 cells were grown to saturation in MOPS EZ Rich Defined Medium (Teknova), with kanamycin selection and 2% glycerol in place of the typical 0.2% glucose as the carbon source. Cells were diluted to OD600 0.1 in 5 L fresh media containing 20 mM (4 g/L) tryptophan and grown in a 14-L reactor vessel on a BioFlo/CelliGen 115 bioreactor (New Brunswick Scientific). The temperature was maintained at 37 °C and the dissolved oxygen level was maintained at 20%. The pH was maintained at 6.8, using 14% ammonium hydroxide as the base source. 1 mM IPTG was added at 3.5 h. Aliquots were taken at time points to monitor metabolite concentration and cell count.
At 40 h, the culture was evacuated from the bioreactor and spun down twice. The pellets were discarded, and the pH of the media was adjusted to 8 using NaOH, to minimize acid hydrolysis during the concentration process. The media was then boiled gently on a stir plate set to 150 °C until the volume decreased from 5 L to 1 L.
Dyeing with indican vs. indigo
Swatches
Concentrated indican media was sprayed onto both sides a 4 cm × 4 cm square of white cotton denim fabric (gift from Levi Strauss & Co.) until the cloth was wet. 500 μL BglA lysate was applied to the swatch for 1 min, then the cloth was let drip dry and allowed to oxidize in air. A sodium dithionite-reduced indigo vat was made as a comparison, using a protocol adapted from McKee and Zanger70. Three pellets of NaOH were dissolved with 140 mg indigo (Spectrum Chemical) in 10 mL water and heated to boiling on a stir plate. 2 mL 10% sodium dithionite was added, and the mixture was stirred until the indigo reduced and dissolved. This solution was poured into 88 mL room temperature water and stirred to mix. The denim fabric swatches were dipped into this dye bath for 1 min, let drip dry, and let oxidize in air until dry. All swatches were vigorously washed in water and dried before photographing.
Scarf
Concentrated indican media and BglA lysate were placed in separate spray bottles. A 23” × 72” piece of white cotton gauze scarf was draped crosswise on a string and pre-wet by lightly spraying with water. Indican was sprayed onto the scarf until both sides were evenly covered, and then BglA was similarly sprayed onto the fabric until covered. The indican was allowed to hydrolyze and oxidize for 30 min before repeating the sprays. After three such dye cycles, the scarf was flipped on the string so the inside surface was on the outside, and three more dye cycles took place. The scarf was dried overnight and washed vigorously in water (Fig. 5b). The scarf was then laundered in a commercial laundry machine with a full capsule of All Laundry Detergent (Sun Products) (Supplementary Fig. 10).
Statistics
Sample sizes are described in the figure legend of the appropriate experiment. Error bars represent the mean ± 1 s.d. Individual data points represent biological replicates.
Life sciences reporting summary
Further information on experimental design and reagents is available in the Life Sciences Reporting Summary.
Data availability
DNA sequences in this work have been deposited in GenBank with accession codes MF688770, MF688771, MF688772, MF688773, MF688774, and MF688775. The crystal structure of PtUGT1 has been deposited in the RCSB Protein Data Bank with accession code 5NLM.
Supplementary Material
Acknowledgments
The authors thank the rest of the Berkeley iGEM 2013 team, C. Somerville, N. Sorek, S. Bauer, N. Harris, D. Savage, B. Sights, S. Wagner, and the Dueber Lab, especially S. Bhakta, D. Stanley, L. Latimer, P. Grewal, and S. Halperin, for valuable discussions and experimental assistance. The UC Berkeley Vincent J. Coates Genomics Sequencing Laboratory and Proteomics/Mass Spectrometry Laboratory provided transcriptome and protein sequencing. The M. Chang laboratory (University of California, Berkeley) provided the base E. coli strain. This work was supported by Bakar Fellows Program (fellowship to J.E.D.), NSF CBET 1605465 (J.E.D.), a generous gift from Levi Strauss & Co. (J.E.D.), the US Department of Defense (fellowship to T.M.H. and Z.N.R.) and Agilent (iGEM undergraduate fellowships). Crystallographic experiments were performed as part of the DOE Joint BioEnergy Institute (http://www.jbei.org) which is supported by the US Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the US Department of Energy. We thank the Berkeley Center for Structural Biology beamline staff for technical assistance during data collection. The BCSB is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the US Department of Energy under Contract No. DE-AC02-05CH11231.
Footnotes
Author contributions
T.M.H., Z.N.R., and J.E.D. designed the research. T.M.H., D.H.W., Z.N.R., B.C., and R.L.P. collected data. D.H.W. solved the crystallographic structure. T.M.H., D.H.W., Z.N.R., P.D.A., and J.E.D. analyzed the data. T.M.H., D.H.W., and J.E.D. wrote the paper.
Competing financial interests
The authors declare competing financial interests: details accompany the online version of the paper.
Additional information
Any supplementary information, chemical compound information and source data are available in the online version of the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations. Correspondence and requests for materials should be addressed to J.E.D.
References
- 1.Splitstoser JC, Dillehay TD, Wouters J, Claro A. Early pre-Hispanic use of indigo blue in Peru. Sci Adv. 2016;2:e1501623. doi: 10.1126/sciadv.1501623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balfour-Paul J. Indigo. Firefly Books; 2011. [Google Scholar]
- 3.Wolf LK. What’s That Stuff? Blue Jeans. Chem Eng News. 2011;89:44. [Google Scholar]
- 4.Schimper CB, Ibanescu C, Bechtold T. Surface activation of dyed fabric for cellulase treatment. Biotechnol J. 2011;6:1280–1285. doi: 10.1002/biot.201100002. [DOI] [PubMed] [Google Scholar]
- 5.Pfleger J. Process of making indoxyl derivatives. 680,395. US Patent. 1901
- 6.Paul R. Denim. Elsevier Ltd; 2015. [Google Scholar]
- 7.Blackburn RS, Bechtold T, John P. The development of indigo reduction methods and pre-reduced indigo products. Color Technol. 2009;125:193–207. [Google Scholar]
- 8.Ensley BD, et al. Expression of naphthalene oxidation genes in Escherichia coli results in the biosynthesis of indigo. Science. 1983;222:167–169. doi: 10.1126/science.6353574. [DOI] [PubMed] [Google Scholar]
- 9.Murdock D, Ensley BD, Serdar C, Thalen M. Construction of metabolic operons catalyzing the de novo biosynthesis of indigo in Escherichia coli. Nat Biotechnol. 1993;11:381–386. doi: 10.1038/nbt0393-381. [DOI] [PubMed] [Google Scholar]
- 10.Berry A, Dodge TC, Pepsin M, Weyler W. Application of metabolic engineering to improve both the production and use of biotech indigo. J Ind Microbiol Biotechnol. 2002;28:127–133. doi: 10.1038/sj.jim.7000228. [DOI] [PubMed] [Google Scholar]
- 11.Han GH, et al. Bio-indigo production in two different fermentation systems using recombinant Escherichia coli cells harboring a flavin-containing monooxygenase gene (fmo) Process Biochem. 2011;46:788–791. [Google Scholar]
- 12.Padden AN, et al. An indigo-reducing moderate thermophile from a woad vat, Clostridium isatidis sp. nov. Int J Syst Bacteriol. 1999;49:1025–1031. doi: 10.1099/00207713-49-3-1025. [DOI] [PubMed] [Google Scholar]
- 13.Yumoto I, et al. Alkalibacterium psychrotolerans sp nov., a psychrotolerant obligate alkaliphile that reduces an indigo dye. Int J Syst Evol Microbiol. 2004;54:2379–2383. doi: 10.1099/ijs.0.63130-0. [DOI] [PubMed] [Google Scholar]
- 14.Gäng M, Krüger R, Miederer P. Concentrated leucoindigo solutions. 6,428,581. US Patent. 2002
- 15.Roessler A, Crettenand D, Dossenbach O, Rys P. Electrochemical reduction of indigo in fixed and fluidized beds of graphite granules. J Appl Electrochem. 2003;33:901–908. [Google Scholar]
- 16.Minami Y, Nishimura O, Hara-Nishimura I, Nishimura M, Matsubara H. Tissue and intracellular localization of indican and the purification and characterization of indican synthase from indigo plants. Plant Cell Physiol. 2000;41:218–225. doi: 10.1093/pcp/41.2.218. [DOI] [PubMed] [Google Scholar]
- 17.Minami Y, et al. β-Glucosidase in the indigo plant: intracellular localization and tissue specific expression in leaves. Plant Cell Physiol. 1997;38:1069–1074. doi: 10.1093/oxfordjournals.pcp.a029273. [DOI] [PubMed] [Google Scholar]
- 18.Dang TTT, Chen X, Facchini PJ. Acetylation serves as a protective group in noscapine biosynthesis in opium poppy. Nat Chem Biol. 2015;11:104–106. doi: 10.1038/nchembio.1717. [DOI] [PubMed] [Google Scholar]
- 19.Chen J, et al. Biosynthesis of the active compounds of Isatis indigotica based on transcriptome sequencing and metabolites profiling. BMC Genomics. 2013;14:857. doi: 10.1186/1471-2164-14-857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang X, et al. High-throughput sequencing and De Novo assembly of the Isatis indigotica transcriptome. PLoS One. 2014;9:e102963–e102968. doi: 10.1371/journal.pone.0102963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Minami Y, Sarangi BK, Thul ST. Transcriptome analysis for identification of indigo biosynthesis pathway genes in Polygonum tinctorium. Biologia. 2015;70:1026–1032. doi: 10.1016/j.gdata.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.John P. In: Handbook of Natural Colorants. Bechtold T, Mussak R, editors. John Wiley and Sons; 2009. Ch 8. [Google Scholar]
- 23.Gilbert KG, et al. Quantitative analysis of indigo and indigo precursors in leaves of Isatis spp. and Polygonum tinctorium. Biotechnol Prog. 2004;20:1289–1292. doi: 10.1021/bp0300624. [DOI] [PubMed] [Google Scholar]
- 24.Grabherr MG, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackenzie PI, et al. The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence. Pharmacogenetics. 1997;7:255–269. doi: 10.1097/00008571-199708000-00001. [DOI] [PubMed] [Google Scholar]
- 26.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:D490–D495. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 28.Osmani SA, Bak S, Møller BL. Substrate specificity of plant UDP-dependent glycosyltransferases predicted from crystal structures and homology modeling. Phytochemistry. 2009;70:325–347. doi: 10.1016/j.phytochem.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 29.Wang X. Structure, mechanism and engineering of plant natural product glycosyltransferases. FEBS Lett. 2009;583:3303–3309. doi: 10.1016/j.febslet.2009.09.042. [DOI] [PubMed] [Google Scholar]
- 30.Loutre C, et al. Isolation of a glucosyltransferase from Arabidopsis thaliana active in the metabolism of the persistent pollutant 3,4-dichloroaniline. Plant J. 2003;34:485–493. doi: 10.1046/j.1365-313x.2003.01742.x. [DOI] [PubMed] [Google Scholar]
- 31.Brazier-Hicks M, et al. Characterization and engineering of the bifunctional N- and O-glucosyltransferase involved in xenobiotic metabolism in plants. Proc Natl Acad Sci USA. 2007;104:20238–20243. doi: 10.1073/pnas.0706421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakamura CE, Whited GM. Metabolic engineering for the microbial production of 1,3-propanediol. Curr Opin Biotechnol. 2003;14:454–459. doi: 10.1016/j.copbio.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 33.Yim H, et al. Metabolic engineering of Escherichia coli for direct production of 1,4-butanediol. Nat Chem Biol. 2011;7:445–452. doi: 10.1038/nchembio.580. [DOI] [PubMed] [Google Scholar]
- 34.Patnaik R, Liao JC. Engineering of Escherichia coli central metabolism for aromatic metabolite production with near theoretical yield. Appl Environ Microbiol. 1994;60:3903–3908. doi: 10.1128/aem.60.11.3903-3908.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malla S, Pandey RP, Kim BG, Sohng JK. Regiospecific modifications of naringenin for astragalin production in Escherichia coli. Biotechnol Bioeng. 2013;110:2525–2535. doi: 10.1002/bit.24919. [DOI] [PubMed] [Google Scholar]
- 36.Lim CG, et al. Development of a recombinant Escherichia coli strain for overproduction of the plant pigment anthocyanin. Appl Environ Microbiol. 2015;81:6276–6284. doi: 10.1128/AEM.01448-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Choi HS, et al. A novel flavin-containing monooxygenase from Methylophaga sp. strain SK1 and its indigo synthesis in Escherichia coli. Biochem Biophys Res Commun. 2003;306:930–936. doi: 10.1016/s0006-291x(03)01087-8. [DOI] [PubMed] [Google Scholar]
- 38.Anderson JC, et al. BglBricks: a flexible standard for biological part assembly. J Biol Eng. 2010;4:1. doi: 10.1186/1754-1611-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li G, Young KD. A cAMP-independent carbohydrate-driven mechanism inhibits tnaA expression and TnaA enzyme activity in Escherichia coli. Microbiology. 2014;160:2079–2088. doi: 10.1099/mic.0.080705-0. [DOI] [PubMed] [Google Scholar]
- 40.Botsford JL, DeMoss RD. Catabolite repression of tryptophanase in Escherichia coli. J Bacteriol. 1971;105:303–312. doi: 10.1128/jb.105.1.303-312.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minami Y, Kanafuji T, Miura K. Purification and characterization of a β-glucosidase from Polygonum tinctorium, which catalyzes preferentially the hydrolysis of indican. Biosci Biotechnol Biochem. 1996;60:147–149. [Google Scholar]
- 42.Song J, Imanaka H, Imamura K, Kajitani K, Nakanishi K. Development of a highly efficient indigo dyeing method using indican with an immobilized β-glucosidase from Aspergillus niger. J Biosci Bioeng. 2010;110:281–287. doi: 10.1016/j.jbiosc.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 43.Kim JY, Lee JY, Shin YS, Kim GJ. Characterization of an indican-hydrolyzing enzyme from Sinorhizobium meliloti. Process Biochem. 2010;45:892–896. [Google Scholar]
- 44.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Paavilainen S, Hellman J, Korpela T. Purification, characterization, gene cloning, and sequencing of a new β-glucosidase from Bacillus circulans subsp. alkalophilus. Appl Environ Microbiol. 1993;59:927–932. doi: 10.1128/aem.59.3.927-932.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen EH, et al. De novo biosynthesis of vanillin in fission yeast (Schizosaccharomyces pombe) and baker’s yeast (Saccharomyces cerevisiae) Appl Environ Microbiol. 2009;75:2765–2774. doi: 10.1128/AEM.02681-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moehs CP, Allen PV, Friedman M, Belknap WR. Cloning and expression of solanidine UDP-glucose glucosyltransferase from potato. Plant J. 1997;11:227–236. doi: 10.1046/j.1365-313x.1997.11020227.x. [DOI] [PubMed] [Google Scholar]
- 48.Etters JN. Advances in indigo dyeing: implications for the dyer, apparel manufacturer and environment. Text Chem Color. 1995;27:17–22. [Google Scholar]
- 49.Sternberg D, Vijayakumar P, Reese ET. β-Glucosidase: microbial production and effect on enzymatic hydrolysis of cellulose. Can J Microbiol. 1977;23:139–147. doi: 10.1139/m77-020. [DOI] [PubMed] [Google Scholar]
- 50.Jäger S, Brumbauer A, Fehér E, Réczey K, Kiss L. Production and characterization of β-glucosidases from different Aspergillus strains. World J Microbiol Biotechnol. 2001;17:455–461. [Google Scholar]
- 51.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Magoč T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crusoe MR, et al. The khmer software package: enabling efficient nucleotide sequence analysis. F1000 Res. 2015;4:900. doi: 10.12688/f1000research.6924.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–1092. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Towns J, et al. XSEDE: accelerating scientific discovery. Comput Sci Eng. 2014;16:62–74. [Google Scholar]
- 56.Tabb DL, McDonald WH, Yates JR., III DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee ME, DeLoache WC, Cervantes B, Dueber JE. A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth Biol. 2015;4:975–986. doi: 10.1021/sb500366v. [DOI] [PubMed] [Google Scholar]
- 58.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene. 1998;212:77–86. doi: 10.1016/s0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 60.Winter G. xia2: an expert system for macromolecular crystallography data reduction. J Appl Crystallogr. 2010;43:186–190. [Google Scholar]
- 61.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCoy AJ, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M. PLIP: fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015;43:W443–W447. doi: 10.1093/nar/gkv315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 2010;38:W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Itaya K, Ui M. A new micromethod for the colorimetric determination of inorganic phosphate. Clin Chim Acta. 1966;14:361–366. doi: 10.1016/0009-8981(66)90114-8. [DOI] [PubMed] [Google Scholar]
- 70.McKee JR, Zanger M. A microscale synthesis of indigo: vat dyeing. J Chem Educ. 1991;68:A242. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA sequences in this work have been deposited in GenBank with accession codes MF688770, MF688771, MF688772, MF688773, MF688774, and MF688775. The crystal structure of PtUGT1 has been deposited in the RCSB Protein Data Bank with accession code 5NLM.